")
Back to Journals » OncoTargets and Therapy » Volume 8
Astroblastoma: beside being a tumor entity, an occasional phenotype of astrocytic gliomas?
Authors Mellai M, Piazzi A, Casalone C, Grifoni S, Melcarne A, Annovazzi L, Cassoni P, Denysenko T, Valentini MC, Cistaro A, Schiffer D
Received 19 July 2014
Accepted for publication 10 September 2014
Published 19 February 2015 Volume 2015:8 Pages 451—460
DOI https://doi.org/10.2147/OTT.S71384
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Faris Farassati
Marta Mellai,1 Angela Piazzi,1 Cristina Casalone,2 Silvia Grifoni,2 Antonio Melcarne,3 Laura Annovazzi,1 Paola Cassoni,4 Tetyana Denysenko,1 Maria Consuelo Valentini,5 Angelina Cistaro,6,7 Davide Schiffer1
1Neuro-Bio-Oncology Center, Policlinico di Monza Foundation/Consorzio di Neuroscienze, University of Pavia, Vercelli, Italy; 2Istituto Zooprofilattico Sperimentale del Piemonte, Liguria e Valle d'Aosta, Turin, Italy; 3Department of Neurosurgery, CTO Hospital/Città della Salute e della Scienza, Turin, Italy; 4Department of Medical Sciences, University of Turin, Turin, Italy; 5Department of Neuroradiology, CTO Hospital/Città della Salute e della Scienza, Turin, Italy; 6Positron Emission Tomography Center IRMET S.p.A, Euromedic Inc., Turin, Italy; 7Institute of Cognitive Sciences and Technologies, National Research Council, Rome, Italy
Abstract: The diagnosis of astroblastoma is based on a typical histological aspect with perivascular distribution of cells sending cytoplasmic extensions to the vessels and vascular hyalinization. These criteria are useful for standardizing the identification of the tumor, but, in spite of this, there are discrepancies in the literature concerning the age distribution and the benign or malignant nature of the tumor. Three cases are discussed in this study: Case 1 was a typical high-grade astroblastoma; Case 2 was an oligodendroglioma at the first intervention and an oligoastrocytoma at the second intervention with typical perivascular arrangements in the astrocytic component; Case 3 was a gemistocytic glioma with malignant features and typical perivascular arrangements. Genetic analysis showed genetic alterations that are typical of gliomas of all malignancy grades. Using the neurosphere assay, neurospheres and adherent cells were found to have developed in Case 1, while adherent cells only developed in Case 2, in line with the stemness potential of the tumors. The cases are discussed in relation to their diagnostic assessment as astroblastoma, and it is hypothesized that the typical perivascular distribution of cells may not indicate a separate and unique tumor entity, but may be a peculiarity that can be acquired by astrocytic gliomas when an unknown cause from the tumor microenvironment influences the relationship between vessels and tumor cells.
Keywords: gliomas, cell lines, histology, genetics
Introduction
Astroblastoma has been, and still is, an often-discussed tumor entity with affinity to ependymoma. Its characteristics have been recognized to be the perivascular arrangement of astrocytic tumor cells with broad processes on the vessel walls and vascular hyalinization. Since its first description by Bailey,1 the literature on brain tumor classification has categorized astroblastoma as follows: as a stage in the process of glioma dedifferentiation2 and as an astrocytoma of large cells producing fibers3 or as a rare tumor, likely originating from tanycytes or ependymal astrocytes, as shown by electron microscopy.4,5 Tumor descriptions in the literature only concern individual cases or small collections of cases.4–7
Astroblastoma has been prevailingly regarded as a high-grade neoplasm,8 but, in a recent review, 5-year survival rates were found in 95% of cases.9 Therefore, it could be more frequently a low-grade tumor.10 As a matter of fact, both benign and malignant neoplasia have been described, with or without mitoses, endothelial proliferations, and necrosis.11 Astroblastoma has been considered to range between astrocytoma and glioblastoma multiforme (GBM).12 The main feature of the tumor is represented by both perivascular processes, specifically unipolar cytoplasmic processes anchoring tumor cells to vessels,13 different from those of ependymal pseudorosettes, and vascular hyalinization. In recently described cases,14–16 the perivascular arrangement of cells and vessel hyalinization have been confirmed as typical features of this tumor. The TP53 and isocitrate dehydrogenase 1 and 2 (IDH1/2) genes have been found to be wild type, and the occurrence of microvilli has been ultrastructurally observed.17 The main differential diagnoses of astroblastoma are ependymoma and angiocentric glioma. Ependymoma can be distinguished because of the lack of fibrillarity10 in astroblastoma; with angiocentric glioma, the distinction is not clear-cut, because this is an ill-defined tumor entity, characterized by perivascular distribution of cells not otherwise specified, which is benign and occurring in children. At worst, astroblastoma might be regarded as an angiocentric glioma.18
Based on the data of 95 patients in the literature, astroblastoma location was found to be prevailingly supratentorial (87 supratentorial versus 8 infratentorial cases). The tumor showed a bimodal age distribution, with one peak in infancy and the other one in young adults. On magnetic resonance imaging (MRI), it typically appeared as a large, lobulated mass.9
From the abovementioned paper, a rather wide range of phenotypic characteristics emerged after analysis of the literature,9 and this means that astroblastomas are collected with different criteria.10 Both the perivascular arrangement of cells and hyalinization are the most crucial characteristics, and the only means to recognize the tumor, provided that it is a stable and diffuse feature and not a sporadic one. As a matter of fact, perivascular formations not otherwise specified are not a rare occurrence in gliomas. Herein, we will discuss three cases in which the perivascular distribution of astrocytic tumor cells, which are distinct from ependymomatous cells, occurred in gliomas with different phenotypic contexts.
Materials and methods
Cases
Case 1: 77-year-old woman
Two months before admission to the hospital, the patient suffered from an epileptic seizure followed by mental confusion. The MRI scan showed a 3 cm left frontal mass with a ring of contrast enhancement and a peritumoral edema (Figure 1). She was operated on at the Department of Neurosurgery, CTO Hospital/Città della Salute e della Scienza (Turin, Italy), and the mass was removed. The diagnosis was World Health Organization (WHO) high-grade astroblastoma. The postsurgical period was uneventful, and the patient underwent radiotherapy (RT) with a total dose of 60 Gy. Ten months later, the MRI with contrast enhancement showed a recurrence in the posterior wall of the surgical cavity, and the patient was treated with temozolomide. The patient died 18 months after surgery.
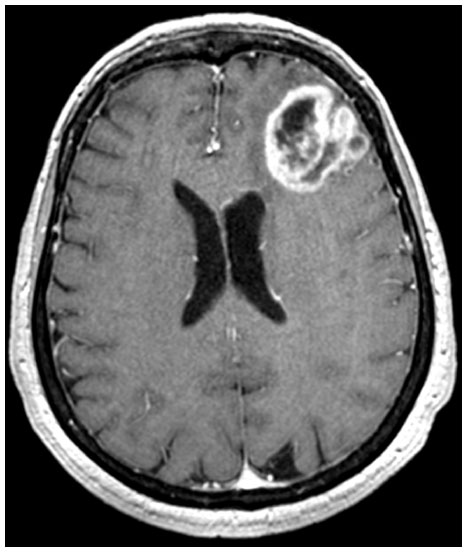 | Figure 1 T1-weighted contrast enhancement magnetic resonance imaging scan of Case 1. |
Case 2: 68-year-old woman
The clinical presentation was motor aphasia and the MRI revealed a hyperintense mass with calcifications in the left basal frontal region. The patient was operated on at the Neurosurgical Unit, University of Turin, and the diagnosis was of WHO grade II oligodendroglioma.
Eight months later, the tumor recurred, and the patient underwent conventional RT with a total dose of 60 Gy. Nine months later, another operation was performed, and the diagnosis was of WHO grade III oligoastrocytoma. The patient died 29 months from the first diagnosis.
Case 3: 60-year-old man
The patient had been treated for papillary eruothelial carcinoma. Two months before intervention, the patient had cephalalgia and ideomotor slowdown. On MRI, a right, hypointense frontal lesion was discovered. The patient was operated on at the Neurosurgical Unit, University of Turin. The diagnosis was uncertain between a giant cell GBM and a gemistocytic astrocytoma with malignant features. The patient underwent conventional RT with a total dose of 60 Gy over 2 months and two cycles of temozolomide. He was still alive 38 months after surgery.
Pathology
All tumor samples were formalin fixed and paraffin embedded (FFPE). From paraffin blocks, 5 μm-thick sections were cut and stained with hematoxylin and eosin and Gomori silver impregnation for reticulum fibers (Bio-Optica, Milan, Italy). Immunohistochemistry with the primary antibodies listed in Table 1 was performed on a Ventana Full BenchMark® XT automated immunostainer (Ventana Medical Systems Inc., Tucson, AZ, USA). The ultraView™ Universal DAB Detection Kit was the revelation system. When indicated, heat-induced epitope retrieval was performed in Tris–ethylenediaminetetraacetic acid, pH 8 (Ventana Medical System Inc.).
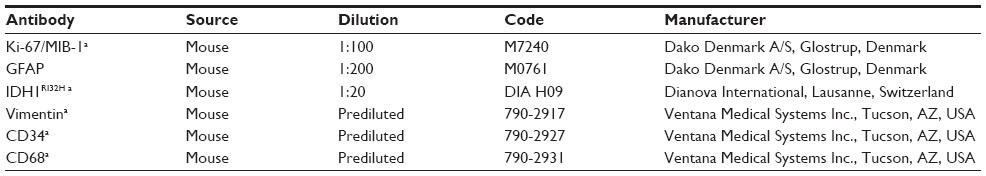 | Table 1 List of primary antibodies used for immunohistochemistry |
The histological diagnosis was in agreement with WHO guidelines.19
Molecular genetics
Genomic DNA (gDNA) from FFPE tumor samples and matched cell lines was extracted using the QIAamp DNA Mini Kit (Qiagen NV, Venlo, the Netherlands), according to the manufacturer’s instructions. Constitutive gDNA was isolated from peripheral blood by a salting-out procedure.
Molecular genetics analyses were performed, as previously published.20–23 Allelic imbalances on the 9p, 10q, and 17p chromosome arms were assessed by loss of heterozygosity (LOH) analysis with microsatellite markers amplified in multiplex reactions by polymerase chain reaction (PCR) with fluorescently labeled primers (Thermo Fisher Scientific Inc., Walthman, MA, USA). The 1p/19q chromosome status was determined by multiplex ligation-dependent probe amplification with the SALSA-MLPA Kit P088 (lot number 0608; MRC-Holland, Amsterdam, the Netherlands). Capillary electrophoresis (CE) was performed on an ABI® 3130 Genetic Analyzer (Thermo Fisher Scientific Inc.).
The EGFR gene amplification status (GenBank sequence NM_005228) was assessed by EGFR co-amplification in PCR reaction with INF-γ (GenBank sequence NM_000619) as reference housekeeping gene, followed by CE.
Quantitative methylation-specific PCR and fragment analysis were used to assess the promoter hypermethylation status of both the MGMT (GenBank sequence NM_002412) and EMP3 (GenBank sequence NM_001425) genes.
TP53 (exons 2 to 11) (GenBank sequence NM_000546), PTEN (exons 1 to 9) (GenBank sequence NM_000314) and IDH1 Arg132 (exon 4) (GenBank sequence NM_005896), IDH2 Arg172 (exon 4) (GenBank sequence NM_002168), and BRAF Val600 (exon 15) (GenBank sequence NM_004333) hot-spot codons were amplified by PCR and searched for mutations by Sanger direct sequencing. Cycle sequencing was performed using the BigDye® Terminator v1.1 Cycle Sequencing Kit (Thermo Fisher Scientific Inc.). The analysis software programs used were Sequencing Analysis v 5.3.1 and GeneMapper v 4.0 (Thermo Fisher Scientific Inc.).
Neurosphere assay
Fragments from fresh surgical tumor samples from Cases 1 and 2 (first intervention) were minced and put in culture for the neurosphere (NS) assay.24 Culture conditions were Dulbecco’s Modified Eagle’s Medium (DMEM)/F-12 supplemented with 20 ng/mL epidermal growth factor (EGF) and 10 ng/mL basic fibroblast growth factor (bFGF) for NS development and DMEM supplemented with 10% fetal bovine serum for adherent cell (AC) development. Cell cultures were maintained in a 5% O2/CO2 atmosphere.
To assess NS multipotency, a differentiation assay was carried out by mitogen withdrawal and the addition of 3% fetal bovine serum to the culture. NS abilities for self-renewal, clonogenicity, and tumorigenicity were assessed as described elsewhere.21
Immunofluorescence
Immunofluorescence on cell lines was performed with the primary antibodies listed in Table 2. The secondary antibodies used were Alexa Fluor® 488-conjugated AffiniPure goat anti-rabbit IgG and Alexa Fluor® 594-conjugated AffiniPure rabbit anti-mouse IgG antibodies (Jackson ImmunoResearch Laboratories Inc, West Grove, PA, USA). Cell nuclei were stained with 4′,6-diamidino-2′-phenylindole, dihydrochloride (DAPI). Images were obtained on a Zeiss Axioskop fluorescence microscope (Carl Zeiss Meditec AG, Jena, Germany) equipped with an AxioCam5MR5c and coupled to an imaging system (AxioVision Release 4.5; Carl Zeiss Meditec AG).
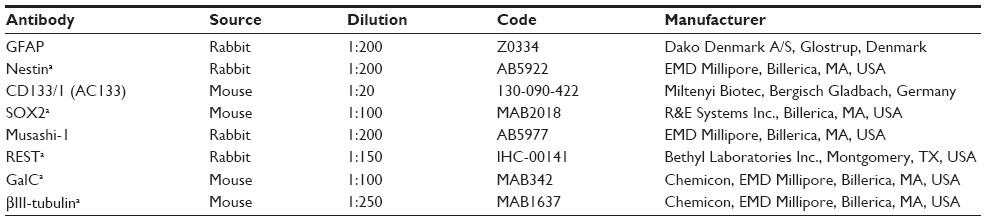 | Table 2 List of primary antibodies used for immunofluorescence |
For confocal microscopy of glial fibrillary acidic protein (GFAP) and nestin, the secondary antibodies used were Alexa Fluor® 555 goat anti-rabbit IgG and Alexa Fluor® 488 goat anti-mouse IgG (Thermo Fisher Scientific Inc.), respectively. High-resolution multichannel images were obtained on a Leica SP8 laser-scanning confocal microscope (Leica Microsystems, Wetzlar, Germany).
Results
Pathology
Case 1
The tumor showed a high cell density with slightly polymorphic nuclei and mitoses (Figure 2A and B). GFAP- and vimentin-positive staining were observed (Figure 2C). Cells sent thick and short processes without end feet on vessels (Figure 2A and B) with no fibrillary component. The perivascular formations were diffuse in the tumor and were recognized as different from perivascular pseudorosettes of ependymoma. The vessel walls were thickened, with mild hyalinization. Microvascular proliferations and glomeruli were frequent, especially at the periphery. Large and circumscribed necroses with pseudopalisading were present (Figure 2B). The Ki-67/MIB-1 labeling index (LI) was approximately 30% (Figure 2D). IDH1R132H staining was negative. The diagnosis was GBM.
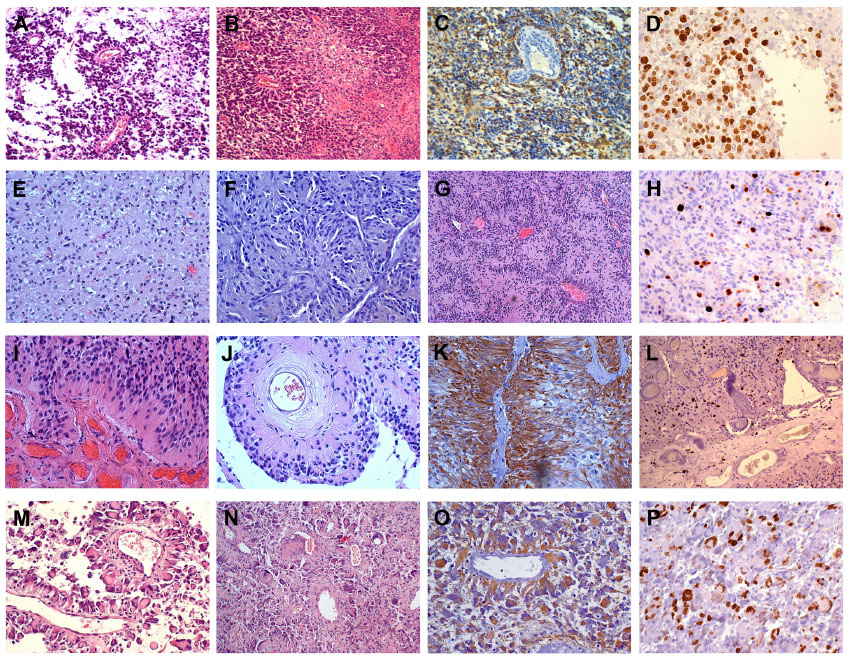 | Figure 2 Immunohistochemistry. |
Case 2
At the first intervention, the tumor showed a high density of GFAP- and vimentin-negative cells with round nuclei (Figure 2E and F). In scattered areas, there was a honeycomb appearance with clear halos around the nuclei. In a wide area, there were typical nuclear palisades, similar to those that were described in polar spongioblastoma, associated with a typical small vessel branching (Figure 2G). Many capillaries and small vessels showed calcification. The Ki-67/MIB-1 LI was 8% (Figure 2H), and IDH1R132H staining was negative. The diagnosis was WHO grade II oligodendroglioma.
At the second intervention, many GFAP-positive large cells of the gemistocytic type were present. Diffusely, there was a perivascular disposition of large cells that sent thick processes on the vessel walls with no fibrillary aspect (Figure 2I–K). The aspect of the tumor was found to be disrupted, with dissociation areas, polymorphic nuclei, evident mitoses, and a Ki-67/MIB-1 LI >20% (Figure 2L). Oligodendroglial features were almost absent. IDH1R132H staining was negative. The diagnosis was WHO grade III oligoastrocytoma.
Case 3
The tumor was composed of rather large, irregularly shaped, frequently GFAP-positive cells with polymorphic and monstrous nuclei. The general architecture of the tumor was composed of cells arranged around vessels sending large and thick cytoplasmic extensions to the vessel walls with no fibrillarity (Figure 2M–O). Vessels of various sizes showed thick, homogeneous, hyalinized walls, often with scanty endothelial hyperplasia. Large and circumscribed necroses were present, with or without pseupalisading. Normal and pathological mitoses were frequent with a Ki-67/MIB-1 LI >20% (Figure 2P). IDH1R132H staining was negative. Since the sample for histological diagnosis was very small, the diagnosis was tentatively that of gemistocytic astrocytoma with malignant features.
In all of the cases, CD34 was expressed, together with vimentin, in endothelial cells and their proliferation. CD68-positive cells were irregularly scattered in all of the cases. Reticulin production, as revealed by the Gomori method, was confined to vessels.
Molecular genetics
In Case 1, a typical glioma/GBM molecular signature was found. By LOH analysis, a common allelic loss was detected on the 10q chromosome arm (D10S212 and D10S562), whereas 9p and 17p chromosomes were intact. The MGMT gene (but not EMP3) displayed promoter hypermethylation. Direct sequencing revealed a stop mutation, c.822G>A (p.Trp274*), in PTEN exon 8, as well as a missense mutation, specifically c.824G>A (p.Cys275Tyr), in TP53 exon 8 (Figure 3A and B). Each mutation was verified to be of somatic origin and previously described in gliomas or glioma cell lines in the Catalogue of Somatic Mutations in Cancer (COSMIC database, v 67 release, October 2013; http://www.sanger.ac.uk/genetics/CGP/cosmic/). The IDH1/2 and BRAF hot-spot codons were wild type, and EGFR gene amplification was not found (Table 3).
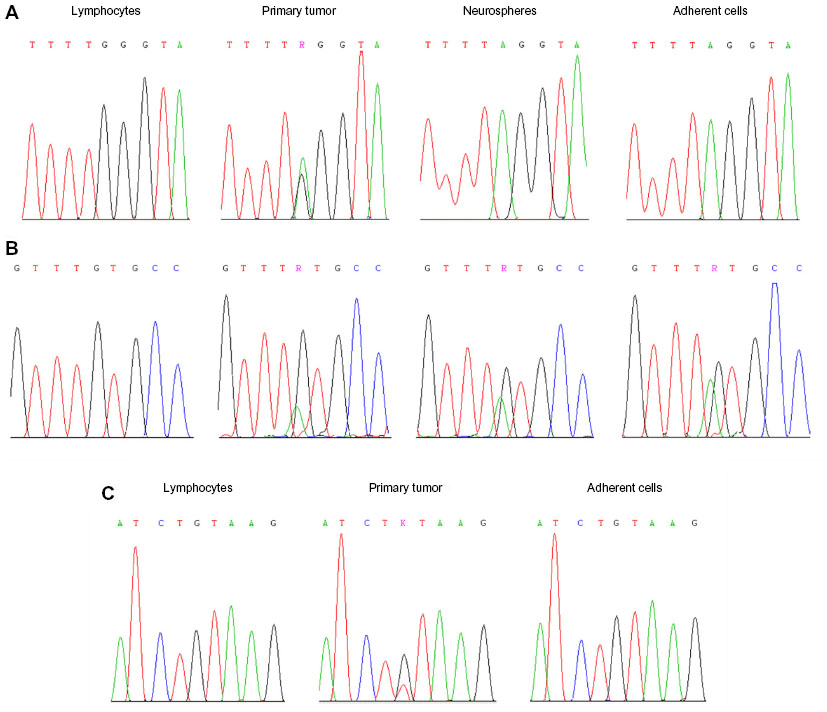 | Figure 3 Sanger direct sequencing. |
 | Table 3 Genetic and epigenetic alterations in tumors and matched cell lines |
In Case 2 (first intervention), LOH on 10q (D10S212, D10S562, and D10S190) was the only allelic imbalance identified. Direct sequencing revealed a somatic nucleotide sequence variation, c.209 +1G>T, in PTEN intron 3, which was responsible for aberrant splicing (Figure 3C). The TP53 gene and the IDH1/2 and BRAF hot-spot codons were wild type. The EMP3 gene (but not MGMT) displayed promoter hypermethylation, and EGFR gene amplification was not found (Table 3).
In Case 3, genetic analyses revealed the absence of any genetic/epigenetic alterations, with the exception of MGMT promoter hypermethylation (Table 3).
Cell lines
NS and AC developed in Case 1, but only AC in Case 2. After confocal microscopy, NS expressed stemness markers (nestin, CD133, SOX2, Musashi-1, and REST) (Figure 4A and B), whereas AC expressed differentiation antigens (GFAP, GalC, and βIII-tubulin) (Figure 4C and D).
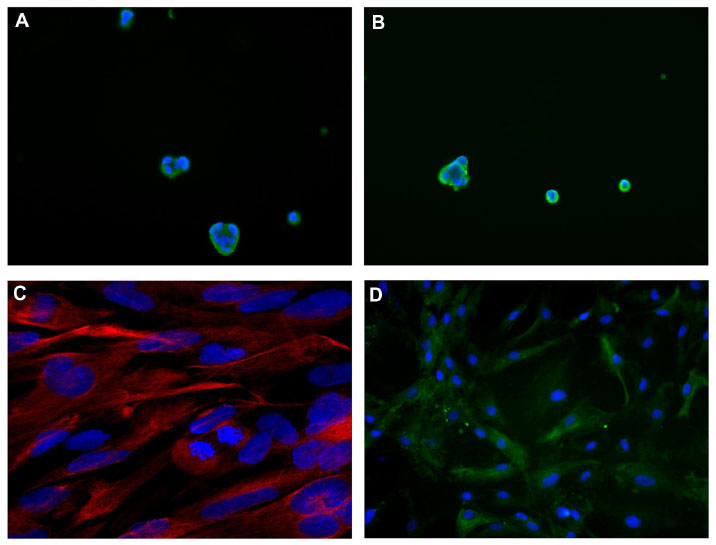 | Figure 4 Immunofluorescence. |
Molecular genetics analysis applied to the cell lines showed that both NS and AC from Case 1 totally mimicked the tumor genotype (Figure 3A and B). In contrast, AC from Case 2 (first intervention) did not display any genetic/epigenetic alterations common to the respective primary tumor (Figure 3C).
Discussion
The three presented cases were chosen as examples of glial tumors with perivascular formations that could not be recognized as radiated crowns or perivascular pseudorosettes of ependymoma, of which, on the other hand, other features were lacking. Perivascular cells sent thick, broad, and small processes on vessels. In Case 1, there was a general aspect of high-grade astrocytic glioma with high cell proliferation, circumscribed necroses, and microvascular proliferations, and the tumor was diagnosed as such. Perivascular arrangement of cells was an additional feature, occurring diffusely in the tumor, which precisely recalled that of astroblastoma. The tumor occurred in an elderly woman and showed MRI features consistent with those of astroblastoma.
Case 2 was a WHO grade II oligodendroglioma at the first intervention, with round nuclei, honeycomb appearance of cells, and, limited to a single area, palisades of cells intermingled with capillaries similar to that previously described in polar spongioblastoma.25 At the second intervention, the tumor phenotype was different, with reduction of the oligodendroglial component and the acquisition of an astrocytic one. It was diagnosed as an anaplastic oligoastrocytoma. In the tumor, there were many GFAP-positive cells of the gemistocytic type sending thick processes on the vessel walls without a fibrillary component. The formation recalled astroblastoma and not ependymoma. This finding was not diffusely present, but limited to the astrocytic component that appeared in the tumor removed at the second intervention; in this component, it was diffusely distributed, meaning the tumor fell within the narrow diagnostic criteria of astroblastoma. Alternatively, it could have been already present at the first intervention, but not removed. The occurrence of the perivascular asset of tumor astrocytic cells could be interpreted as the consequence of a peculiar tumor cell/vessel relationship, possibly linked to microenvironment conditioning, as in the other two cases and, in general, in astroblastoma.26
Case 3 was a malignant gemistocytic tumor with a diffuse perivascular arrangement of the cells without both end feet and a fibrillary component, similar to that of astroblastoma. It is not clear from the literature whether the criteria for recognizing astroblastoma include the dimensions of the cells, whether they are broad or small. The general consensus is that both have been accepted. Therefore, this case also might fall into the category of astroblastoma.
In all three cases, the typical features (perivascular cell disposition and hyalinization) of astroblastoma were present; in the first case, these features were the only characteristics of the tumor, while, in the other two cases, these features appeared alongside other phenotypic features that are typical of astrocytic tumors. In a previous study, data for 116 patients operated on for astroblastoma were collected from the literature, following inclusion criteria deduced from 62 references.9 The patients’ clinical, surgical, and radiological features were defined, and two main peaks of age distribution were observed, due to a bimodal distribution of the tumor: specifically, one peak in infancy and the other one in adult age. Astroblastoma was, therefore, considered to be a rare tumor described in the literature as individual cases or as small collections of cases, with the criteria of astroblastic pseudorosettes and vascular hyalinization.11 In review of the literature, it can be seen that, contrarily to the diagnosis of astroblastoma in the strict sense, tumors with a wider range of features could be accepted. Possibly, different criteria are used in the interpretation of the perivascular distribution of tumor cells.
Two of our cases were malignant gliomas, and one was benign, turning in malignant at recurrence. This is in line with recent publications,13,15,17,27,28 confirming that the nature of the tumor can be both benign or malignant.26 No ependymomatous features were present in our cases, and, unfortunately, it was not possible to perform an electron microscopy study. It is intriguing that, in a recently published case, microvilli were observed in the inter- and intracellular lumina,17 suggesting a relationship with ependymoma. As a matter of fact, this has been previously observed and, besides tanycytes, ependymal astrocytes have also been hypothesized to be the source of the tumor29,30 without identifying the tumor as an ependymoma.
Data on the molecular genetics of astroblastoma are rare and only recently available from the literature. The observed aberrations include gains on the 20q chromosome arm and chromosome 19, and losses on 9q, 10, and X chromosome, which have been revealed by comparative genomic hybridization.27 TP53 and IDH1/2 somatic mutations have been seen to be absent.17 The molecular genetic profiles showed typical alterations of WHO grade III–IV gliomas with LOH on 10q and wild-type IDH1/2 and BRAF genes. PTEN and TP53 somatic mutations, as well as MGMT promoter hypermethylation, occurred in Case 1, consistently with a diagnosis of high-grade astrocytic glioma. PTEN somatic mutations and EMP3 promoter hypermethylation were found in Case 2, whereas MGMT promoter hypermethylation was the only molecular alteration identified in Case 3. EGFR gene amplification, as well as LOH on the 9p chromosome arm, was not found in our three cases. The presence of EMP3 promoter hypermethylation in Case 2 is remarkable, as this epigenetic alteration is typical of oligodendrogliomas, where it is mainly associated with the 1p/19q co-deletion.23 As such, its occurrence in Case 2 could be in line with the diagnosis of oligodendroglioma at the first intervention. However, the lack of association with the 1p/19q co-deletion in Case 2 may support the diagnosis of oligoastrocytoma at the second intervention. The absence of a specific genetic/epigenetic signature in our cases may indicate that our tumors either were not astroblastomas or that they do not support astroblastoma as a specific tumor entity. It must be taken into account that a genetic signature of astroblastoma has never been described in the literature.
In Case 1, the stemness potential was typical of a primary GBM, in that generation of both NS expressing stemness antigens and AC expressing differentiation antigens occurred. In Case 2, AC only developed, denouncing a reduced stemness potentiality. The stemness asset of cell lines denounced a stemness spectrum of the tumor cells, which, in turn, corresponded to a spectrum of phenotypic malignancy. Out of the great amount of knowledge on tumor stem cells currently available,34–36 the interpretation of cancer stem cells as the expression of a functional status21,31–33 must not be forgotten. This interpretation depends on the tumor microenvironments,26,37 as well as on epigenetic influences.31
Our conclusion is that the perivascular formations found in our three gliomas, not identifiable with those of ependymoma, may be expressions of an astroblastic trend of the tumors. It may indicate that astroblastoma does not exist as an established and unique tumor entity, or that it exists, but that its main feature can occasionally appear as a special phenotype in astrocytic gliomas. The only reason to discuss its existence in relation to ependymoma may be its possible origin from tanycytes or ependymal astrocytes.14
Conclusion
Astroblastoma may represent a tumor phenotype either diffused in the entire tumor mass or only focally represented. It is exclusive of astrocytic gliomas, independently of the histopathologic features that they can show. The perivascular cell distribution should be conceived as the expression of the molecular machinery regulated by the tumor genetics and epigenetics, id, microenvironment.
Acknowledgments
This work was supported by grant number 4011 SD/cv 2011-0438 from Compagnia di San Paolo, Turin, Italy.
Disclosure
The authors report no conflicts of interest in this work.
References
Bailey P. Cellular types in primary tumors of the brain. In: Penfield W, editor. Cytology and Cellular Pathology of The Nervous System. New York, PA: Hoeber; 1932:905–951. | |
Kernohan JW, Mabon RF, Svien HJ, et al. A simplified classification of the gliomas. Proc Staff Meet Mayo Clin. 1949;24(3):71–75. | |
Zülch KJ. Biologie und Pathologie der Hirn Geschwülste [Biology and pathology of brain tumors]. In: Zülch KJ, Christensen E, editors. Pathologische Anatomie der Raumbeengenden Intrakraniellen Prozesse. Berlin: Springer; 1956:1–702. German. | |
Russell DS, Rubinstein LJ. Pathology of Tumours of the Nervous System. 5th ed. London: Edward Arnold; 1989. | |
Kubota T, Hirano A, Sato K, Yamamoto S. The fine structure of astroblastoma. Cancer. 1985;55(4):745–750. | |
Eng LF, Rubinstein LJ. Contribution of immunohistochemistry to diagnostic problems of human cerebral tumors. J Histochem Cytochem. 1978;26(7):513–522. | |
Velasco ME, Dahl D, Roessmann U, Gambetti P. Immunohistochemical localization of glial fibrillary acidic protein in human glial neoplasms. Cancer. 1980;45(3):484–494. | |
Fuller GN, Scheithauer BW. The 2007 Revised World Health Organization (WHO) Classification of Tumours of the Central Nervous System: newly codified entities. Brain Pathol. 2007;17(3):304–307. | |
Sughrue ME, Choi J, Rutkowski MJ, et al. Clinical features and post-surgical outcome of patients with astroblastoma. J Clin Neurosci. 2011;18(6):750–754. | |
Bonnin JM, Rubinstein LJ. Astroblastomas: a pathological study of 23 tumors, with a postoperative follow-up in 13 patients. Neurosurgery. 1989;25(1):6–13. | |
Husain AN, Leestma JE. Cerebral astroblastoma: immunohistochemical and ultrastructural features. Case report. J Neurosurg. 1986;64(4):657–661. | |
Burger PC, Scheithauer BW. Tumor of the central nervous system. In: Washington, DC: Armed Forces Institute of Pathology, editor. Atlas of Tumor Pathology. Washington, DC: Armed Forces Institute of Pathology, 3rd series, fascicle 10; 1994:146–148. 77–96. | |
Aldape KD, Rosenblum IK. Astroblatoma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC; 2007:88–89. | |
Kujas M, Faillot T, Lalam T, Roncier B, Catala M, Poirier J. Astroblastomas revisited. Report of two cases with immunocytochemical and electron microscopic study. Histogenetic considerations. Neuropathol Appl Neurobiol. 2000;26(3):295–298. | |
Port JD, Brat DJ, Burger PC, Pomper MG. Astroblastoma: radiologic-pathologic correlation and distinction from ependymoma. AJNR Am J Neuroradiol. 2002;23(2):243–247. | |
Alaraj A, Chan M, Oh S, Michals E, Valyi-Nagy T, Hersonsky T. Astroblastoma presenting with intracerebral hemorrhage misdiagnosed as dural arteriovenous fistula: review of a rare entity. Surg Neurol. 2007;67(3):308–313. | |
Fu YJ, Taniguchi Y, Takeuchi S, et al. Cerebral astroblastoma in an adult: an immunohistochemical, ultrastructural and genetic study. Neuropathology. 2013;33(3):312–319. | |
Shakur SF, McGirt MJ, Johnson MV, et al. Angiocentric glioma: a case series. J Neurosurg Pediatr. 2009;3(3):179–202. | |
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK. WHO Classification of Tumours of the Central Nervous System. 4th ed. Lyon: IARC; 2007. | |
Mellai M, Piazzi A, Caldera V, et al. IDH1 and IDH2 mutations, immunohistochemistry and associations in a series of brain tumors. J Neurooncol. 2011;105(2):345–357. | |
Caldera V, Mellai M, Annovazzi L, et al. Antigenic and genotypic similarity between primary glioblastomas and their derived neurospheres. J Oncol. 2011;2011:314962. | |
Mellai M, Monzeglio O, Piazzi A, et al. MGMT promoter hypermethylation and its associations with genetic alterations in a series of 350 brain tumors. J Neurooncol. 2012;107(3):617–631. | |
Mellai M, Piazzi A, Caldera V, et al. Promoter hypermethylation of the EMP3 gene in a series of 229 human gliomas. Biomed Res Int. 2013;2013:756302. | |
Galli R. The neurosphere assay applied to neural stem cells and cancer stem cells. Methods Mol Biol. 2013;986:267–277. | |
Schiffer D, Cravioto H, Giordana MT, Migheli A, Pezzulo T, Vigliani MC. Is polar spongioblastoma a tumor entity? J Neurosurg. 1993;78(4):587–591. | |
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60(3):502–514. | |
Brat DJ, Hirose Y, Cohen KJ, Feuerstein BG, Burger PC. Astroblastoma: clinicopathologic features and chromosomal abnormalities defined by comparative genomic hybridization. Brain Pathol. 2000;10(3):342–352. | |
Navarro R, Reitman AJ, de León GA, Goldman S, Marymont M, Tomita T. Astroblastoma in childhood: pathological and clinical analysis. Childs Nerv Syst. 2005;21(3):211–220. | |
Rubinstein LJ, Herman MM. The astroblastoma and its possible cytogenic relationship to the tanycyte. An electron microscopic, immunohistochemical, tissue- and organ-culture study. Acta Neuropathol. 1989;78(5):472–483. | |
Kubota T, Sato K, Arishima H, Takeuchi H, Kitai R, Nakagawa T. Astroblastoma: immunohistochemical and ultrastructural study of distinctive epithelial and probable tanycytic differentiation. Neuropathology. 2006;26(1):72–81. | |
Facchino S, Abdouh M, Bernier G. Brain cancer stem cells: current status on glioblastoma multiforme. Cancers (Basel). 2011;3(2):1777–1797. | |
de Almeida Sassi F, Lunardi Brunetto A, Schwartsmann G, Roesler R, Abujamra AL. Glioma revisited: from neurogenesis and cancer stem cells to the epigenetic regulation of the niche. J Oncol. 2012;2012:537861. | |
Olar A, Aldape KD. Using the molecular classification of glioblastoma to inform personalized treatment. J Pathol. 2014;232(2):165–177. | |
Schiffer D, Mellai M, Annovazzi L, Piazzi A, Monzeglio O, Caldera V. Glioblastoma cancer stem cells: basis for a functional hypothesis. Stem Cell Discovery. 2012;2(3):122–131. | |
Zipori D. The nature of stem cells: state rather than entity. Nat Rev Genet. 2004;5(11):873–878. | |
Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6(6):425–436. | |
Filatova A, Acker T, Garvalov BK. The cancer stem cell niche(s):the crosstalk between glioma stem cells and their microenvironment. Biochim Biophys Acta. 2013;1830(2):2496–2508. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.