")
Back to Journals » Clinical Epidemiology » Volume 9
Assessment of channeling bias among initiators of glucose-lowering drugs: A UK cohort study
Authors Ankarfeldt MZ, Thorsted BL, Groenwold RHH , Adalsteinsson E, Ali MS, Klungel OH
Received 6 October 2016
Accepted for publication 10 December 2016
Published 18 January 2017 Volume 2017:9 Pages 19—30
DOI https://doi.org/10.2147/CLEP.S124054
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Henrik Sørensen
Mikkel Z Ankarfeldt,1,2 Brian L Thorsted,1 Rolf HH Groenwold,2,3 Erpur Adalsteinsson,1 M Sanni Ali,2–4 Olaf H Klungel2,3
1Novo Nordisk A/S, Bagsvaerd, Denmark; 2Julius Center for Health Sciences and Primary Care, University Medical Center Utrecht, Utrecht, the Netherlands; 3Division of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, University of Utrecht, Utrecht, the Netherlands; 4Nuffield Department of Orthopaedics, Rheumatology, Musculoskeletal Sciences, University of Oxford, Oxford, UK
Background: Channeling bias may occur when a newly marketed drug and an established drug, despite similar indications, are prescribed to patients with different prognostic characteristics (ie, confounding).
Aim: To investigate channeling bias and its impact on relative effectiveness of glucagon-like peptide-1 (GLP-1) analogs versus basal insulin and dipeptidyl peptidase-4 inhibitors (DPP-4i) versus sulfonylurea.
Methods: In the UK Clinical Practice Research Datalink, patients with type 2 diabetes initiating treatment between 2006 and 2015 were included. Analyses were stratified by years since first prescription of GLP-1 and DPP-4i, respectively. The characteristics of GLP-1 versus insulin and DPP-4i versus sulfonylurea initiators were compared over time. After propensity score matching, the relative effectiveness regarding 6-month changes in glycated hemoglobin (HbA1c) and body weight was estimated.
Results: In total, 8,398 GLP-1, 14,807 insulin, 24,481 DPP-4i, and 33,505 sulfonylurea initiators were identified. No major channeling was observed. Considerable overlap in distributions of characteristics allowed for propensity score-matched analyses. Relative effectiveness was similar across time. The overall relative effect of GLP-1 versus insulin showed no difference for HbA1c and relative increase in body weight (3.57 kg [95% confidence interval {CI}: 3.21, 3.92]) for insulin. The overall relative effect of DPP-4i versus sulfonylurea showed relative decrease in HbA1c (–0.34% [95% CI: –0.38, –0.30]) and increase in body weight (1.58 kg [95% CI: 1.38, 1.78]) for sulfonylurea.
Conclusion: No major channeling was identified in the investigated glucose-lowering drugs. Relative effectiveness could be estimated already in the first year after launch and was consistent in the years thereafter.
Keywords: channelling bias, channeling bias, glucose-lowering drugs, DPP-4i, GLP-1, type 2 diabetes, observational study, relative effectiveness
Corrigendum for this paper has been published
Introduction
Randomized controlled trials are primarily designed and conducted to meet the needs of regulatory bodies, so as to provide evidence on the efficacy and the safety of new drugs or other health care interventions. However, these studies are generally insufficient by themselves to meet the evidentiary needs of many health technology assessment agencies, that is, evidence on the drug’s effectiveness. Similarly, clinicians and payers desire evidence on comparative effectiveness of new drugs immediately after launch to take informed decisions.1 The sooner valid comparative effectiveness research results can be generated, the more useful they are to patients, clinicians, and payers. Comparative effectiveness research using secondary health care data (including electronic medical records, longitudinal claims data, and registries) provides evidence on the benefits and risks of drugs in routine medical practice.1 However, channeling bias is a potential risk when relative effectiveness of a newly marketed drug compared to an established drug is investigated in observational data.
Channeling bias may occur when a newly marketed drug and an established drug, despite similar therapeutic indications, are prescribed to patients with different prognostics characteristics.2 Over time, the prognostic characteristics of the patients who prescribed the two drugs may become more balanced as the newly marketed drug becomes more established. Reasons for channeling bias could be a belief in extra advantages of the new drug compared to the established drug, or simply because doctors do not know how else to treat a subgroup of patients due to intolerance or low response to established drugs. It is possible that patients with a better prognostic are channeled to the newly marketed drug, but it is often theorized that a newly marketed drug is predominantly prescribed to patients with worse prognostics compared to the established drug being prescribed to patients with better prognostics.1,2 When channeling bias occurs, comparative effectiveness research becomes challenging because comparison with a drug will be confounded and knowledge of comparative effectiveness close to market entry may be biased or absent.
The first glucagon-like peptide-1 analogs (GLP-1), exenatide, was approved by the European Medicines Agency (EMA) in November 2006, and the first dipeptidyl peptidase-4 inhibitor (DPP-4i), sitagliptin, was approved by the EMA in March 2007. A US study on prescription pattern of exenatide during the first half year after approval showed that a higher proportion of obese patients used exenatide compared to patients with other glucose-lowering drugs, indicating an awareness of weight-lowering effects of GLP-1.3 Three US studies4–6 compared the characteristics of sitagliptin users with patient receiving other oral glucose-lowering drugs; two of the studies4,5 compared 2006–2007 versus 2008–2010, and the third study6 investigated the first 2½ years after approval of sitagliptin. The three studies4–6 found that DPP-4i users were older and have more comorbidities than patients receiving other oral glucose-lowering drugs, but with no change over time. These studies3–6 were all descriptive and did not investigate if the differences in characteristics could be taken into account in an outcome analysis.
The objectives of this study were, first, to compare the characteristics of patients initiating GLP-1 to those initiating basal insulin and of patients initiating DPP-4i to those initiating sulfonylurea stratified on calendar time and, second, to assess the relative effectiveness over time since the market entry of GLP-1 and DPP-4i.
Methods
The United Kingdom Clinical Practice Research Datalink (CPRD) was used for this study. The CPRD is derived from UK primary care and holds longitudinal patient-level data such as diagnoses, mortality, laboratory results, and prescription data. It is the representative for the UK general population, and validation ensures high-quality data.7–10 Anonymized data are available after protocol approval from the Independent Scientific Advisory Committee (ISAC).11
For this study, data about patients aged ≥18 years with a diagnosis of type 2 diabetes and a prescription of glucose-lowering medication between January 1, 2006, and February 9, 2015, were extracted. Validation studies have shown that diagnosis of type 2 diabetes alone is not always correct, so to ensure identification of patients with type 2 diabetes, patients were included if they fulfilled one of the following criteria:12 1) first diagnosis (either type 1 or type 2) before age 35 years and no continual insulin treatment within six months of diagnosis, or 2) first diagnosis (either type 1 or type 2) after age 35 years and not on continual insulin treatment from diagnosis.
Exposure, cohorts, and time blocks
Two comparison groups were used throughout the study; initiators of GLP-1 were compared with initiators of basal insulin, whereas initiators of DPP-4i were compared with initiators of sulfonylurea. The chosen comparator groups were to compare second-line (DPP-4i and sulfonylurea) and third-line (GLP-1 and insulin) treatments, respectively.13 The glucose-lowering drugs were handled at the drug-class level, and combination products of metformin and DPP-4i and of rosiglitazone and sulfonylurea were considered as DPP-4i and sulfonylurea. Prescriptions of drugs were used to identify episodes of treatment. For each patient, the end of a prescription was estimated from the daily dose and amount prescribed (typically available for GLP-1, DPP-4i, and sulfonylurea), or as the patient’s median duration of prescription of the drug (typically the case for insulin). If there were <120 days between the estimated end date of one prescription and the start date of the next prescription, the prescriptions were considered to make up a continuous episode of drug treatment. Initiation was defined as patients with a prescription of one of the four glucose-lowering drugs after minimum 180 days without a prescription of the drug (washout period). Two cohorts were created, by analogy with the terms used in clinical trials: the intention-to-treat (ITT) cohorts comprise subjects with a treatment duration of at least one day, whereas the per-protocol (PP) cohorts comprise subjects with a treatment duration of at least 180 days. Patients could appear on both the ITT and the PP cohorts. With the two comparisons, this makes up four cohorts in total. If patients were identified as incident users more than one time for the same drug, only the first initiation was considered. If patients initiated GLP-1 and insulin, or DPP-4i and sulfonylurea at the same time, this initiation was censored. Within each of the four cohorts, time blocks of 365-day duration were defined with start date of the first initiation of GLP-1 or DPP-4i, respectively. If patient initiated the comparison drugs within the same time block, only the first initiation was used and the second initiation was censored.
Outcome
Measures of glycated hemoglobin (HbA1c) and body weight were used to establish outcome measures. The goal was to observe change in HbA1c and in body weight during a 180-day period. However, because measurements were not always available at the exact date of treatment initiation and 180 days after initiation, the following were deployed when measurements were not available: If a measurement was available within 90 days prior to initiation, this was used as the measurement at initiation, and if a measurement was available between 90 and 179 days after initiation, this was used as the measurement 180 days after initiation. If several measurements were available within these periods, the measurement closest to the initiation date and 180 days after initiation, respectively, was used.
Covariates
The following covariates were used: sex; age at initiation; weight at initiation; body mass index (BMI) at initiation; HbA1c at initiation; duration of diabetes at initiation; number of oral glucose-lowering drugs at initiation; any use of glucose-lowering drugs (sulfonylurea, basal insulin, DPP-4i, metformin, and other oral glucose-lowering drugs) during the washout period; any use of antihypertensive, statins, and anticoagulants during the washout period; Charlson Comorbidity Index14 at initiation; and any diagnosis of hypertension, end-stage renal disease, myocardial infarction, or stroke prior to initiation. The covariate number of oral glucose-lowering drugs at index was a count of prescriptions at index of any of the following glucose-lowering drugs; acarbose, canagliflozin, canagliflozin combined with metformin, dapagliflozin, dapagliflozin combined with metformin, empagliflozin, pioglitazone, pioglitazone combined with metformin, rosiglitazone, rosiglitazone combined with metformin, nateglinide, repaglinide, troglitazone, guar gum and metformin in the comparison of DPP-4i and sulfonylurea, and in the comparison of GLP-1 and basal insulin number of other oral glucose-lowering drugs at index also included DPP-4i and sulfonylurea. The covariate other oral glucose-lowering drugs during washout was a binary response if at least one of the following glucose lowering drugs were prescribed during the washout period; acarbose, canagliflozin, canagliflozin combined with metformin, dapagliflozin, dapagliflozin combined with metformin, empagliflozin, pioglitazone, pioglitazone combined with metformin, rosiglitazone, rosiglitazone combined with metformin, nateglinide, repaglinide, troglitazone or guar gum.
Missing data
Information on HbA1c, BMI, weight, and diabetes duration was missing for some individuals. Under the assumption that these missing data were missing at random, missing values were imputed. Multiple imputation was applied, with 10 imputed datasets.15 In addition to imputation of missing measurements at initiation, missing measurements 180 days after initiation were also imputed.16 We used the listed covariates, treatment group and change in HbA1c and weight for the imputation. The Charlson comorbidity index was categorized into four groups: 0–1, 2, 3, and more than 3 points. Number of OADs was categorized into four groups: 0 OADs, 1 OADs, 2 OADs, 3 OADs, and more than 3 OADs. Results for both the descriptive and the outcome analyses were pooled across the imputed datasets using Rubin’s rule.17 Post hoc we noticed that a fraction of HbA1c measurements was very low for GLP-1 and insulin initiators, and that most of these measurements were located around the fourth year since GLP-1 approval. As we suspected these measurements were incorrect, all HbA1c measurements below 4% were treated as missing and were imputed as described above.
Propensity score and matching
Propensity scores were estimated as the probability of initiating GLP-1 in the comparison of GLP-1 and insulin initiators and the probability of initiating DPP-4i in the comparison of DPP-4i and sulfonylurea initiators. The propensity score models included the covariates mentioned earlier, except the covariate describing use of the comparison drug during washout. These variables were not included because they were collinear with the initiated drug. Standardized difference was used to assess balance on covariates and to select the appropriate propensity score model.18 The propensity score was estimated twice: in the whole cohort for the descriptive purpose of characteristics over time and in the subgroup that did not receive the comparison drug during washout to be used for matching in the outcome analyses. Based on the propensity score, patients were matched in order to create treatment groups that were comparable with respect to the propensity score and thus the potential confounders. Greedy matching was done in a 1:1 ratio, stratified by time block and within each imputed dataset. The greedy matching was done in up to eight iterations; first the data were searched for matches based on eight digits of the propensity score, then seven digits, and continued to matches according to one digit (see Parson19 for details).
Descriptive analyses: characteristics over time
To assess changes in patient characteristics over time, the covariates and propensity score were plotted stratified by time blocks and treatment groups. This shows both general differences between treatment groups and potential channeling, where characteristics change over time. The propensity score serves as a single variable to describe differences in characteristics across comparison groups. Both the propensity score for all individuals and the propensity score for individuals suitable for matching (without use of the comparison drug during washout) were assessed. This was done in both the ITT and the PP cohorts. Because of the many covariates available, only the propensity score over time will be shown in the Result section, whereas graphs for each individual variable can be found in the supplementary material.
Outcome analyses: relative effect estimates over time
In the subgroup of the PP cohort that did not receive the comparison drug during washout, linear regression analyses were used to estimate the relative effect regarding change in HbA1c and change in body weight after 180 days of follow-up. Again, the comparison groups were GLP-1 versus basal insulin and DPP-4i versus sulfonylurea. These analyses were stratified by time blocks. Crude analyses were performed based on all identified initiators, in addition to analyses of the propensity score-matched initiators, to take confounding into account. In the analyses of change in HbA1c and body weight, baseline levels of HbA1c and body weight, respectively, were included as covariates in the outcome model.20 The relative effect estimate and their 95% confidence intervals (CIs) were plotted for graphical inspections of changes in effect estimates over time.
SAS software 9.4 was used to carry out analyses. Data management was done using template programs developed by the department of epidemiology at Novo Nordisk for importing CPRD data into SAS format. Access to CPRD data was gained through approval of protocol by the ISAC (https://www.cprd.com/isac/).
Results
In total, 304,953 patients with diabetes and a prescription of a glucose-lowering drug were identified. The first initiation of GLP-1 was observed on June 14, 2007, whereas the first initiation of DPP-4i was observed on June 22, 2007. For the descriptive analyses of characteristics over time, 10,633 and 19,243 initiators of GLP-1 and insulin, respectively, and 30,048 and 41,346 initiators of DPP-4i and sulfonylurea, respectively, were included in the ITT cohorts. The numbers of individuals in the PP cohorts were a little smaller. For the outcome analyses, 8,398 and 13,286 initiators of GLP-1 and insulin, respectively, and 10,847 and 30,643 initiators of DPP-4i and sulfonylurea, respectively, were identified. The exact number of propensity score-matched individuals varies within each imputed dataset, but within each time block nearly half of the initiators of GLP-1 were matched with an initiator of insulin and nearly all initiators of DPP-4i were matched with an initiator of sulfonylurea. It was possible to create seven time blocks of 365 days in both the GLP-1 and insulin, and the DPP-4i and sulfonylurea comparisons. Figure S1 provides a full flowchart detailing this.
Trends over time
The distributions of propensity score look similar for both the ITT and PP cohorts of all identified initiators of GLP-1 and insulin, and DPP-4i and sulfonylurea, respectively, and also for the PP cohort of initiators with no use of the comparison drug during washout, which is the subgroup used for propensity score matching. Time blocks 1–7 are demonstrated as histograms of the propensity score of the latter in Figures 1 and 2. Other propensity score plots are found in the supplementary material Figure S2. In general, the distribution of propensity score of the GLP-1 and insulin initiators indicate an overlap in propensity score for all time blocks considered. The propensity score of DPP-4i and sulfonylurea initiators has an even greater overlap in all time blocks, and for both DPP-4i and sulfonylurea, the distribution moves from being centered on lower propensity scores in the early time blocks to being centered on 0.4 in later time blocks, which indicate comparable patients with a similar change in characteristics over time.
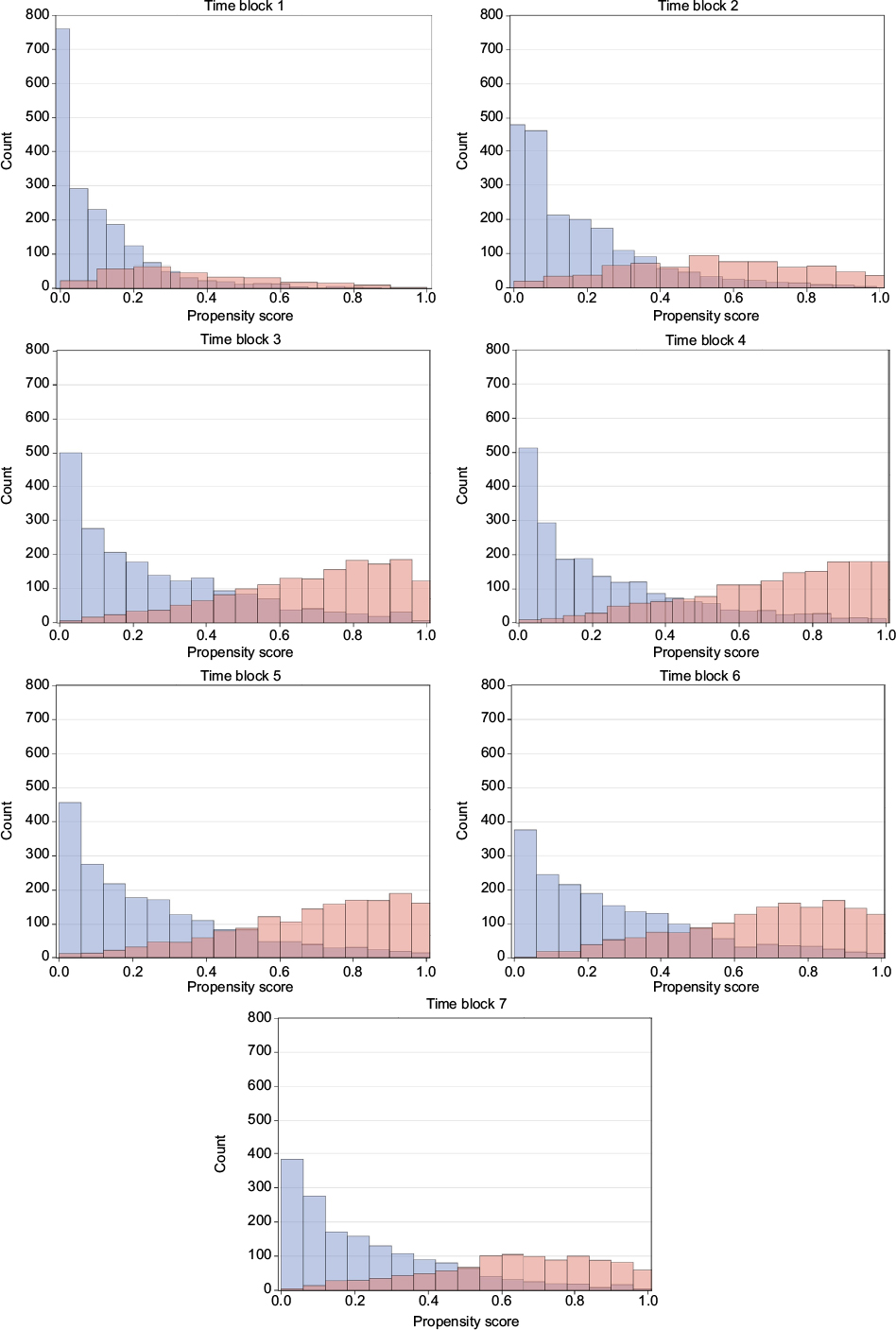 | Figure 1 Propensity score over time for GLP-1 versus basal insulin initiators. Notes: Blue: GLP-1, red: insulin. Abbreviation: GLP-1, glucagon-like peptide-1 analogs. |
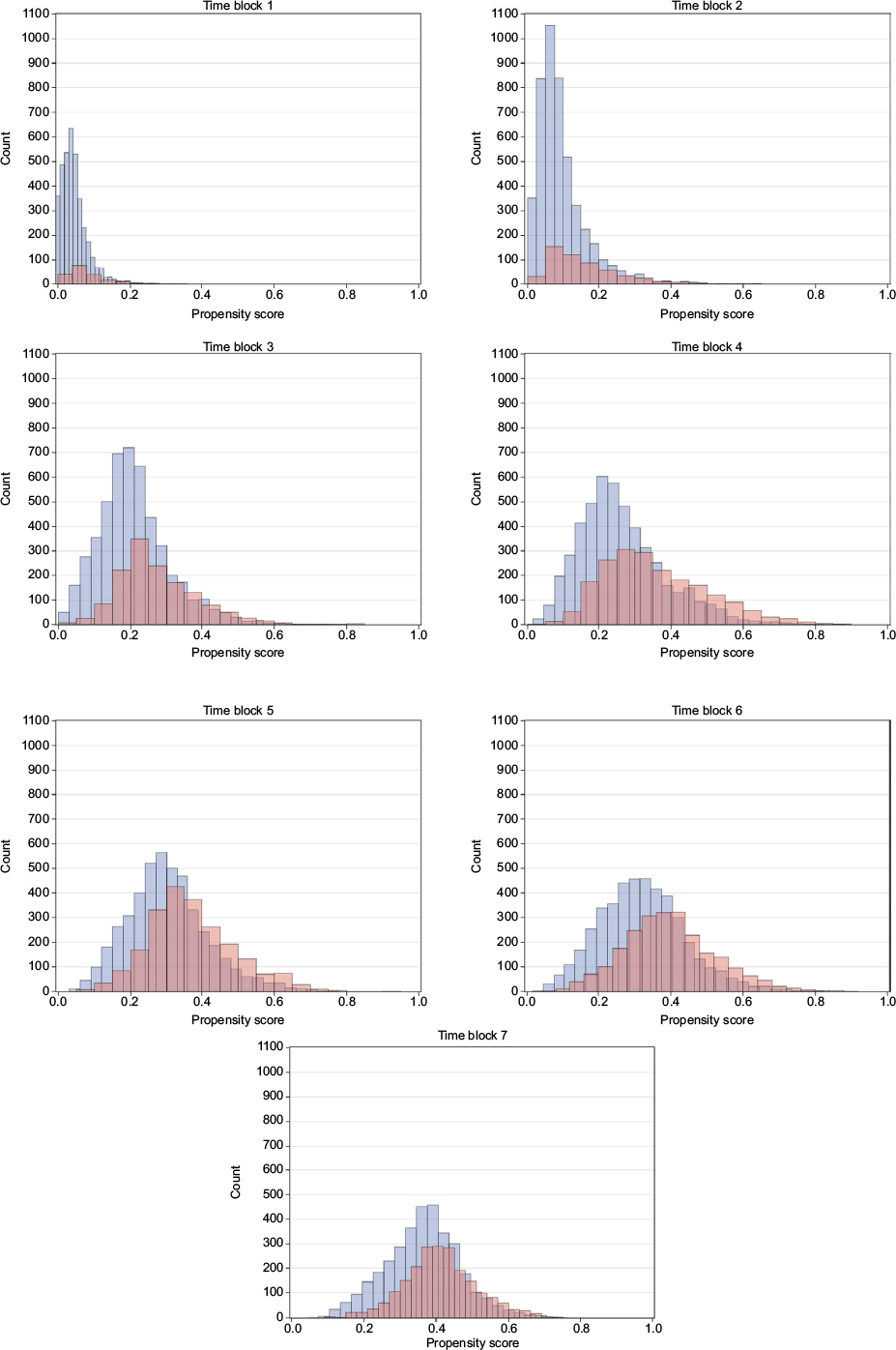 | Figure 2 Propensity score over time for DPP-4i versus sulfonylurea initiators. Notes: Blue: DPP-4i, red: insulin. Abbreviation: DPP-4i, dipeptidyl peptidase-4 inhibitors. |
Figures of the individual covariates are found in Figure S3–S4. In general, trends for the ITT and PP cohort were similar. Hence, there will be no distinction of the ITT and PP cohort in the following text. For initiators of GLP-1 and insulin, minor changes in HbA1c at initiation were observed; however, all were within the range of 0.5%. Both GLP-1 and insulin initiators had an increase in use of DPP-4i during washout and a decrease in use of other oral glucose-lowering drugs during washout. For insulin initiators only, an increase in use of sulfonylurea, GLP-1, and metformin during washout was seen. Plots of the individual covariates also show some general differences of the comparison groups across time. Compared to initiators of insulin, the initiators of GLP-1 tended to have higher BMI; more number of glucose-lowering drugs at initiation; increased use of metformin, other glucose-lowering drugs, DPP-4i, and statins during washout, increased proportion of with hypertension diagnoses; lower age; higher Charlson Comorbidity Index, increased proportion of myocardial infarction and end-stage renal disease diagnoses; and shorter duration of diabetes.
For DPP-4i and sulfonylurea initiators, the plots of the individual covariates showed that initiators of both drugs had an increase in duration of diabetes and use of metformin during washout and a decrease in other oral glucose-lowering drugs during washout. For sulfonylurea initiators only, a minor increase in HbA1c at initiation and an increase in use of DPP-4i and GLP-1 during washout were seen. Across the different time blocks, the initiators of DPP-4i, compared to initiators of sulfonylurea, tended to have higher BMI, more prevalent use of other glucose-lowering drugs and statins during washout, longer duration of diabetes, lower HbA1c, and, in the late time blocks, higher Charlson Comorbidity Index.
Outcome analyses
The considerable overlap in propensity scores allowed for propensity score matching and thus propensity score-matched relative effectiveness estimation. Standardized differences suggested balanced comparison groups after matching (Tables S1–S2).
Figure 3A and 3B shows the difference in change in HbA1c and body weight, respectively, for the comparison of GLP-1 and insulin; nonsignificant differences in change in HbA1c, except from a peak in time block 4, and a relative increase in body weight for the insulin initiators compared to GLP-1 initiators in all time blocks were seen. Across time blocks, the relative increase in body weight was 3.57 kg (95% CI: 3.21, 3.92) for insulin initiators. Similarly, Figure 4A and 4B shows the difference in change in HbA1c and body weight, respectively compared to DPP-4i and sulfonylurea; a relative decrease in HbA1c and a relative increase in body weight for sulfonylurea initiators compared to DPP-4i initiators in all time blocks were seen. Across time blocks, the relative decrease in HbA1c was –0.34% (95% CI: –0.38, –0.30) and the relative increase in body weight was 1.58 kg (95% CI: 1.38, 1.78) for sulfonylurea initiators.
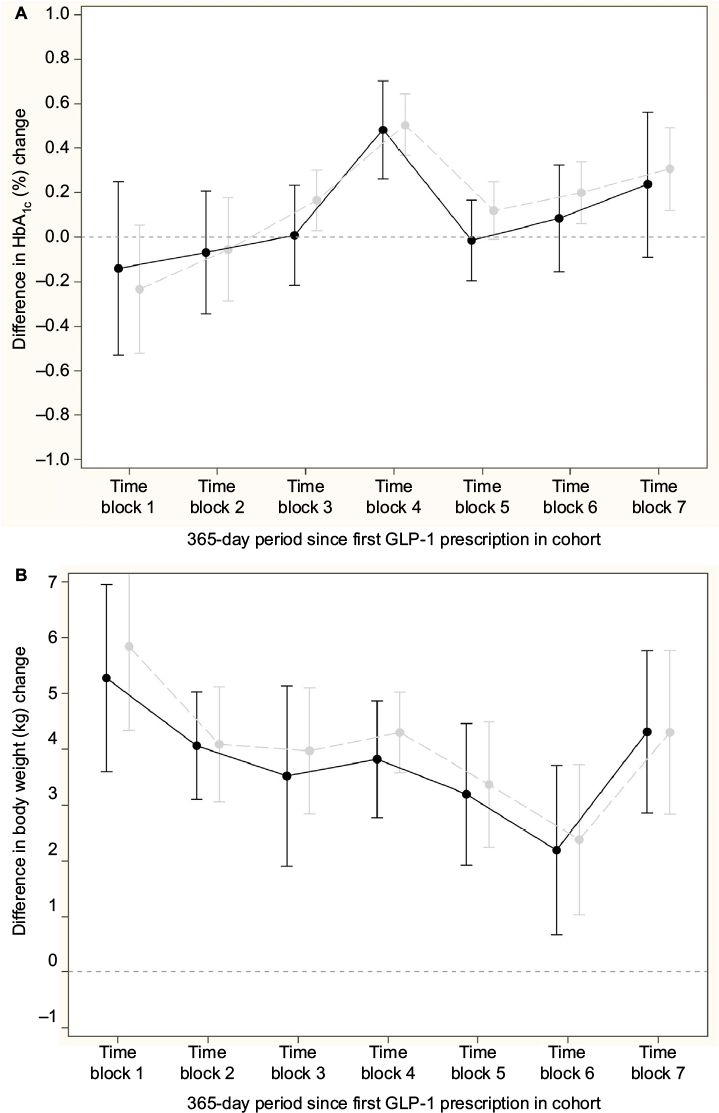 | Figure 3 Relative mean difference in change in HbA1C and weight (95% CI), GLP-1 (reference group) versus basal insulin initiator in the PP cohort adjusted for baseline HbA1c and weight, respectively. Notes: Gray: crude analyses, black: propensity score-matched patients. (A) HbA1c. (B) weight. Abbreviations: CI, confidence interval; GLP-1, glucagon-like peptide 1 analogs; HbA1c, glycated hemoglobin; PP, per-protocol. |
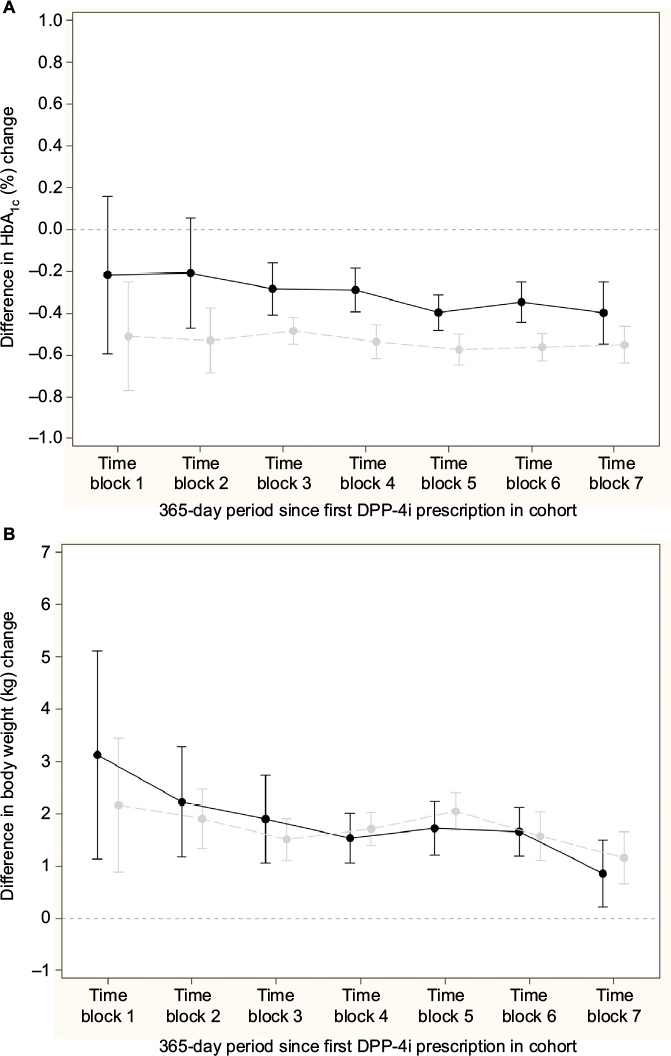 | Figure 4 Relative mean difference in change in HbA1C and weight (95% CI), DPP-4i (reference group) versus sulfonylurea initiator in the PP cohort adjusted for baseline HbA1c and weight, respectively. Notes: Gray: crude analyses, black: propensity score-matched patients. (A) HbA1c. (B) weight. Abbreviations: CI, confidence interval; DPP-4i, dipeptidyl peptidase-4 inhibitors; HbA1c, glycated hemoglobin; PP, per-protocol. |
Discussion
In this study, change in characteristics over time since beginning treatment with GLP-1 and DPP-4i in comparison with basal insulin and sulfonylurea, respectively, was investigated with focus on potential channeling bias. Propensity score matching was applied to assess the relative effect on change in HbA1c and weight. Channeling bias was not seen in the sense that the characteristics of GLP-1 and DPP-4i initiators changed over time after market entry, whereas the characteristics of the initiators of the established drugs, insulin and sulfonylurea, were stable. Distribution of propensity scores, used as an overall measure of differences in characteristics, showed that initiators of GLP-1 and insulin were somewhat different, although with a substantial overlap, in all time blocks. Initiators of DPP-4i and sulfonylurea were more comparable with an even greater overlap across time blocks and with a similar change in propensity scores over time. For the individual covariates, changes in characteristics were observed in both groups of GLP-1 and insulin initiators, and among DPP-4i and sulfonylurea initiators, as well as among initiators of insulin or sulfonylurea only.
There was sufficient overlap in distribution of characteristics so that propensity score matching was possible to conduct relative effectiveness analysis, even in the first year after launch. The outcome analyses suggest similar change in HbA1c for GLP-1 and insulin, except for the peak in time block 4. Overall, we consider this peak as a result of random variation given the absence of a difference in effects in the other time blocks. Insulin leads to weight gain compared to GLP-1 across time. The outcome analyses suggest a greater reduction in HbA1c and weight gain for sulfonylurea compared to DPP-4i.
It is hypothesized that patients initiating a newly launched drug are those most ill and with more use of comedications, but such differences will disappear over time when the new drug becomes more established.1,2 In this study, such a time trend was not observed. Also, it is hypothesized that patients initiating a newly launched drug have a history of suboptimal drug response or intolerance. Direct information on drug response was not available in data, but HbA1c level indicated how well-regulated patients with diabetes are. From the measurements of HbA1c at initiation, there was not much difference over time between initiators of GLP-1 and insulin, and DPP-4i initiators had lower HbA1c at initiation compared to initiators of sulfonylurea. This indicates that patients initiating the newly launched drugs had similar response profiles as those who initiated the established drugs. The findings in this study are in line with US studies comparing Sitagliptin (Merck & Co., Inc., Whitehouse Station, NJ, USA) with other oral glucose-lowering drugs where there were no changes in characteristics over time in the first years after launch.4–6
The use of real-world data is a necessity to investigate channeling bias, but it also brings along some general problems. The following potential limitations of the study were identified: real-world data are data of real-world patients, yet may suffer from missing information, which in this study was imputed by means of multiple imputation under the assumption that the missing data are missing at random. The quality of real-world data may be questioned as the data seldom are collected primarily for research purposes. This study concerns a period after implementation of quality outcome framework in CPRD, through which the highest possible data quality is achieved. It was a concern that for the majority of patients in CPRD, body weight was measured only once, and that measurement was carried forward. Consequently, weight change will be zero for those patients. This was, however, not the case in the data available for this study. The risk of confounding is present in observational studies of the effects of medical treatments; this study applied propensity score matching to take confounding into account. Not all relevant potential confounding factors are captured in the CPRD database (eg, diet and ethnicity), but the propensity score model included a diverse set of covariates describing demographics, clinical measurements, comedication including glucose-lowering drugs, and comorbidities. Immortal time bias, which is used to denote misclassification of observation time to exposure groups in survival analyses, was not an issue in this study since follow-up started at the first prescription made and was continued for the same duration for all subjects (ie, 180 days). However, the risk of selection bias in the PP cohort in this study might be an issue because individuals who have “survived” or stayed in the dataset at least 180 days were selected. However, the analyses of a PP cohort are relevant because our outcome measure of HbA1c is highly affected by exposure of the investigated drugs, which is more likely in the PP cohort. In this study, we looked at drug-class levels, and combination products of sulfonylurea and rosiglitazone, as well as products of DPP-4i and metformin, were classified as sulfonylurea and DPP-4i. Change in characteristics over time may be affected by launch of specific brands and the use of monotherapy and combination products. Future studies may investigate channeling related to individual drugs. Given the size of CPRD, we decided to compare and match patients within time blocks of 365-day duration. It is possible that time blocks of shorter duration would reveal channeling in the very early period after launch, but the duration of 365 days was suitable as this will ensure some level of statistical power.
Studies in other medical areas have observed channeling bias in CPRD data. In a study comparing new nonsteroidal anti-inflammatory drugs (NSAIDs) with old NSAIDs on the risk of gastrointestinal hemorrhage, baseline characteristics suggested that the new NSAID population had a higher baseline risk (possibly explained by new NSAIDs having a safer profile with regard to gastrointestinal toxicity). Outcome analyses seemed to adjust for confounding, including channeling bias, and showed a lower risk among patients treated with new NSAIDs.21 In the treatment of chronic obstructive pulmonary disease, corticosteroids have been compared with long-acting bronchodilators. More severe patients were prescribed corticosteroids, which indicated channeling. The adjusted analyses showed higher risk of pneumonia among corticosteroid users, but it was not clear to the authors if this was related to the effect of the drug alone, or if channeling bias had occurred.22
The use of a sequential cohort design, where analyses are done at several time points as in this study, is generally proposed to take channeling bias into account.23–27 Despite the fact that no major channeling bias was observed in this study, some variation over time was seen. Also, the characteristics of the initiators of the established drugs (basal insulin and sulfonylurea) changed over time, despite the time period observed (2006–2015) being more or less arbitrary for these drugs. This generally pleads for stratifying analyses on time. The sequential cohort design in this study used the same propensity score model across time blocks, as standardized differences suggested that balance across treatment groups were established after matching. However, individual propensity score models may be needed within each time block in other settings.
Conclusion
To conclude, we did not identify major channeling so that patients initiating the new glucose-lowering drugs differed importantly from patients initiating the established glucose-lowering drugs. In this study, the differences in patient characteristics and changes over time were at a magnitude where it was still possible to estimate propensity score and conduct matching to control for confounding in the outcome analyses, even in the first year after market entry. Change in the characteristics of initiators of established drugs generally advocates for time-dependent analyses.
Acknowledgment
The research leading to these results was conducted as part of the GetReal consortium. For further information, please refer to www.imi-getreal.eu. This article only reflects the personal views of the stated authors. The authors thank Vinay Mehta, Center for Observational & Real World Evidence (CORE), Merck Research Labs, North Wales, PA, USA, for his comments on the protocol of this study. Preliminary results of this work were presented as a poster at the 32nd International Conference on Pharmacoepidemiology & Therapeutic Risk Management (ICPE) in Dublin (August 25–28, 2016).
Author contributions
MZA, RHHG, MSA and OHK conceived the study. MZA did the coding with inputs from BLT and EA. All authors analyzed data and interpreted the results. MZA drafted the manuscript. All authors revised the manuscript. All authors read and approved the final manuscript and agreed to be accountable for all aspects of the work.
Disclosure
The work leading to these results has received support from the Innovative Medicines Initiative Joint Undertaking (IMI JU) under grant agreement number (115546), resources of which are composed of financial contribution from the European Union’s Seventh Framework Programme (FP7/2007–2013) and the European Federation of Pharmaceutical Industries and Association (EFPIA) companies in kind contribution. In addition, as a special form of the IMI JU grant, University Medical Center, Utrecht, the Netherlands, received a direct financial contribution from Novo Nordisk A/S to support work on this article. MZA, BLT, and EA belong to EFPIA member companies in the IMI JU, and costs related to their part in the research were carried by the respective company as in kind contribution under the IMI JU scheme.
MZA is employed by Novo Nordisk A/S as a postdoc in the IMI GetReal project. BLT and EA are employed by Novo Nordisk A/S and are shareholders in Novo Nordisk A/S. RHHG, MSA, and OHK have no conflicts of interest. The authors report no other conflicts of interest in this work.
References
Schneeweiss S, Gagne JJ, Glynn RJ, Ruhl M, Rassen JA. Assessing the comparative effectiveness of newly marketed medications: methodological challenges and implications for drug development. Clin Pharmacol Ther. 2011;90(6):777–790. | ||
Petri H, Urquhart J. Channeling bias in the interpretation of drug effects. Stat Med. 1991;10(4):577–581. | ||
Segal JB, Dy SM, Millman EA, Herbert R, Bass EB, Wu A. Diffusion into use of exenatide for glucose control in diabetes mellitus: a retrospective cohort study of a new therapy. Clin Ther. 2007;29(8):1784–1794. | ||
Brodovicz KG, Kou TD, Alexander CM, et al. Recent trends in the characteristics of patients prescribed sitagliptin and other oral antihyperglycaemic agents in a large U.S. claims database. Int J Clin Pract. 2013;67(5):449–454. | ||
Cai B, Katz L, Alexander CM, Williams-Herman D, Girman CJ. Characteristics of patients prescribed sitagliptin and other oral antihyperglycaemic agents in a large US claims database. Int J Clin Pract. 2010;64(12):1601–1608. | ||
Zhang Q, Rajagopalan S, Mavros P, et al. Baseline characteristic differences between patients prescribed sitagliptin vs. other oral antihyperglycemic agents: analysis of a US electronic medical record database. Curr Med Res Opin. 2010;26(7):1697–1703. | ||
Khan NF, Harrison SE, Rose PW. Validity of diagnostic coding within the General Practice Research Database: a systematic review. Br J Gen Pract. 2010;60(572):e128–e136. | ||
Williams T, van Staa T, Puri S, Eaton S. Recent advances in the utility and use of the General Practice Research Database as an example of a UK Primary Care Data resource. Ther Adv Drug Saf. 2012;3(2):89–99. | ||
Tate AR, Beloff N, Al-Radwan B, et al. Exploiting the potential of large databases of electronic health records for research using rapid search algorithms and an intuitive query interface. J Am Med Inform Assoc. 2014;21(2):292–298. | ||
Herrett E, Gallagher AM, Bhaskaran K, et al. Data resource profile: Clinical Practice Research Datalink (CPRD). Int J Epidemiol. 2015;44(3):827–836. | ||
The Clinical Practice Research Datalink (CRPD) [homepage on the Internet]. Available from: https://www.cprd.com. Accessed December 5, 2016. | ||
Mulnier HE, Seaman HE, Raleigh VS, et al. Mortality in people with type 2 diabetes in the UK. Diabet Med. 2006;23(5):516–521. | ||
Managing blood glucose in adults with type 2 diabetes [homepage on the Internet]. The National Institute for Health and Care Excellence (NICE). Available from: https://pathways.nice.org.uk/pathways/type-2-diabetes-in-adults#path=view%3A/pathways/type-2-diabetes-in-adults/managing-blood-glucose-in-adults-with-type-2-diabetes.xml&content=view-node%3Anodes-insulin-based-treatments/. Accessed November 25, 2016. | ||
Khan NF, Perera R, Harper S, Rose PW. Adaptation and validation of the Charlson Index for Read/OXMIS coded databases. BMC Fam Pract. 2010;11:1. | ||
Sterne JA, White IR, Carlin JB, et al. Multiple imputation for missing data in epidemiological and clinical research: potential and pitfalls. BMJ. 2009;338:b2393. | ||
Groenwold RH, Donders AR, Roes KC, Harrell FE Jr, Moons KG. Dealing with missing outcome data in randomized trials and observational studies. Am J Epidemiol. 2012;175(3):210–217. | ||
Little RJA, Rubin DB. Statistical Analysis with Missing Data. 2nd ed. New York (NY): Wiley-Interscience; 2002. | ||
Austin PC. Balance diagnostics for comparing the distribution of baseline covariates between treatment groups in propensity-score matched samples. Stat Med. 2009;28(25):3083–3107. | ||
Parsons LS. Performing a 1:N Case-Control match on propensity score. Paper 165–129 presented at: SUGI 29 Proceedings; May 9–12; 2004; Montreal Canada. | ||
Lepage B, Lamy S, Dedieu D, Savy N, Lang T. Estimating the causal effect of an exposure on change from baseline using directed acyclic graphs and path analysis. Epidemiology. 2015;26(1):122–129. | ||
MacDonald TM, Morant SV, Goldstein JL, Burke TA, Pettitt D. Channelling bias and the incidence of gastrointestinal haemorrhage in users of meloxicam, coxibs, and older, non-specific non-steroidal anti-inflammatory drugs. Gut. 2003;52(9):1265–1270. | ||
DiSantostefano RL, Sampson T, Le HV, Hinds D, Davis KJ, Bakerly ND. Risk of pneumonia with inhaled corticosteroid versus long-acting bronchodilator regimens in chronic obstructive pulmonary disease: a new-user cohort study. PloS One. 2014;9(5):e97149. | ||
Helin-Salmivaara A, Lavikainen P, Aarnio E, Huupponen R, Korhonen MJ. Sequential cohort design applying propensity score matching to analyze the comparative effectiveness of atorvastatin and simvastatin in preventing cardiovascular events. PloS one. 2014;9(3):e90325. | ||
Dusetzina SB, Mack CD, Sturmer T. Propensity score estimation to address calendar time-specific channeling in comparative effectiveness research of second generation antipsychotics. PLoS One. 2013;8(5):e63973. | ||
Mack CD, Glynn RJ, Brookhart MA, et al. Calendar time-specific propensity scores and comparative effectiveness research for stage III colon cancer chemotherapy. Pharmacoepidemiol Drug Saf. 2013;22(8):810–818. | ||
Reams BD, O’Malley CD, Critchlow CW, Lauffenburger JC, Brookhart MA. Changing patterns of use of osteoporosis medications in the years after launch: implications for comparative effectiveness research. Pharmacoepidemiol Drug Saf. 2014;23(3):251–260. | ||
Gagne JJ, Bykov K, Willke RJ, Kahler KH, Subedi P, Schneeweiss S. Treatment dynamics of newly marketed drugs and implications for comparative effectiveness research. Value Health. 2013;16(6):1054–1062. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.