")
Back to Journals » Open Access Emergency Medicine » Volume 14
Assessment and Management of Immune Thrombocytopenia (ITP) in the Emergency Department: Current Perspectives
Authors Zitek T , Weber L , Pinzon D, Warren N
Received 9 November 2021
Accepted for publication 20 January 2022
Published 29 January 2022 Volume 2022:14 Pages 25—34
DOI https://doi.org/10.2147/OAEM.S331675
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Hans-Christoph Pape
Tony Zitek,1,2 Luke Weber,1 Dominique Pinzon,1 Nicole Warren1– 3
1Department of Emergency Medicine, Mount Sinai Medical Center, Miami Beach, FL, USA; 2Department of Emergency Medicine, Herbert Wertheim College of Medicine at Florida International University, Miami, FL, USA; 3Department of Medical Education, University of Miami School of Medicine, Miami, FL, USA
Correspondence: Tony Zitek
Mount Sinai Medical Center, Department of Emergency Medicine, 4300 Alton Road, Miami Beach, FL, 33140, USA
, Tel +1 305-674-2121 x56632
, Email [email protected]
Abstract: Immune thrombocytopenia (ITP) is characterized by a platelet count less than 100 × 10^9/L without anemia or leukopenia. Patients with ITP may be asymptomatic, or they may have mild bleeding like petechiae, purpura, or epistaxis. In rare cases, they may present to the emergency department (ED) with life-threatening bleeding as a result of their thrombocytopenia. The emergency physician should thus be prepared to diagnose ITP and treat the bleeding that can result from it. The diagnosis of ITP requires excluding secondary causes of thrombocytopenia, and in the ED, the bare minimum workup for ITP includes a complete blood count and a peripheral blood smear. The peripheral blood smear should show a small number of large platelets with normal morphology, and there should not be an increased number of schistocytes. Many patients with ITP require no emergent treatment. However, if a patient with suspected ITP presents to the ED with critical hemorrhage, the emergency physician should initiate treatment with a platelet transfusion, corticosteroids, and intravenous immune globulin (IVIG) as soon as possible. For less severe bleeding, platelet transfusions are not recommended, and the treatment consists of corticosteroids by themselves or in conjunction with IVIG.
Keywords: ITP, symptoms, diagnosis, treatment
Introduction
Immune thrombocytopenia (ITP), formerly known as immune thrombocytopenic purpura, is a common cause of thrombocytopenia, but cases requiring diagnosis or treatment in the emergency department (ED) are sufficiently rare that some emergency physicians may feel uncomfortable with this disease. The name of the disease was changed to keep the commonly utilized abbreviation (“ITP”), but to clarify that many patients do not have purpura at the time of diagnosis.1 ITP is characterized by platelet counts less than 100 × 10^9/L. Previously, the low platelet counts of ITP were attributed to autoantibody-mediated destruction of platelets. However, more recent evidence suggest that immune-mediated decreased platelet production also plays a role in the pathogenesis of ITP.2,3
ITP is generally divided into two categories: primary ITP and secondary ITP. Primary ITP is an acquired immune thrombocytopenia, whereas secondary ITP is thrombocytopenia triggered by another disorder. These inciting disorders include other autoimmune diseases (systemic lupus erythematosus, rheumatoid arthritis, etc.), endocrine disorders (Addison’s disease, hypothyroidism, etc), chronic infections (human immunodeficiency virus, hepatitis C, H. pylori, etc.), lymphoproliferative neoplasms (chronic lymphocytic leukemia, lymphoma), as well as a variety of medications and vaccines.4
As per a consensus statement from an international working group on ITP, the classification of ITP involves 3 phases.5 Newly diagnosed ITP defines the first 3 months post-diagnosis. Persistent ITP indicates symptoms remaining for 3–12 months. Chronic ITP refers to symptoms lasting longer than 12 months. An additional phase is termed refractory ITP, which consists of cases that fail to resolve with splenectomy and require further therapeutic intervention.4–6
ITP is a diagnosis of exclusion after other causes of thrombocytopenia are deemed unlikely. A complete blood count (CBC) should demonstrate isolated thrombocytopenia without presence of anemia or leukopenia.1 Treatment goals must first consider severity of bleeding. In cases of critical hemorrhage, standard management includes platelet transfusion, corticosteroids, and intravenous immune globulin (IVIG).7
This paper presents an updated review of current literature and guidelines regarding ITP including an analysis of epidemiology, clinical presentation, diagnosis, and treatment in both pediatric and adult populations. We focus on what is most relevant for the emergency physician.
Epidemiology
ITP affects both children and adults. In children, the incidence of ITP is about 1.9–6.4 per 100,000 children per year.8 However, this figure is likely an underestimate of the affected population, as ITP may be asymptomatic, and these numbers primarily reflect patients with symptomatic thrombocytopenia who are seeking treatment. In a large prospective study, 78% of pediatric ITP patients were diagnosed between ages 0–7 with a slight male predominance.9 The male to female ratio in children decreases with increasing age, and reverses in adulthood. Although many children will experience spontaneous resolution of ITP without medical intervention, around 25% will progress to chronic ITP.9
Regarding adults, one large review of published articles reported the incidence of ITP to be around 3.3 per 100,000 per year.8 As ITP is frequently a chronic disease in adults, the prevalence is higher than the incidence, and one study reported the prevalence of ITP to be 12.1 per 100,000 adults per year.10 Female predominance is well documented in adults with ITP. Increased overall incidence of autoimmune diseases in adult females is thought to be a factor. Both male and female incidence of ITP increases with increasing age.11 Without medical intervention, most adults will progress to chronic ITP.6
Clinical Presentation
Bleeding
The symptoms of ITP are dependent upon the degree of thrombocytopenia. Signs and symptoms typically begin occurring with platelet counts less than 100 × 109/L, but bleeding is rare until platelet counts drop below 30 × 109/L.11 Cutaneous and mucosal bleeding usually occurs with platelet numbers below 20 × 109/L, while severe hemorrhagic cases such as intracranial, intestinal, or other internal bleeding typically occurs with platelet counts less than 10 × 109/L.4 Many patients with ITP are asymptomatic, and their reduced platelet count is often discovered incidentally.
Adult and pediatric with symptomatic ITP have similar clinical presentations, but there are also some notable differences. Both adult and pediatric patients often present with symptoms related to bleeding. Cutaneous bleeding (such as petechiae or purpura) is the most common bleeding manifestation in both adults and children — 63% of adults with bleeding-related symptoms from ITP have cutaneous bleeding12 while 86% of children do.13 Purpura that occur as a result of thrombocytopenia are not palpable, not pruritic, and preferentially develop in dependent areas of the body (Figure 1A). This contrasts with purpura from a vasculitis, which are palpable, and less likely to occur in dependent areas of the body.14 While disorders of coagulation may cause cutaneous bleeding, they tend to cause more bleeding into joints and muscles rather than purpura.14
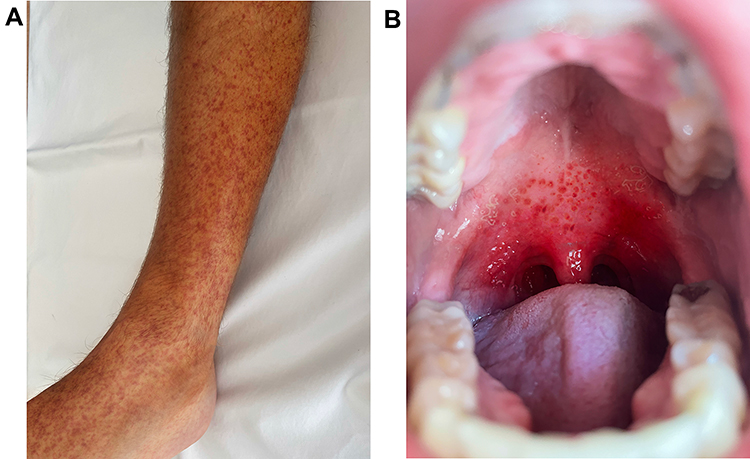 |
Figure 1 (A) Example of cutaneous purpura. (B) Example of oral (“wet”) purpura. Note: Original picture; consent obtained from subject for publication. |
After cutaneous bleeding, the next most common bleeding symptoms in both adults and children with ITP are oral bleeding and nasal bleeding. These symptoms may range from very mild such that the patient does not seek medical care (such as epistaxis only with nose blowing) to severe (such as epistaxis requiring surgery). One notable type of oral bleeding is “oral purpura”, also called “wet purpura”. These are hemorrhagic blisters of the oral mucosa (Figure 1B). Some prior data suggest that the presence of oral purpura predict subsequent major bleeding from ITP.15
While minor bleeding is common in ITP, severe or critical bleeding is rare in both adult and pediatric patients. Indeed, patients with ITP may occasionally present with heavy bleeding from hematuria, melena, rectal bleeding, menorrhagia, spontaneous intra-abdominal bleeding, or intracerebral hemorrhage (ICH). One systematic review reported that severe bleeding occurs more commonly in children with ITP than adults — with reported rates of 9.6% for adults and 20.2% for children.16 Most of the studies included in this systematic review considered extensive mucosal bleeding as the minimum definition of “severe bleeding,” but the reported rates of severe bleeding should be interpreted with caution due to variability in definitions.
Amongst the types of bleeding that can occur from ITP, cases of ICH are the most deadly. Although severe or critical bleeding (in general) may occur more frequently in pediatric patients with ITP than in adults, ICH seems to occur more commonly in adults than in children (1.4% versus 0.4%, respectively).16 In particular, ICH is identified more frequently in adults with chronic ITP and less frequently in those with acute ITP.16
Symptoms Not Due to Bleeding
Outside of bleeding-related symptoms, a couple of other symptoms are worth mentioning. Fatigue is a common symptom in both adults and pediatric patients with ITP, but it is a more common symptom in adults. One study reported that for adult patients diagnosed with ITP more than 1 month after onset of symptoms, fatigue was the most common symptom and was noted in 58% of patients.13 Meanwhile fatigue has been reported to occur in around 22% of pediatric patients.4
Despite the fact that the primary symptoms of ITP involve bleeding, thrombosis is more common in patients with ITP as compared to controls.17–19 Thrombosis in ITP, however, is still uncommon, so the emergency physician should not search for thrombosis unless there is already a clinical suspicion for thrombosis.
Compared with the general population, patients with ITP have a significantly increased risk of infection, as well as a 1.5-fold higher overall mortality rate.20
ITP in Pregnancy
ITP may occur during pregnancy with an estimated incidence of 1 in 1000 to 1 in 10,000 pregnancies.21 Gestational thrombocytopenia of pregnancy (GT) remains the most common cause of low platelet numbers in pregnancy, and it can be difficult to distinguish from ITP. Both disorders are diagnoses of exclusion, but the platelet count in GT rarely falls below 70 × 10^9/L, and typically does not cause bleeding. In contrast, ITP can cause a much lower platelet count and increases the risk of maternal and neonatal bleeding.21 Cases of maternal or neonatal bleeding are rare when patients are properly medically managed. However, women diagnosed with ITP prior to pregnancy appear to have a higher incidence of fetal loss (11.2% vs 3.9% in women diagnosed during pregnancy) and lower fetal birth weight (17.9% vs 9.7%). A higher incidence of premature birth has also been reported.22
Thrombocytopenia and COVID-19
Patients with COVID-19 may develop thrombocytopenia as a result of disseminated intravascular coagulation, sepsis, or medication reactions.23 Additionally, COVID-19 has become a well-documented cause of ITP.23–25 In a systemic review, Bhattacharjee and Banerjee noted that 71% of COVID-19-induced ITP were found in elderly patients, and that 75% of cases had moderate to severe COVID-19.25 In most cases of thrombocytopenia in patients associated with COVID-19, the platelet count does not fall below 100 × 10^9/L.26 Although lower platelet counts have been associated with increased risk of severe disease and mortality in patients with COVID-19, severe bleeding has been uncommon in affected patients.27,28 The vast majority of ITP cases related to COVID-19 have a favorable prognosis, with most patients responding well to standard ITP treatments discussed below.24,25,27
Diagnosis
The diagnosis of ITP is one of exclusion. According to an International Working Group consensus panel of both adult and pediatric experts, ITP is defined as a platelet count less than 100 × 109/L in the absence of other causes of thrombocytopenia.5 A broad differential diagnosis should be maintained, and focus should be placed on identifying and excluding other conditions that may cause thrombocytopenia.
Additionally, amongst patients with ITP, it is important to distinguish primary and secondary ITP. In particular, the physician should thoroughly review the patient’s recent medications as drug-induced immune thrombocytopenia is often clinically indistinguishable from primary ITP, so it may be missed even if the appropriate diagnostic tests are ordered. Quinine was the first drug to be identified as causing immune-mediated thrombocytopenia,29 and is thus frequently mentioned in textbooks as a cause of drug-induced immune thrombocytopenia.30,31 However, many emergency physicians rarely see a patient on this medication. Some more widely used medications that are known to cause drug-induced immune thrombocytopenia include carbamazepine, ceftriaxone, ibuprofen, mirtazapine, penicillin, trimethoprim-sulfamethoxazole, and vancomycin.29 If a patient has received heparin within the last 10 days, heparin-induced thrombocytopenia should be considered.32
Recommended Diagnostic Tests in the ED
After performing a careful history and physical examination, the emergency physician will likely instinctively order a CBC if considering ITP. As mentioned above, the CBC should demonstrate normal cell lines other than thrombocytopenia. In addition to a CBC, a peripheral blood smear is a necessary test for a patient with possible ITP.33 The peripheral smear should show large, well-granulated platelets, few in number. Platelet clumping on a peripheral smear indicates pseudothrombocytopenia. Morphologic platelet abnormalities may indicate a hereditary platelet disorder. An elevated schistocyte count may indicate disseminated intravascular coagulation, thrombotic thrombocytopenic purpura, or another microangiopathic hemolytic anemia.
While a history, physical exam, CBC, and peripheral smear may be sufficient for the emergency physician to make a presumptive diagnosis of ITP and appropriately disposition the patient, the American Society of Hematology recommends testing all adult patients with suspected ITP for hepatitis C virus and human immunodeficiency virus as specific treatment of these diseases may substantially increase the platelet count.34 Therefore, if available, we recommend these viral tests be performed in the ED on adults with suspected ITP. These viral tests are not necessary in pediatric patients, unless clinically indicated.
Additionally, in practice, we typically order a comprehensive metabolic panel along with the above recommended diagnostic tests. No specific abnormalities are expected on a comprehensive metabolic panel for patients with primary ITP, but a number of abnormalities might be identified for secondary etiologies. Similarly, while not mandatory, we frequently order coagulation studies (a prothrombin time and an activated partial thromboplastin time) which may be helpful in that they should be normal in ITP but prolonged in liver disease or disseminated intravascular coagulation.34,35
There are no high quality data to recommend when a head CT scan should be ordered in a patient with ITP who has a headache. However, based on the fact that the risk of ICH is substantially increased when the platelet count is less than 10 × 109/L,4 we recommend a head CT scan be routinely ordered in ITP patients with a headache and a platelet count less than 10 × 109/L. Additionally, given the potential severity of a missed ICH in a patient with thrombocytopenia, we recommend the consideration of a head CT scan in patients with a headache and a platelet count of 10–30 × 109/L.
Diagnostic Tests to Avoid or Defer in the ED
In general, if the diagnostic tests described above are consistent with primary ITP, no additional testing is warranted in the ED. One specific test to avoid in the ED is antiplatelet antibody testing. A prospective study designed to detect platelet-bound autoantibodies reactive against GPs IIb/IIIa, Ib/IX, or Ia/IIa found the sensitivity of antiplatelet antibody testing to be 53%36 — which is likely too low for the test to be useful. Additionally, while bone marrow examination may sometimes be utilized to exclude malignancy or myelodysplastic syndrome in atypical or refractory cases of suspected ITP, it is not recommended in the vast majority of cases and need not be arranged in the ED.
Patients who have joint pain, malar rash, or other symptoms that may suggest a rheumatologic condition might benefit from immunologic studies (like antinuclear antibodies), but since the definitive diagnosis of a rheumatologic condition is unlikely in the ED, it may be best for the emergency physician to defer immunologic studies to the outpatient setting or a specialist.
Treatment
Management of ITP differs depending on if it is primary ITP or secondary ITP. In the remainder of this section, we will discuss treatments that could be used for either primary or secondary ITP. However, if a patient has secondary ITP, the critical issue is treating the underlying disorder, or if drug-induced, discontinuing the medication. Regardless of the cause of the ITP, the goal of treatment is to prevent or treat significant bleeding, not to normalize the platelet count. This is especially true in the ED. In general, the treatment of ITP is based on low quality evidence and expert opinion as large randomized trials are lacking. As such, our management of ITP follows the 2019 clinical practice guidelines from the American Society of Hematology and a 2019 international consensus report.37,38
When considering the management of ITP in the ED, the first thing to consider is which patients require emergent treatment, and which can be observed or referred for outpatient treatment. In general, the approach to treatment in the ED is the same for children and adults with ITP, but a less aggressive treatment strategy might be chosen for children with ITP because they have higher rates of spontaneous resolution than adults.11 If there is no significant bleeding and the platelet count is greater than 30,000, typically no emergent treatment is indicated at that time. Therefore, many cases of ITP can be managed as an outpatient, optimally under the care of a hematologist. If significant bleeding is present, the platelet count is less than 30,000, or the patient needs an increased platelet count prior to a procedure, then emergent treatment in the ED may be indicated. For those without significant bleeding and platelet count less than 30,000, we recommend a hematology consultation (if available) prior to treatment, as the indications to initiate treatment are multifactorial and decided on an individual basis. On the other hand, for patients with ITP who have critical bleeding, the emergency physician should initiate treatment for ITP immediately. After initiation of treatment, hematology consultation or transfer to a facility with hematology services should proceed.
For the purposes of guiding treatment decisions, we categorize the severity of bleeding into minor, severe and critical bleeding. As per expert panel consensus, critical bleeding is defined as bleeding in a critical anatomical site (intracranial, intraspinal, intraocular, pericardial, etc.), bleeding that results in hemodynamic instability, or bleeding that results in respiratory compromise.39 Severe bleeding is less formally defined, but we consider it to be extensive mucosal bleeding or heavier bleeding (such as gastrointestinal, intra-abdominal, or uterine) that does not meet the definition of critical bleeding. Minor bleeding is bleeding that does not meet the definition for severe or critical and would be more consistent with petechiae, ecchymoses, and non-severe mucous membrane bleeding that is more easily controlled.
Emergency physicians need to be most familiar with how to treat ITP patients who have critical bleeding. For these patients, platelet transfusions, corticosteroids, and IVIG are indicated. We will also discuss some other therapeutic options, including anti-D immune globulin, hemostatic agents, thrombopoietin receptor agonists, and splenectomy, However, these are not standard ED therapies for ITP. A summary of our recommended treatment strategies is shown in Table 1.
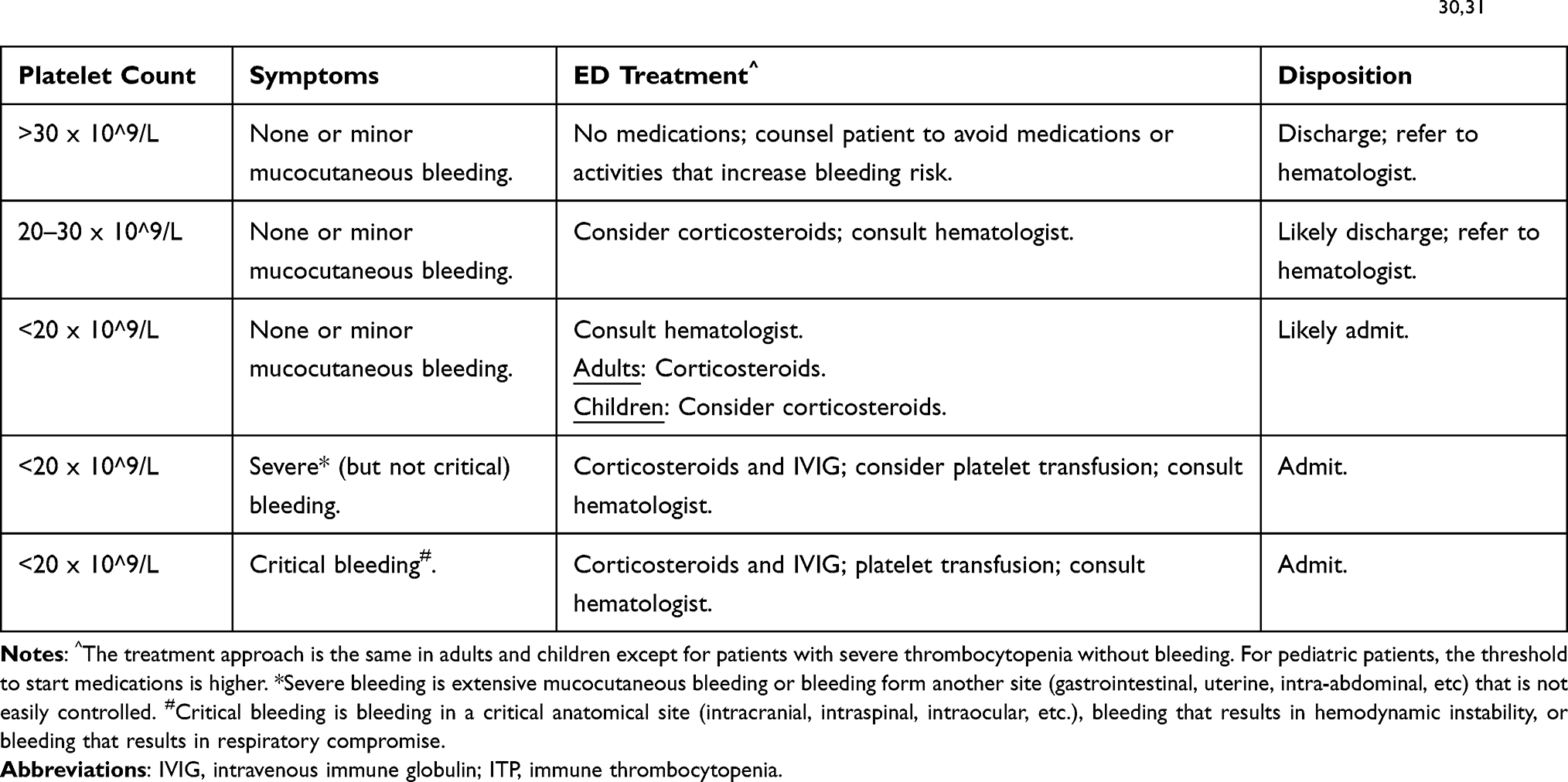 |
Table 1 Treatment Strategies for ITP Based on Platelet Count and Symptoms. These Recommendations are Primarily Based on the 2019 Clinical Practice Guidelines from the American Society of Hematology and a 2019 International Consensus Report.30,31 |
Platelet Transfusion
Platelet transfusions should only be routinely initiated if critical bleeding is present, or if an emergent major surgery is needed. In some cases, it may be reasonable to administer a platelet transfusion for severe bleeding that is not quite critical, if there is evidence of clinical deterioration.
Platelet transfusion is the fastest way to increase platelet counts. The typical dosing is 1 apheresis unit (preferred), or 4–6 units of pooled platelets. Transfused platelets will not result in disease progression, as many once believed; however, they will likely be quickly destroyed by the disease process and are thus only a temporizing measure.40 Platelet transfusions should not be used solely, and other systemic therapies are necessary. IVIG may also augment the response to platelet transfusion.41
Corticosteroids
For patients who need treatment in the ED, corticosteroids are the preferred first-line treatment option. Corticosteroids will raise the platelet count; however, the initial response usually takes at least a couple of days.42 Possible corticosteroids options include prednisone 1mg/kg PO per day for one to two weeks, dexamethasone 0.6 mg/kg (maximum 40 mg) PO/IV per day for four days, and methylprednisolone 30 mg/kg (maximum 1 g) IV per day for three days. With regards to the choice of corticosteroids, a systematic review of previously untreated ITP patients found that platelet count responses were higher after 14 days of treatment with dexamethasone as compared to prednisone. However, after 6 months, there was no difference in platelet counts between the groups.43 Given the potential benefits of a faster rise in platelet counts and the shorter duration of treatment, we recommend dexamethasone over prednisone for patients with non-critical bleeding. For ITP patients with critical bleeding, we prefer intravenous methylprednisolone, but dexamethasone may also be appropriate as there are no clinical trials comparing the two.
IVIG
We recommend emergency physicians initiate IVIG in addition to corticosteroids for patients with critical or major bleeding. As per guideline recommendations,37 some patients with mild bleeding or no bleeding might benefit from IVIG, but we recommend deferring that decision to a hematologist, if possible. IVIG increases platelet counts faster than corticosteroids, and the initial increase in platelet count usually occurs in 1 to 3 days.44 Common dosing of IVIG is 1 g/kg IV. This dose may be repeated daily until the platelet count is greater than 50 x 10^9/L.
Anti-D Immune Globulin
Some practitioners use anti-d immune globulin as an alternative to IVIG for Rh positive patients. However, IVIG has more research data to support its use in ITP than anti-D immune globulin. Additionally, the use of anti-d immune globulin is associated with an increased risk of severe hemolytic transfusion reactions.45–47 Thus, if IVIG is available, we recommend using IVIG rather than anti-D immune globulin.
Other Therapies
For patients with ITP, the emergency physician should focus on whether to administer the primary treatments: platelet transfusions, corticosteroids, and IVIG. However, in special circumstances or refractory cases, other therapies may be considered. These include hemostatic products, thrombopoietin receptor agonists, splenectomy, other immunosuppressive agents, and addressing other bleeding risk factors.
Hemostatic products include tranexamic acid (TXA) and epsilon aminocaproic acid (EACA). TXA is a common hemostatic product that is likely familiar to many emergency physicians. While some cases series have reported a benefit of TXA in ITP,48 there are no high quality clinical data to support its routine use in ITP. Additionally, a randomized trial found no benefit to the use of TXA in patients with thrombocytopenia associated with hematologic malignancies.49 Therefore, we recommend against the routine use of TXA in ITP. That being said, there are some instances when TXA may be useful in patients with ITP who have bleeding. For example, there is some (weak) data to support the use of topical TXA in patients with epistaxis who do not have ITP.50 Therefore, we believe it is reasonable to use topical TXA in patients with epistaxis associated with ITP.
EACA may be used in those with critical bleeding from ITP. However, data to support this practice are based on case series data.51–53 There are no high quality clinical trials to support its use. Thus, we recommend against the routine use of EACA for patients with ITP.
Thrombopoietin receptor agonists such as romiplostim or eltrombopag do not work fast enough to impact the outcomes of acute bleeding events, but may play an important role in the management of chronic ITP.54 Therefore, we recommend the initiation of these medications in the ED only if instructed by a hematologist.
For patients with severe or refractory cases of ITP, splenectomy, rituximab, mycophenolate mofetil, and other immunosuppressive agents may be considered. However, these treatments are nonstandard, and we do not recommend emergency physicians initiate these treatments without the close guidance of a hematologist. Of note, a recently published study found that the addition of mycophenolate mofetil to corticosteroids for stable ITP patients resulted in greater increases in platelet counts as compared to the use of corticosteroids alone, but those who received mycophenolate mofetil also had worse quality-of-life outcomes.55 As such, we do not recommend emergency physicians initiate this medication unless prompted by a hematologist.
Finally, another component of the treatment of ITP involves addressing other bleeding risk factors. Patients with ITP may be taking antiplatelet or anticoagulant medications. There are no high quality data to support a platelet threshold at which these medications should be discontinued. However, the risks and benefits of continuing these medications needs to be assessed, and consideration should be made for stopping these medications. For patients with severe or critical bleeding, they should be stopped and anticoagulants should be reversed. Patients with ITP should also be counselled to avoid alcohol and nonsteroidal anti-inflammatory drugs, as they may increase the risk of bleeding. Additionally, ITP patients should be counselled to avoid activities that might put them at increased risk of a traumatic injury (such as contact sports).
Conclusion
Emergency physicians occasionally need to diagnose and treat patients with ITP. ITP can be presumptively diagnosed in the ED with a CBC and a peripheral blood smear. The CBC should demonstrate thrombocytopenia without leukopenia or anemia, and the blood smear should show morphologically normal platelets. While many patients with ITP require no emergent treatment and can be managed by a hematologist outside the ED, the emergency physician should be prepared to treat bleeding ITP patients even when a hematologist is not available for consultation. Treatment strategies are generally not based on high quality clinical strategies, but based on clinical guidelines, we recommend emergency treatment with corticosteroids, IVIG, and platelet transfusions for patients with ITP and critical bleeding.
Disclosure
The authors report no conflicts of interest in this work.
References
1. McCrae K. Immune thrombocytopenia: no longer ‘idiopathic’. Cleve Clin J Med. 2011;78(6):358–373. doi:10.3949/ccjm.78gr.10005
2. Nuent D, McMillan R, Nichol JL, Slichter SJ. Pathogenesis of chronic immune thrombocytopenia: increased platelet destruction and/or decreased platelet production. Br J Haematol. 2009;146(6):585. doi:10.1111/j.1365-2141.2009.07717.x
3. Toltl LJ, Arnold DM. Pathophysiology and management of chronic immune thrombocytopenia: focusing on what matters. Br J Haematol. 2011;152(1):52. doi:10.1111/j.1365-2141.2010.08412.x
4. Onisâi M, Vlădăreanu A-M, Spînu A, Găman M, Bumbea H. Idiopathic thrombocytopenic purpura (ITP) – new era for an old disease. Rom J Intern Med. 2019;57(4):273–283.
5. Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113(11):2386. doi:10.1182/blood-2008-07-162503
6. Zufferey A, Kapur R, Semple J. Pathogenesis and therapeutic mechanisms in immune thrombocytopenia (ITP). JCM. 2017;6(2):16. doi:10.3390/jcm6020016
7. Samson M, Fraser W, Lebowitz D. Treatments for primary immune thrombocytopenia: a review. Cureus. 2019;11(10):e5849. doi:10.7759/cureus.5849
8. Terrell DR, Beebe LA, Vesely SK, Neas BR, Segal JB, George JN. The incidence of immune thrombocytopenic purpura in children and adults: a critical review of published reports. Am J Hematol. 2010;85(3):174–180. doi:10.1002/ajh.21616
9. Zeller B, Rajantie J, Hedlund-Treutiger I, et al. Childhood idiopathic thrombocytopenic purpura in the Nordic countries: epidemiology and predictors of chronic disease: childhood ITP in the Nordic countries. Acta Paediatrica. 2007;94(2):178–184. doi:10.1111/j.1651-2227.2005.tb01887.x
10. Terrell DR, Beebe LA, Neas BR, Vesely SK, Segal JB, George JN. Prevalence of primary immune thrombocytopenia in Oklahoma. Am J Hematol. 2012;87(9):848–852. doi:10.1002/ajh.23262
11. Despotovic JM, Grimes AB. Pediatric ITP: is it different from adult ITP? Hematology Am Soc Hematol Educ Program. 2018;2018(1):405–411. doi:10.1182/asheducation-2018.1.405
12. Cooper N, Kruse A, Kruse C, et al. Immune thrombocytopenia (ITP) World Impact Survey (iWISh): patient and physician perceptions of diagnosis, signs and symptoms, and treatment. Am J Hematol. 2021;96(2):188–198. doi:10.1002/ajh.26045
13. Kuhne T, Berchtold W, Michaels LA, et al. Newly diagnosed immune thrombocytopenia in children and adults: a comparative prospective observational registry of the Intercontinental Cooperative Immune Thrombocytopenia Study Group. Haematologica. 2011;96(12):1831–1837. doi:10.3324/haematol.2011.050799
14. Charrot SC, Sellar RS, Manson JJ. Purpura. Br J Hosp Med. 2017;78(10):C147–C151. doi:10.12968/hmed.2017.78.10.C147
15. Mithoowani S, Cervi A, Shah N, et al. Management of major bleeds in patients with immune thrombocytopenia. J Thromb Haemost. 2020;18(7):1783. doi:10.1111/jth.14809
16. Neunert C, Noroozi N, Norman G, et al. Severe bleeding events in adults and children with primary immune thrombocytopenia: a systematic review. J Thromb Haemost. 2015;13(3):457–464. doi:10.1111/jth.12813
17. Sarpatwari A, Bennett D, Logie JW, et al. Thromboembolic events among adult patients with primary immune thrombocytopenia in the United Kingdom general practice research database. Haematologica. 2010;95(7):1167–1175. doi:10.3324/haematol.2009.018390
18. Nørgaard M, Severinsen MT, Lund Maegbaek M, Jensen AØ, Cha S, Sørensen HT. Risk of arterial thrombosis in patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol. 2012;159(1):109. doi:10.1111/j.1365-2141.2012.09231.x
19. Nørgaard M, Cetin K, Maegbaek ML, et al. Risk of arterial thrombotic and venous thromboembolic events in patients with primary chronic immune thrombocytopenia: a Scandinavian population-based cohort study. Br J Haematol. 2016;174(4):639. doi:10.1111/bjh.13787
20. Frederiksen H, Maegbaek ML, Nørgaard M. Twenty-year mortality of adult patients with primary immune thrombocytopenia: a Danish population-based cohort study. Br J Haematol. 2014;166(2):260–267. doi:10.1111/bjh.12869
21. Care A, Pavord S, Knight M, Alfirevic Z. Severe primary autoimmune thrombocytopenia in pregnancy: a national cohort study. BJOG. 2018;125(5):604–612. doi:10.1111/1471-0528.14697
22. Lambert MP, Gernsheimer TB. Clinical updates in adult immune thrombocytopenia. Blood. 2017;129(21):2829–2835. doi:10.1182/blood-2017-03-754119
23. Bomhof G, Mutsaers PGNJ, Leebeek FWG, et al. COVID‐19‐associated immune thrombocytopenia. Br J Haematol. 2020;190(2). doi:10.1111/bjh.16850
24. Murt A, Eskazan AE, Yılmaz U, Ozkan T, Ar MC. COVID‐19 presenting with immune thrombocytopenia: a case report and review of the literature. J Med Virol. 2021;93(1):43–45. doi:10.1002/jmv.26138
25. Bhattacharjee S, Banerjee M. Immune thrombocytopenia secondary to COVID-19: a systematic review. SN Compr Clin Med. 2020;2(11):2048–2058. doi:10.1007/s42399-020-00521-8
26. Sahu KK, Siddiqui AD, Rezaei N, Cerny J. Challenges for management of immune thrombocytopenia during COVID‐19 pandemic. J Med Virol. 2020;92(11):2277–2282. doi:10.1002/jmv.26251
27. Mahévas M, Moulis G, Andres E, et al. Clinical characteristics, management and outcome of COVID‐19‐associated immune thrombocytopenia: a French multicentre series. Br J Haematol. 2020;190(4). doi:10.1111/bjh.17024
28. Lippi G, Plebani M, Henry BM. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta. 2020;506:145–148. doi:10.1016/j.cca.2020.03.022
29. Vayne C, Guery E, Rollin J, Baglo T, Petermann R, Gruel Y. Pathophysiology and diagnosis of drug-induced immune thrombocytopenia. J Clin Med. 2020;9(7):2212. doi:10.3390/jcm9072212
30. Arnold DM, Cuker A. Immune thrombocytopenia (ITP) in adults: clinical manifestations and diagnosis. UpToDate. Available from: https://www.uptodate.com/contents/immune-thrombocytopenia-itp-in-adults-clinical-manifestations-and-diagnosis?search=immune%20thrombocytopenia&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1#H21.
31. Tintinalli JE. Tintinalli’s Emergency Medicine.
32. Warkentin TE, Kelton JG. Temporal aspects of heparin-induced thrombocytopenia. N Engl J Med. 2001;344(17):1286. doi:10.1056/NEJM200104263441704
33. Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346:995. doi:10.1056/NEJMra010501
34. Neunert C, Lim W, Crowther M, Cohen A, Solberg L
35. Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115:168. doi:10.1182/blood-2009-06-225565
36. Davoren A, Bussel J, Curtis BR, Moghaddam M, Aster RH, McFarland JG. Prospective evaluation of a new platelet glycoprotein (GP)-specific assay (PakAuto) in the diagnosis of autoimmune thrombocytopenia (AITP). Am J Hematol. 2005;78(3):193–197. doi:10.1002/ajh.20309
37. Neunert C, Terrell DR, Arnold DM, et al. American Society of Hematology 2019 guidelines for immune thrombocytopenia. Blood Adv. 2019;3:3829. doi:10.1182/bloodadvances.2019000966
38. Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3:3780. doi:10.1182/bloodadvances.2019000812
39. Sirotich E, Guyatt G, Gabe C, et al. Definition of a critical bleed in patients with immune thrombocytopenia: communication from the ISTH SSC subcommittee on platelet immunology. J Thromb Haemost. 2021;19:2082. doi:10.1111/jth.15368
40. McCullough J. Overview of platelet transfusion. Semin Hematol. 2010;47(3):235–242. doi:10.1053/j.seminhematol.2010.04.001
41. Baumann MA, Menitove JE, Aster RH, Anderson T. Urgent treatment of idiopathic thrombocytopenic purpura with single-dose gammaglobulin infusion followed by platelet transfusion. Ann Intern Med. 1986;104:808. doi:10.7326/0003-4819-104-6-808
42. Praituan W, Rojnuckarin P. Faster platelet recovery by high-dose dexamethasone compared with standard-dose prednisolone in adult immune thrombocytopenia: a prospective randomized trial. J Thromb Haemost. 2009;7(6):1036–1038. doi:10.1111/j.1538-7836.2009.03359.x
43. Mithoowani S, Gregory-Miller K, Goy J, et al. High-dose dexamethasone compared with prednisone for previously untreated primary immune thrombocytopenia: a systematic review and meta-analysis. Lancet Haematol. 2016;3(10):e489. doi:10.1016/S2352-3026(16)30109-0
44. Godeau B, Chevret S, Varet B, et al. Intravenous immunoglobulin or high-dose methylprednisolone, with or without oral prednisone, for adults with untreated severe autoimmune thrombocytopenic purpura: a randomised, multicentre trial. Lancet. 2002;359(9300):23. doi:10.1016/S0140-6736(02)07275-6
45. Tarantino MD, Bussel JB, Cines DB, et al. A closer look at intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). Blood. 2007;109:
46. Gaines AR. Response: further examination of intravascular hemolysis (IVH) following intravenous anti-D for immune thrombocytopenic purpura (ITP). Blood. 2007;109:5528. doi:10.1182/blood-2007-02-070706
47. Garratty G. What is the mechanism for acute hemolysis occurring in some patients after intravenous anti-D therapy for immune thrombocytopenic purpura? Transfusion. 2009;49:1026.
48. Mayer B, Salama A. Successful treatment of bleeding with tranexamic acid in a series of 12 patients with immune thrombocytopenia. Vox Sang. 2017;112(8):767–772. doi:10.1111/vox.12549
49. Gernsheimer TB, Brown SP, Triulzi DJ, et al. Effects of tranexamic acid prophylaxis on bleeding outcomes in hematologic malignancy: the a-TREAT trial. Blood. 2020;136(1):1–2. doi:10.1182/blood-2020-138920
50. Gottlieb M, DeMott JM, Peksa GD. Topical tranexamic acid for the treatment of acute epistaxis: a systematic review and meta-analysis. Ann Pharmacother. 2019;53(6):652. doi:10.1177/1060028018820625
51. Gardner FH, Helmer RE
52. Bartholomew JR, Salgia R, Bell WR. Control of bleeding in patients with immune and nonimmune thrombocytopenia with aminocaproic acid. Arch Intern Med. 1989;149(9):1959. doi:10.1001/archinte.1989.00390090039008
53. Kalmadi S, Tiu R, Lowe C, Jin T, Kalaycio M. Epsilon aminocaproic acid reduces transfusion requirements in patients with thrombocytopenic hemorrhage. Cancer. 2006;107(1):13. doi:10.1002/cncr.21958
54. Giordano P, Lassandro G, Barone A, et al. Use of eltrombopag in children with chronic immune thrombocytopenia (ITP): a real life retrospective multicenter experience of the Italian Association of Pediatric Hematology and Oncology (AIEOP). Front Med. 2020;7:66. doi:10.3389/fmed.2020.00066
55. Bradbury CA, Pell J, Hill Q, et al. Mycophenolate Mofetil for first-line treatment of immune thrombocytopenia. N Engl J Med. 2021;385:885–895. doi:10.1056/NEJMoa2100596
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.