")
Back to Journals » Journal of Inflammation Research » Volume 15
Aspirin Ameliorates Pancreatic Inflammation and Fibrosis by Inhibiting COX-2 Expression in Experimental Chronic Pancreatitis
Authors Xu XF, Fan JW, Xin JQ, Wu N, Gao H, Duan LF, Zou WB, Zhang H, Li ZS
Received 23 May 2022
Accepted for publication 3 August 2022
Published 19 August 2022 Volume 2022:15 Pages 4737—4749
DOI https://doi.org/10.2147/JIR.S375383
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xiao-Fan Xu,1– 3 Jian-Wei Fan,2 Jia-Qi Xin,4 Nan Wu,4 He Gao,2 Li-Fang Duan,2 Wen-Bin Zou,3 Hong Zhang,2,5 Zhao-Shen Li3
1School of Clinical Medicine, Ningxia Medical University, Yinchuan, 750004, People’s Republic of China; 2Basic Medical Academy, Shaanxi University of Chinese Medicine, Xianyang, People’s Republic of China; 3Department of Gastroenterology, Changhai Hospital, Shanghai, People’s Republic of China; 4Department of Gastroenterology, Internal Medicine II, Klinikum Rechts der Isar, Technical University of Munich (TUM), Munich, Germany; 5Shaanxi International Cooperation Base, Shaanxi University of Chinese Medicine, Xianyang, People’s Republic of China
Correspondence: Zhao-Shen Li; Hong Zhang, Email [email protected]; [email protected]
Aim: Chronic pancreatitis (CP) is a complex and intractable disease mainly manifested as chronic inflammation and fibrosis. Aspirin(acetylsalicylic acid, ASA) has been reported to be used in the treatment of acute pancreatitis (AP), but its effectiveness on CP is unclear. This study aimed to investigate the therapeutic effects of ASA in CP mice.
Methods: A murine model of CP was induced by intraperitoneal injection with 20% L-arginine. After one week of L-arginine administration, mice in the ASA treatment group were administered aspirin (100mg/kg/d) by intragastric gavage. At two, four, and six weeks after the first injection of L-arginine, mice were euthanized and the pancreas was collected for histological and molecular analysis. A second model of CP (caeruelin-induced) was used as a validation experiment to test the effect of ASA.
Results: L-arginine-induced CP resulted in over-expression of the inflammatory enzyme cyclooxygenase (COX)-2. COX-2 expression decreased after ASA treatment. Pancreatic-injury inflammatory response (measured by changes in amylase, CK-19, F4/80, CD3, MCP-1, IL-6) and fibrosis degree (measured by expression of COL1A1, MMP-1 and TIMP-1) was reduce in ASA -treated mice model. The therapeutic effect of ASA was also observed in caeruelin-induced CP.
Conclusion: ASA has an ameliorating effect in murine models of CP through inhibition of pancreatic inflammation and fibrosis, which may be a promising option for clinical treatment.
Keywords: chronic pancreatitis, fibrosis, inflammation, aspirin, COX-2
Introduction
Chronic pancreatitis (CP) is a progressive inflammatory and fibrotic disease in which the pancreatic parenchyma is destroyed and replaced by fibrotic tissue.1 The pathogenesis of CP remains obscure, thus current therapeutic strategies are limited and focus mainly on alleviating clinical symptoms.2
Studies have found that cyclooxygenase (COX)-2 is strongly increased in pancreatic specimens from patients with CP.3,4 As an important pathway regulating inflammation, COX-2 participates in the synthesis of prostaglandins in a variety of inflammatory diseases and can be elevated through the stimulation of inflammatory mediators and oxidative stress.5,6 These studies revealed that COX-2 was localized to atrophic pancreatic acinar cells, islets, and duct cells. Moreover, the overexpression of COX-2 in patients with CP is significantly related to the frequency of acute pancreatitis (AP), which suggests that COX-2 is involved in the inflammatory response and disease progression of CP in patients. Reding et al7 explored the role of COX-2 in animals with CP and found a different response between WBN/Kob rats and C57/Bl6 mice. In rats, the pancreatic pathological lesions were ameliorated by a specific COX-2 inhibitor (rofecoxib), but there was no obvious improvement for histopathological alteration in caerulein-induced CP mice treated with rofecoxib, even with a specific knockout of COX-2 in the pancreas. Thus, the researchers concluded that COX-2 modulates the inflammatory process during the development of pancreatitis in a species-specific manner. Huang et al8 produced mice that overexpressed COX-2 specifically in pancreatic acinar cells via a conditional transgenic mouse model and observed interesting results: the mice with elevated COX-2 levels in the pancreas spontaneously initiated the processes of fibrosis, which eventually developed into chronic pancreatitis with preneoplastic lesions.
Aspirin(acetylsalicylic acid, ASA) is the oldest and most widely used non-steroidal anti-inflammatory drug (NSAID) and is one of the inhibitors of the COX enzymes.9,10 Recent research has found that ASA can be used to treat AP. Akyazi et al11 reported that ASA pretreatment for 100 days could ameliorate acute histopathological alterations caused by caerulein in rats. Lu et al12 showed that ASA could reduce the severity of pathological lesions in the pancreas and lungs of mice with severe acute pancreatitis. Several other studies on cancer, including pancreatic cancer (PC), indicated that ASA had beneficial effects in preventing cancer.13–16 The occurrence of CP may be associated with recurrent episodes of acute pancreatitis,17 and CP is considered one of the risk factors for PC.18 However, the pathogenesis of CP is not exactly the same as that of AP. The pathological manifestations of CP are more concentrated in acinar atrophy, chronic inflammatory cell infiltration and irreversible fibrosis. Therefore, although ASA is effective in the treatment of AP and prevention of PC, its role in CP must be verified. In view of this, it is worth exploring whether ASA is also effective in ameliorating CP.
L-arginine has been reported as an inexpensive and stable inducer of CP. In mice, this L-arginine-induced CP mimics features of human CP including pancreatic atrophy, fibrosis, inflammation, exocrine insufficiency and loss of architecture.19 In this study, we induced Kunming and C57BL/6 mouse models with CP using L-arginine and caerulein, respectively, and treated the mice with ASA, aiming to explore the role of ASA in the progression of CP, and to identify a potential mechanism.
Materials and Methods
Animals
Male C57BL/6 mice (18–23g) were obtained from the Animal Experiment Center of the Medical College of Xi’an Jiaotong University (Certificate SCXK [Shaan] 2018–001). Male Kunming mice (20–25 g) were obtained from the Experimental Animal Center of the Fourth Medical University of PLA (Certificate No. 0014945). Mice experiments were approved by the Animal Care and Use Committee of Shaanxi University of Chinese Medicine (YXSY (Shaanxi) 2017–0002). All experiments were performed in accordance with the principles and guidelines of YXSY (Shaanxi) 2017–0002. The mice were maintained in a temperature-controlled room (22 ± 3°C; relative humidity: 60% ± 5%) under a 12 h light/dark cycle.
CP Models and Experimental Design
Mice were randomly divided into three groups (n = 8 per time point in each group): control group, CP group, CP+ aspirin group. To induce CP, mice were intraperitoneally injected with 20% L-arginine (3 g/kg, twice per day with an interval of 1 h, one day per week) for six weeks, or caerulein (50 μg/kg, six times per day, three days per week) for six weeks. A control group comprised mice injected with the same volume of saline. After one week of L-arginine or caerulein administration, the mice in the aspirin group were administered ASA (100 mg/kg/d, this dose correspond to the low dose for humans, 11mg/kg per day) by intragastric gavage based on Ibrahim’s and Lu’s experiment for AP.11,12 The dosage of ASA was determined through a pre-experiment where CP-induced mice were treated with 175 mg/kg and 100 mg/kg ASA respectively. Both doses had the effect of alleviating CP, and there was no significant difference between the two doses. Considering the side effects of ASA, we chose a lower dose for treatment in this experiment. ASA was purchased from Bayer Inc. and was dissolved in ethyl alcohol (1 g/mL) and further diluted to appropriate concentrations in saline solution (4 mg/mL). The mice in the control group were administered only normal saline at the same time point.
H&E and Masson’s Staining
For morphological analyses, the mice were sacrificed at two, four, and six weeks after treatment with caerulein (Toronto Research Chemicals, Darmstadt, Germany) or L-arginine (Sigma–Aldrich), and the pancreases of the mice were immediately immersed in 4% neutral phosphate-buffered paraformaldehyde for 12 hours, embedded in paraffin, and sectioned (3 μm). The sections of the specimens were stained with H&E or Masson’s staining, and morphological changes and fibrosis in the pancreas were observed under a light microscope. Pancreatic sections were assessed at 20× objective magnification over 10 separate fields to determine the severity of pancreatitis by scoring for oedema, inflammation, atrophy and fibrosis. Lesion size scores on edema, fibrosis, and acinar atrophy were evaluated as follows: 0 = absent; 1 = only a limited part of section; 2 = slightly shown in lobule or intralobular region; 3 = a lesion across lobule and intralobular region or with destruction of lobular architecture. Inflammatory cell infiltration scores were determined by counting the cell number: 0 = 0–10 per field; 1=11–30 per field; 2 = 31–100 per field; 3 = 101~ per field.8,20
Immunohistochemistry and Immunofluorescence
Immunohistochemistry (IHC) and immunofluorescence analyses were performed according to standard protocols. IHC was performed using a ready-to-use StreptAvidin Biotin-Complex (SABC) kit (Boster Biological Technology Co. Ltd., Wuhan, China). Sections were incubated with a rabbit anti-F4/80 antibody (Santa Cruz Biotechnology Inc., Dallas, TX, USA; 1:500 dilution), a rabbit anti-CK-19 antibody (Bioss; 1:200 dilution), a rabbit anti-amylase antibody (Bioss; 1:200 dilution), a rabbit anti-CD3 antibody (Bioss; 1:200 dilution), and a goat anti-COX-2 antibody (Santa Cruz; 1:200 dilution) for 48 h at 4°C. The corresponding secondary antibodies were subsequently incubated for 1 h at room temperature. Images were captured using a ZEISS Imager A1 microscope (Carl Zeiss AG, Oberkochen, Germany).
For double staining of immunofluorescence, a 1:200 dilution of COX-2 antibody and a 1:400 dilution of F4/80 antibody or CK-19 antibody was incubated in a humid chamber for 48 h at 4°C, and a 1:500 dilution of FITC-labeled anti-goat or CY3-labeled anti-rabbit secondary antibodies was incubated for 1.5 h. Nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) (Roche) for 5 min, dehydrated, and mounted on a coverslip with anti-fade mounting medium. Images were captured with a ZEISS Imager A2 fluorescence microscope (Carl Zeiss AG).
Western Blotting
For Western blotting analysis, proteins were prepared from the mouse pancreas using standard methods. Protein concentrations were determined using the BCA method (Boster Biological Technology) and adjusted to 4 ug/ul. Samples were boiled at 95°C for 4 min before loading and then subjected to SDS-polyacrylamide gel electrophoresis (40 ug/sample) and blotted to PVDF membranes. The membranes were probed with the following antibodies: anti-COL1A1 (Cell Signaling Technology; 1:1000), anti-COX-2 (Santa Cruz; 1:500), and anti-β-actin (Boster Biological Technology; 1:2000). A low–molecular weight protein marker was used to determine the size of the detected bands in the Western blot. Detection and image capturing were performed using an enhanced chemiluminescence illuminating system.
Total RNA Extraction and Real-Time Polymerase Chain Reaction
Total RNA was extracted from pancreatic tissue using a commercial RNA extraction kit (Takara Biotechnology, Dalian, China) and transcribed into complementary DNA using oligo primer and reverse transcriptase kits (Takara Biotechnology) according to the manufacturer’s guidelines. For real-time polymerase chain reaction (RT-PCR), 100 ng of complementary DNA was used as a template for each reaction and amplified. The RT-PCR was performed using a SYBR Green kit (Takara Biotechnology). Amplification and detection were accomplished using an ABI 7500 Fast RT-PCR system (Thermo Fisher Scientific). All experiments were performed in triplicate, and expression levels were normalized to β-actin and calculated using the2–ΔΔCt method. The primer sequences are shown in Table 1.
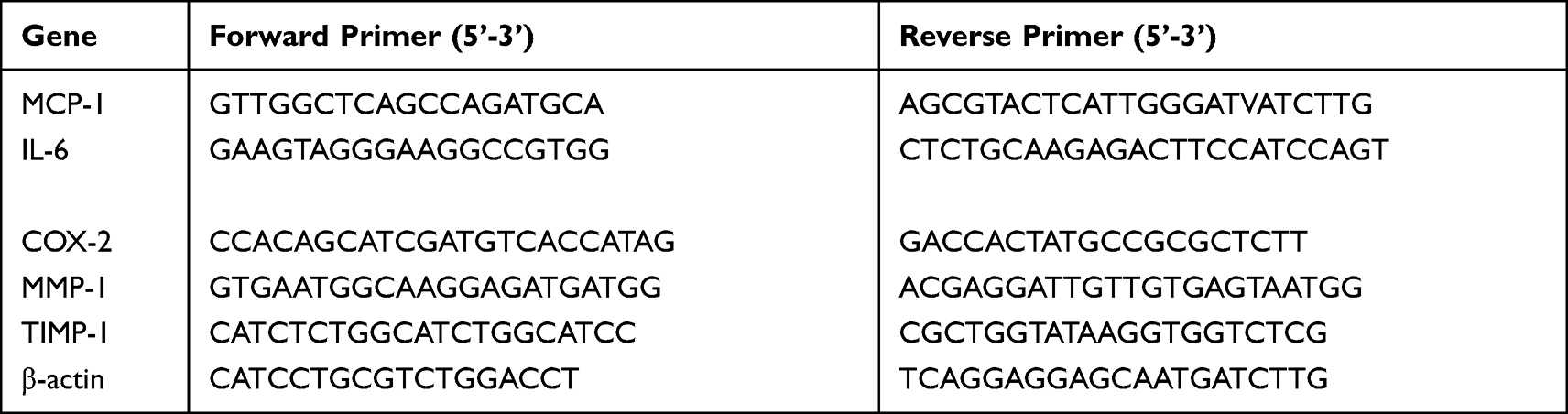 |
Table 1 Primer Sequences Used for Real Time-PCR |
Statistical Analysis
Our statistical analyses were performed using GraphPad Prism and SPSS. Data were expressed with mean ± standard deviation (SD) and compared by the unpaired Student’s t-test and the one-way analysis of variance (ANOVA). T-tests were used to determine differences between CP mice and control mice and ANOVAs were used to assess differences between three or more groups. A P value of < 0.05 was considered statistically significant.
Results
L-Arginine Can Induce CP and Overexpression of COX-2 in Mice
As shown in Figure 1A and B, 20% L-arginine can successfully induce CP in Kunming mice, in which the typical histopathological changes of chronic pancreatitis can be observed, including pancreatic acinar cell necrosis, inflammatory cell infiltration, tubular complexes, and a large amount of fibrotic tissue formation. Immunohistochemical staining showed that the number of COX-2 positive cells significantly increased in the pancreas of the CP mice, and the positive staining of COX-2 was mainly found in infiltrating inflammatory cells and tubular complexes (Figure 1C). The Western blot showed that the protein level of COX-2 increased continuously with the process of modeling (Figure 1D). To further ascertain the cell location of COX-2, we performed double immunofluorescence labeling with the following markers: F4/80, as a monocyte-macrophage marker, and COX-2, or CK-19, as a pancreatic acinar tubularization marker, and COX-2. The results showed that co-expression of F4/80 and COX-2 as well as that of CK-19 and COX-2 could be observed in the pancreas (Figure 1E and F). These results demonstrated that L-arginine-induced CP results in the over-expression of COX-2 in the pancreatic tissues of mice, and that COX-2 was also predominantly expressed by infiltrating inflammatory cells and tubular complexes.
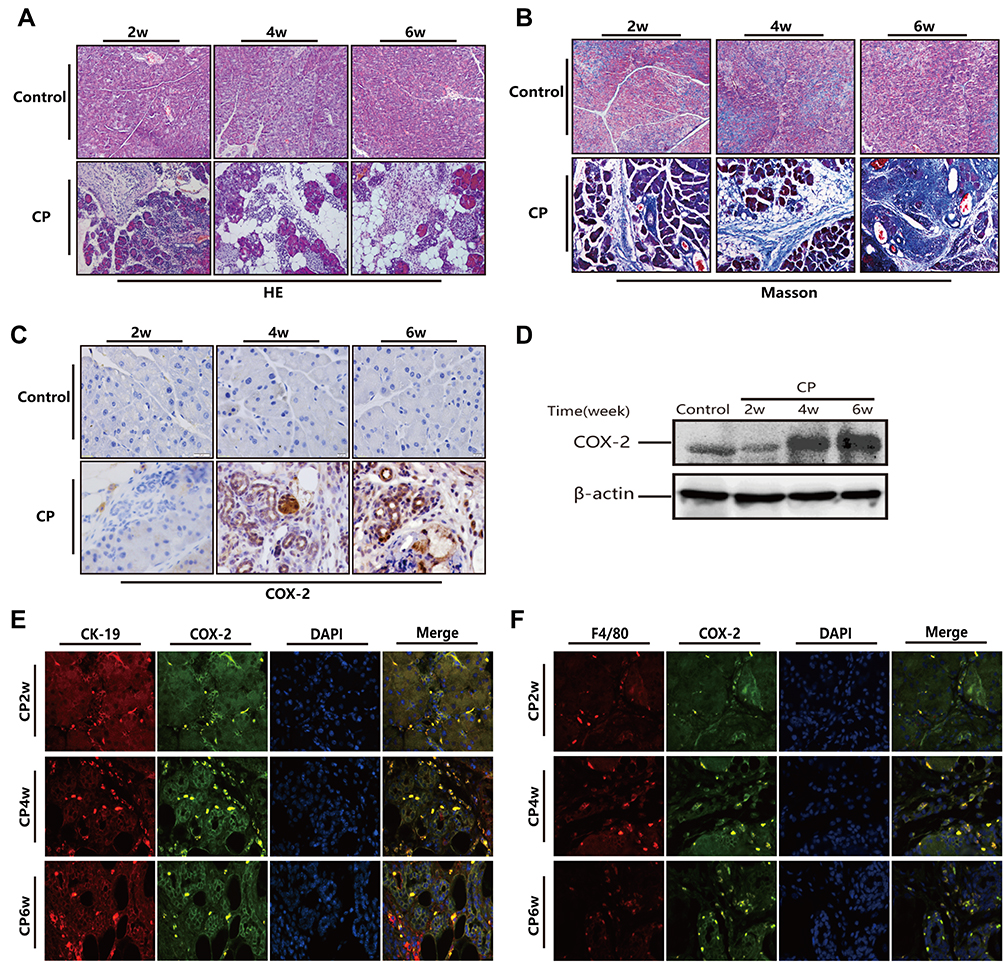 |
Figure 1 Over expression of COX-2 in pancreatic tissue of mice following treatment with 20% L-arginine to induce CP. (A and B) H&E and Masson’s trichrome staining in the pancreas following L-arginine treatment (original magnification: 200×). (C) The expression of COX-2 in the pancreas by IHC staining (original magnification: 400×). (D) COX-2 protein expression in pancreatic tissues as detected by Western blot. β-actin served as a loading control (representative blot). (E and F) Double-labeled immunofluorescence of incorporated COX-2 and CK-19 (E) or COX-2 and F4/80 (F). COX-2 showed green, CK-19 or F4/80 showed red (Original magnification, 400×). |
Aspirin Inhibits the Pancreatic Overexpression of COX-2 in L-Arginine-Induced CP
As shown in Figure 2A and B, there was no COX-2 expression in the control group, but protein and mRNA levels of COX-2 in the CP-induced group progressively increased at two, four and six weeks. However, after intervention with ASA, the elevated COX-2 levels were significantly reduced. COX-2 IHC staining showed similar results (Figure 2C).
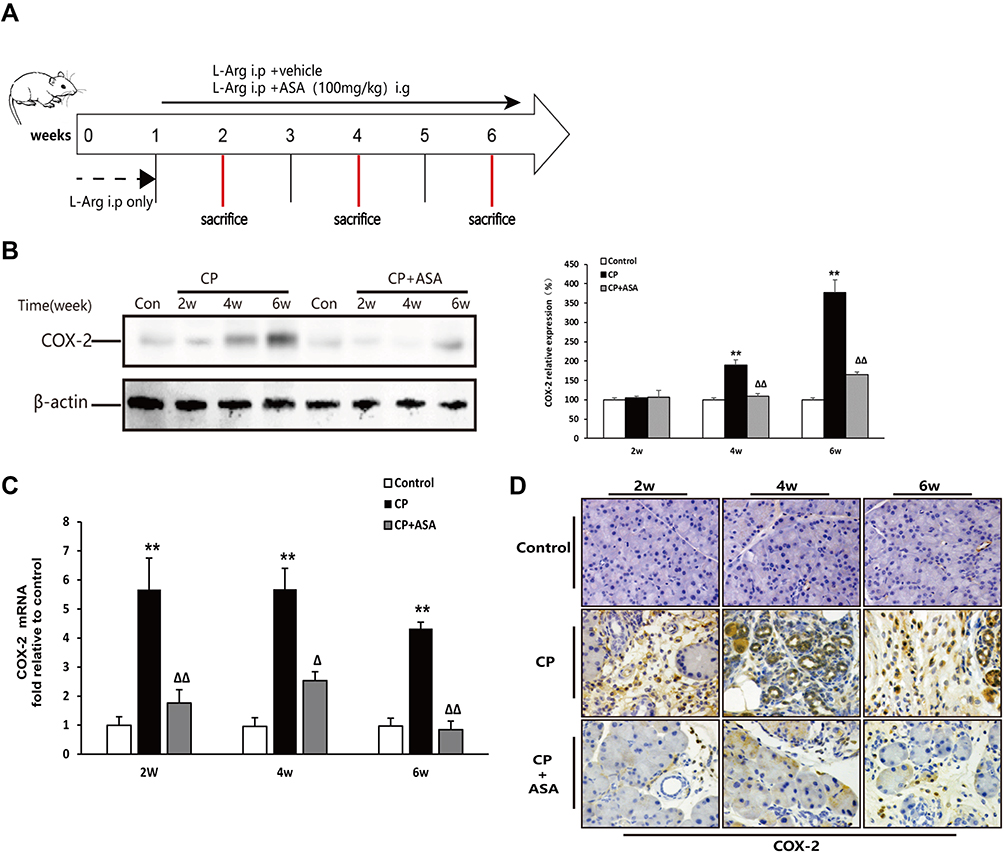 |
Figure 2 Aspirin inhibits the expression of COX-2 in mice with chronic pancreatitis induced by L-arginine. (A) Schematic diagram of animal experiment with 20% L-arginine induced CP. (B) COX-2 protein expression in pancreatic tissues was detected by Western blot. β-actin served as a loading control (representative blot). (C) COX-2 mRNA expression in pancreas was determined by real time PCR. Fold change values were normalized to β-actin mRNA (each group, n=6). Compared with control: **P<0.01; Compared with CP model group: ΔP<0.05, ΔΔP<0.01. (D) The expression of COX-2 in pancreas by immunohistochemistry staining (original magnification: 200×). |
Aspirin Improved the Functional and Morphological Changes in Acinar Cells in L-Arginine-Induced CP
Firstly, our results display the effect of aspirin on the pancreas-to-body weight ratio in mice. As shown in Figure 3A, with the majority of pancreatic lobules loose induced by L-arginine, a reduced relative pancreatic weight was shown in the CP mice and ASA treatment restored this loss of pancreatic weight. We next observed the effect of ASA on pancreatic injury in CP mice. HE staining showed that aspirin treatment ameliorated the pancreatic histopathological changes with decreased histopathological scores in CP mice, including pancreatic oedema, necrosis, inflammatory cell infiltration, acinar atrophy and fibrosis (Figure 3B). Expression of amylase in the serum and pancreatic tissue increased at the initial stage of the disease (at 2 weeks), but reduced by six weeks with the progression of CP, due to considerable pancreatic vacuolization, acinar cell atrophy, and tubular complex formation (Figure 3C and D). Ductal tubular complex is an important pathological manifestation of CP that can be observed by the expression of CK-19, a sensitive duct marker. As shown in Figure 3E, as CP progressed, a large number of acinar cells changed to a flat shape and formed a tube-like structure with strong staining of CK-19. After ASA treatment, the expression of amylase was reversed and the expression of CK-19 decreased. These results further indicated that ASA was able to protect acinar cells from injury and preserve the exocrine function of the pancreas.
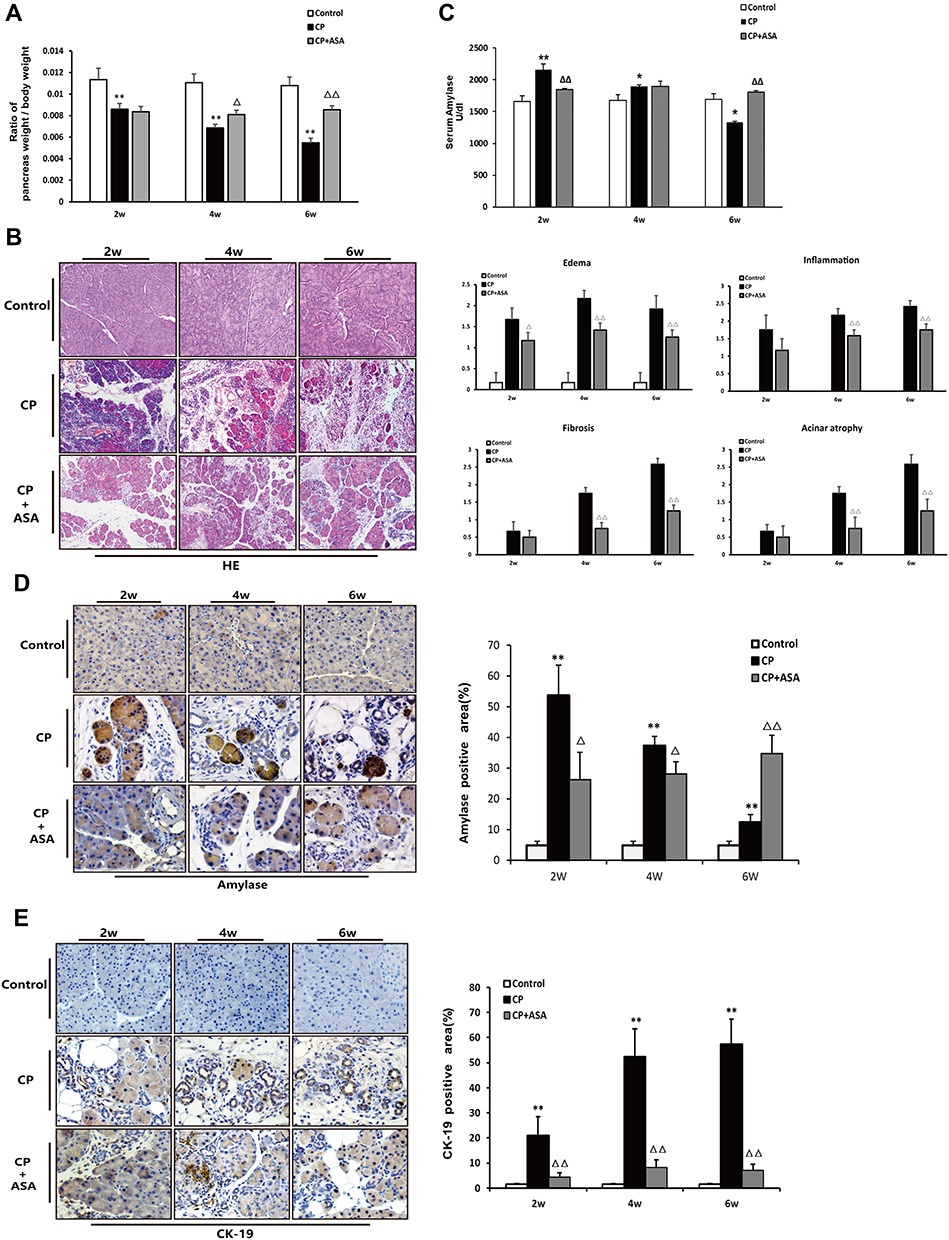 |
Figure 3 Histopathological changes in experimental CP mice and aspirin treatment. (A) Ratio of pancreatic/body weight (n=6). (B) H&E staining in the pancreas of the CP models induced by L-arginine (original magnification: 100×). Histological scoring of edema, inflammation, fibrosis, and acinar atrophy was performed as described in methods. Values are expressed as mean ± SD (n = 6). (C) The amylase level in serum at different time-points. Results are expressed as mean ± SD (n = 6). (D) IHC staining for amylase in the pancreas (original magnification: 400×) and morphometric analysis. (E) Immunohistochemistry staining for CK-19 in pancreas (original magnification: 400×) and morphometric analysis. *P<0.05, **P<0.01 vs control group; ΔP <0.05, ΔΔP <0.01vs. CP group. |
Aspirin Attenuates Inflammatory Cells Infiltration and the Expression of Inflammatory Factors in CP
As shown in Figure 4A and D, two weeks after disease induction, mice showed increased macrophage and lymphocytes infiltration in the pancreas (assessed as F4/80 or CD3 positive staining cells). ASA treatment significantly reduced F4/80 and CD3 positive staining cells in the pancreas, indicating that macrophages formed an important population of inflammatory cells during the development of CP. As shown in Figure 4B and C, the mRNA expression of inflammatory factors, including monocyte chemoattractant protein (MCP)-1 and interleukin (IL)-6, was elevated in the CP group, but after treatment with ASA, these inflammatory factors all decreased. The results suggest that ASA is able to inhibit inflammatory cells infiltration and inflammatory responses in L-arginine-induced CP.
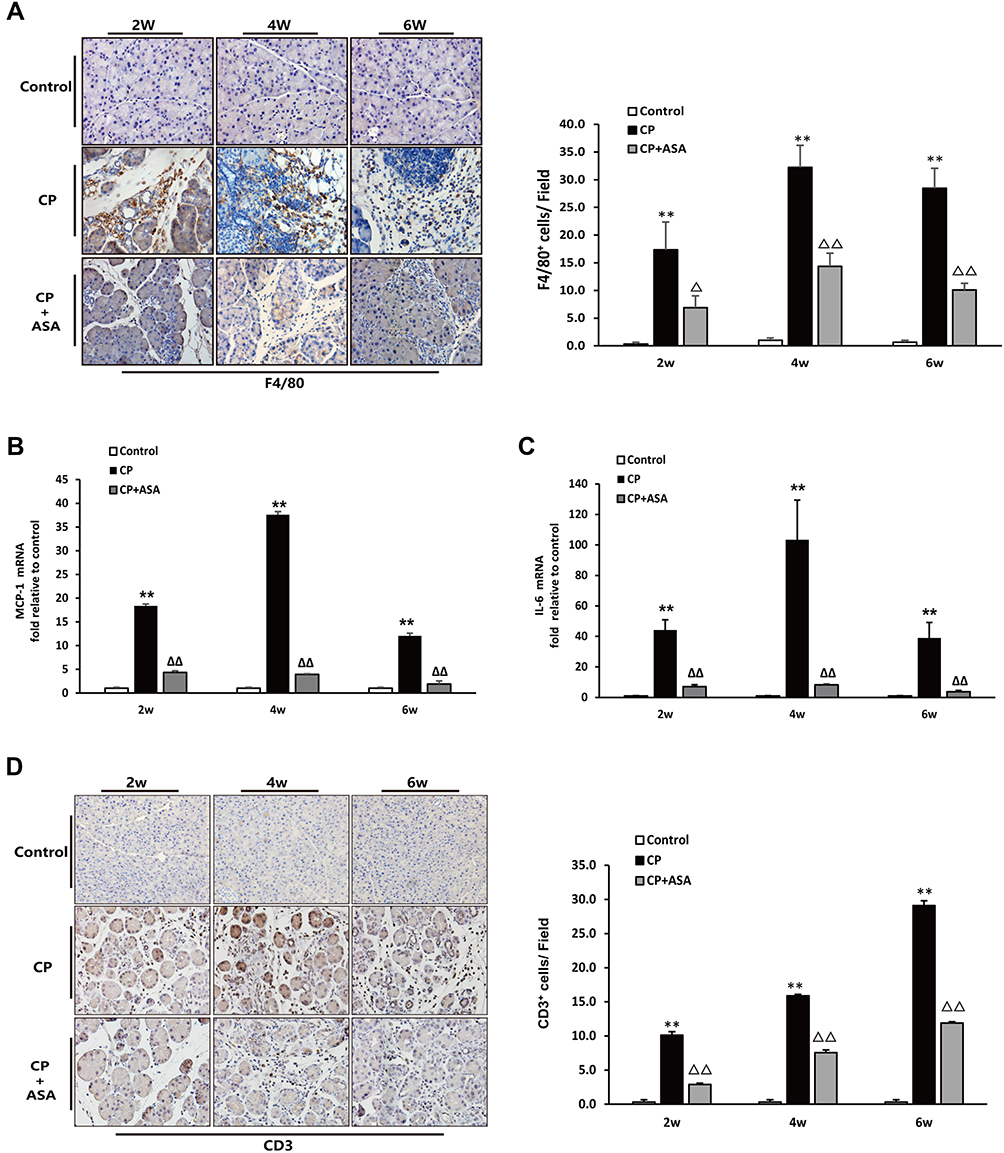 |
Figure 4 Aspirin attenuates inflammatory infiltration in the pancreas of mice treated with 20% L-arginine. (A) IHC staining for F4/80 in pancreas (original magnification: 200×) and F4/80 positive cell count analysis. (B) MCP-1 and (C) IL-6 mRNA expression was analyzed by RT-PCR, fold changes were normalized to β-actin mRNA. Results were expressed as Mean±SD (each group, n=6). **P<0.01 vs control group; ΔP<0.05, ΔΔP<0.01vs. CP group. (D) IHC staining for CD3 in pancreas (original magnification: 200×) and CD3 positive cell count analysis. |
Aspirin Inhibits Pancreatic Fibrosis by Regulating the Balance of Matrix Metalloproteinase (MMP)-1 and Tissue Inhibitor of Metalloproteinase (TIMP)-1
We observed a considerable amount of extracellular matrix (ECM) deposition in the pancreas at six weeks after L-arginine administration by Masson’s trichrome stain (Figure 5A). The expression of collagen type 1 alpha 1 chain (COL1A1), which is known to be predominant in pancreatitis-related fibrosis, was also over-expressed at six weeks post-CP-induction, compared to two weeks L-arginine treatment and control mice (Figure 5B). Analyzing the variation in enzymes regulating ECM production, we detected the expression of MMP-1 and TIMP-1 mRNA in the pancreas after CP. As shown in Figure 5C and D, compared with the control group, the MMP-1 expression level increased at two weeks and four weeks, but showed a significant reduction at six weeks (P < 0.01). The TIMP-1 mRNA level was significantly increased two weeks after modeling (P < 0.01) and fell back at four weeks, but it was still higher than that of the control group (P < 0.05) and sustained this level until six weeks. These results suggest that an imbalance of MMPs/TIMPs may be a relevant factor leading to extensive fibrosis deposition in CP induced by L-arginine. Pancreatic fibrosis of CP mice was alleviated after ASA treatment, compared to the CP group, based on a decrease of ECM deposition, the decrease of COL1A1 protein expression, the increase of MMP-1 expression and the decrease of TIMP-1 expression.
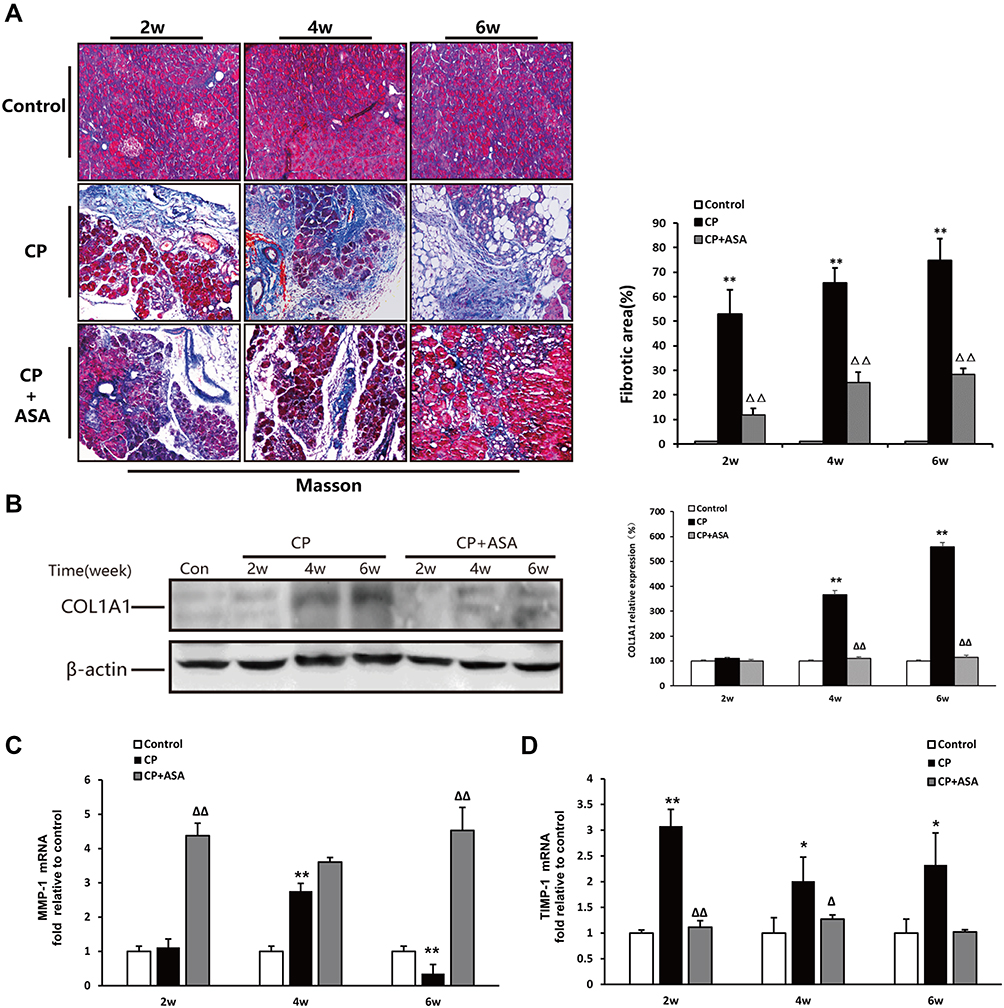 |
Figure 5 Aspirin inhibits pancreatic fibrosis in pancreas of mice treated with 20% L-arginine. (A) Masson’s trichrome staining (original magnification: 100×), histograms showing the quantitative analysis of the % fibrotic area. (B) COL1A1 protein level in pancreatic tissues was detected by Western blot. β-actin served as loading control (representative blot). The relative protein abundance was quantified through densitometry analysis normalized against β-actin. (C and D) MMP-1 and TIMP-1 mRNA expression were determined by real time PCR. Fold change values were normalized to β-actin mRNA (each group, n=6). Results are expressed as mean ± SD. Compared with control: *P<0.05, **P<0.01; Compared with CP model group: ΔP<0.05, ΔΔP<0.01. |
Aspirin Attenuates Caerulein-Induced Chronic Pancreatic Injury in C57BL/6 Mice
The above results indicate that aspirin has the effect of improving pancreatic injury in L-arginine-induced chronic pancreatitis in Kunming mice. In the following experiments, we adopted different modeling methods (repeated intraperitoneal injection of caerulein to induce chronic pancreatitis in C57BL/6 mice) to verify the beneficial effect of ASA on CP (Figure 6A). Mouse pancreas-to-body weight ratios showed that the caerulein treated group exhibited lower pancreatic weights due to chronic pancreatic injury, while ASA treatment increased the pancreatic weights (Figure 6B). HE staining showed that ASA could ameliorate caerulein-induced pancreatic histopathological changes in C57BL/6 mice (Figure 6C). Histological staining showed that collagen fiber deposition was significantly reduced in the aspirin-treated CP mice compared with the CP group (Figure 6D). Amylase and CK-19 immunohistochemical staining showed the inhibitory effect of ASA on caerulein-induced pancreatic acinar loss and tubularization (Figure 6E and F). The reduction in the number of F4/80-positive cells further suggested the beneficial effect of ASA in reducing caerulein-induced inflammatory cell infiltration (Figure 6G).
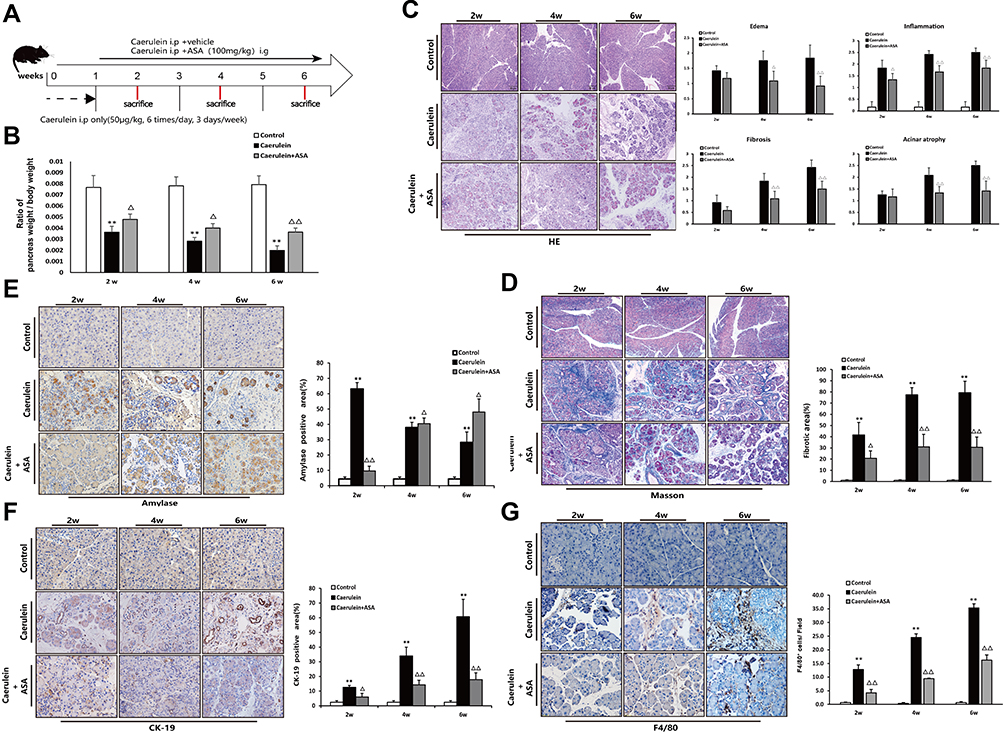 |
Figure 6 Aspirin attenuates caerulein-induced chronic pancreatic injury in C57BL/6 mice. (A) Schematic showing the experimental design of aspirin treatment of caerulein-induced CP in C57Bl/6 mice. (B) Ratio of pancreatic/body weight (n=6). (C) H&E staining in the pancreas of the CP models induced by caerulein (original magnification: 100×). Values of histological scoring were expressed as mean ± SD (n = 6). (D) Masson’s trichrome staining (original magnification: 100×), histograms showing the quantitative analysis of the % fibrotic area. (E) IHC staining for amylase in pancreas (original magnification: 400×) and morphometric analysis. (F) IHC staining for CK-19 in pancreas (original magnification: 400×) and morphometric analysis. (G) IHC staining for F4/80 in pancreas (original magnification: 400×) and morphometric analysis. **P<0.01 vs control group; ΔP<0.05, ΔΔP<0.01 vs CP group. |
Discussion
Chronic pancreatitis (CP) is a progressive and irreversible disease, in which chronic abdominal pain, weight loss, and persistent diarrhea as the main clinical manifestations.2 A recent study showed that low-grade inflammation is a characteristic of CP and an important factor in the development of pancreatic ductal adenocarcinoma.21
As an important pathway regulating inflammation, COX-2 participates in a variety of inflammatory diseases, including pancreatitis.22,23 In CP patients, the overexpression of COX-2 in the pancreatic tissue suggests that it is related to the occurrence of CP and could be a potential target for treatment of CP.3,4 However, the results of inhibiting COX-2 in experimental CP studies have been controversial. Alberto et al24 compared the role of COX-2 in a model of caerulein-induced CP using COX-2-deficient mice (COX-2−/−) and wild-type (WT) mice. They found that the histopathological scores of the juvenile COX-2−/− mice were lower than those of the WT mice, but there was no significant difference in the histological scores of the two groups of adult mice. Therefore, they concluded that the effect of COX-2 on experimental CP is limited and possibly age-dependent. This conclusion is inconsistent with the results of a previous study on the treatment of CP rats with the COX-2 inhibitor rofecoxib,7 which showed that rofecoxib significantly suppressed inflammation and fibrosis and delayed the progression of CP. Recently, Huang et al8 produced mice that overexpressed COX-2 specifically in pancreatic acinar cells (COX-2/Cre mice). They found that COX-2/Cre mice were able to initiate the processes of fibrosis, which eventually developed into spontaneous CP with preneoplastic lesions. The above studies suggested that the role of COX-2 in chronic pancreatitis is controversial and may be different amongst species. In our study, we used Kunming mice to induce a CP model with L-arginine and observed that the expression of COX-2 increased significantly with the progression of CP (Figure 1C and D). In addition, we found COX-2 was predominantly expressed in infiltrating inflammatory cells and damaged acinar cells or tubular complexes during the progression of CP (Figure 1E and F). Based on the above, our study provides evidence that COX-2 plays an important pro-inflammatory role in experimental chronic pancreatitis models, and targeting COX-2 may be an available strategy to both prevent and treat CP.
As a cyclooxygenase inhibitor, ASA has been used for several decades as an anti-inflammatory agent against multiple types of inflammatory disease, and can inhibit the activity of COX-2.7,9,25 In our study, the effect of ASA treatment on reducing COX-2 expression in the pancreatic tissue of CP mice was observed, and it was also found that aspirin could reduce COX-2 expression at both the protein and gene levels (Figure 2B and D). Two studies have reported that ASA pretreatment was valuable in the prevention of pancreatic histopathological alteration and improved some parameters that represent the degree of pancreatic lesion, including high levels of serum amylase and cytokines (IL-6, IL-1β, etc.) in AP in rats and mice.11,12 Moreover, the side effects (inflammation or bleeding) related to ASA administration have not been observed in the gastrointestinal tract in macroscopic and histopathological examinations. As we know, recurrent attack of acute pancreatitis is a risk factor for CP.26–28 However, the pathogenesis of CP is different from that of AP. The pathological manifestations of CP are more concentrated in acinar atrophy, chronic inflammatory cell infiltration and irreversible fibrosis. Therefore, it is worth exploring whether ASA, which is effective in the treatment of AP, can also effectively improve CP.
In the present study, we investigated the effect of ASA in an experimental CP model. The results showed that aspirin significantly alleviated pancreatic injury, as shown by the morphological changes in the mice (Figure 3A and B). At the same time, the parameters related to the function of pancreatic acinar cells, including the level of amylase and the expression of CK-19, were improved after ASA treatment (Figure 3C and E). In addition, we found ASA inhibited the infiltration of inflammatory cells in the pancreas and the mRNA expression levels of MCP-1 and IL-6 in the pancreas (Figure 4), which are critical for creating the inflammatory microenvironment in CP.29 Our study also explored the role of ASA in pancreatic fibrosis, and our results showed that aspirin can reduce the expression of COL1A1, which is known to be highly expressed in pancreatitis-related fibrosis (Figure 5A and B). Pancreatic fibrosis is associated with increased synthesis and decreased degradation of ECM, while ECM degradation is regulated by matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). Our analysis of MMP and TIMP levels in this study suggests that the inhibitory effect of aspirin on collagen deposition may be related to regulating the balance of MMP-1/TIMP-1 in the pancreatic tissue of CP mice (Figure 5C and D) The above results all confirmed that aspirin can reduce pancreatic acinar cell damage, inflammation and fibrotic lesions, and played a beneficial therapeutic role in L-arginine-induced CP in mice. This beneficial effect of aspirin was also verified in another species of mouse CP model, the caerulein-induced CP model (Figure 6), where ASA treatment reduced markers of disease pathogenesis.
In summary, the present study demonstrated that ASA treatment alleviated pancreatic damage and fibrosis in experimental CP. These beneficial effects of ASA can be explained by its elimination effect of inflammatory infiltration and the protective effect of acinar cell loss. Our results may provide a new experimental basis for the clinical promotion of ASA.
Acknowledgment
We would like to thank our colleagues and postgraduates in the Medical Experimental Center and the Department of Pathophysiology for their help in supporting this research.
Funding
This work was supported by the National Natural Science Foundation of China (82174201; 82104815); the Subject Innovation Team of Shaanxi University of Chinese Medicine (2019-LY14); the Special Support Project for High Level Talents in Shaanxi Province (303/141020047). The funders only sponsored the research projects and purchase of materials.
Disclosure
The authors have no conflicts of interest to declare.
References
1. Whitcomb DC, Shimosegawa T, Chari ST. International consensus statements on early chronic pancreatitis. Recommendations from the working group for the international consensus guidelines for chronic pancreatitis in collaboration with The International Association of Pancreatology, American Panc. Pancreatology. 2018;18(5):516–527. doi:10.1016/j.pan.2018.05.008
2. Beyer G, Habtezion A, Werner J, Lerch MM, Mayerle J. Chronic pancreatitis. Lancet. 2020;396(10249):499–512. doi:10.1016/S0140-6736(20)31318-0
3. Schlosser W, Schlosser S, Ramadani M, Gansauge F, Gansauge S, Beger H-G. Cyclooxygenase-2 is overexpressed in chronic pancreatitis. Pancreas. 2002;25(1):26–30. doi:10.1097/00006676-200207000-00008
4. Koliopanos A, Friess H, Kleeff JO, et al. Cyclooxygenase 2 expression in chronic pancreatitis: correlation with stage of the disease and diabetes mellitus. Digestion. 2001;64(4):240–247. doi:10.1159/000048868
5. Koh YC, Liu SY, Wu JC, Chou YC, Pan MH. A new metabolite: the effects of aminated tetrahydrocurcumin on inducible nitric oxide synthase and cyclooxygenase-2. J Cancer Res Pract. 2021;8(2):41. doi:10.4103/JCRP.JCRP_21_20
6. Ahmed M, Hussain AR, Siraj AK. Co-targeting of Cyclooxygenase-2 and FoxM1 is a viable strategy in inducing anticancer effects in colorectal cancer cells. Mol Cancer. 2015;14(1):131. doi:10.1186/s12943-015-0406-1
7. Reding T, Bimmler D, Perren A, et al. A selective COX-2 inhibitor suppresses chronic pancreatitis in an animal model (WBN/Kob rats): significant reduction of macrophage infiltration and fibrosis. Gut. 2006;55(8):1165–1173.
8. Huang H, Chen J, Peng L. Transgenic expression of cyclooxygenase-2 in pancreatic acinar cells induces chronic pancreatitis. Am J Physiol. 2019;316(1):G179–G186. doi:10.1152/ajpgi.00096.2018
9. Wu KK. Aspirin and other cyclooxygenase inhibitors: new therapeutic insights. Semin Vasc Med. 2003;03(2):107–112. doi:10.1055/s-2003-40668
10. Vane JR, Botting RM. Mechanism of action of aspirin-like drugs. Semin Arthritis Rheu. 1997;26:2–10. doi:10.1016/S0049-0172(97)80046-7
11. Akyazi I, Eraslan E, Gülçubuk A. Long-term aspirin pretreatment in the prevention of cerulein-induced acute pancreatitis in rats. World J Gastroentero. 2013;19(19):2894–2903. doi:10.3748/wjg.v19.i19.2894
12. Lu G, Tong Z, Ding Y, et al. Aspirin protects against acinar cells necrosis in severe acute pancreatitis in mice. Biomed Res Int. 2016;2016:1–10. doi:10.1155/2016/6089430
13. Bosetti C, Santucci C, Gallus S, Martinetti M, La Vecchia C. Aspirin and the risk of colorectal and other digestive tract cancers: an updated meta-analysis through 2019. Ann Oncol. 2020;31(5):558–568. doi:10.1016/j.annonc.2020.02.012
14. Jiang W, Yan Y, Chen M. Aspirin enhances the sensitivity of colon cancer cells to cisplatin by abrogating the binding of NF-κB to the COX-2 promoter. Aging. 2020;12(1):611–627. doi:10.18632/aging.102644
15. Alfonso L, Ai G, Spitale RC, Bhat GJ. Molecular targets of aspirin and cancer prevention. Brit J Cancer. 2014;111(1):61–67. doi:10.1038/bjc.2014.271
16. Mohammed A, Madka V, Kumar G, Singh A, Rao CV. Abstract 13: aspirin and metformin combination inhibits PDAC progression and metastasis in KPC mice.
17. Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroen. 2015;31(5):395–399. doi:10.1097/MOG.0000000000000195
18. Korpela T, Udd M, Mustonen H. Association between chronic pancreatitis and pancreatic cancer: a 10-year retrospective study of endoscopically treated and surgical patients. Int J Cancer. 2020;147(5):1450–1460. doi:10.1002/ijc.32971
19. Garg SK, Dawra R, Barlass U, Yuan Z, Dixit AK, Saluja A. Sa1792 development of a new mouse model of chronic pancreatitis induced by administration of L-arginine. Gastroenterology. 2014;146(5):297. doi:10.1016/S0016-5085(14)61062-3
20. Niina Y, Ito T, Oono T, et al. A sustained prostacyclin analog, ONO-1301, attenuates pancreatic fibrosis in experimental chronic pancreatitis induced by dibutyltin dichloride in rats. Pancreatology. 2014;14(3):201–210. doi:10.1016/j.pan.2014.02.009
21. Gukovskaya AS, Gukovsky I, Algül H, Habtezion A. Autophagy, inflammation, and immune dysfunction in the pathogenesis of pancreatitis. Gastroenterology. 2017;153(5):1212–1226. doi:10.1053/j.gastro.2017.08.071
22. Zannikou M, Barbayianni I, Fanidis D, Grigorakaki T, Aidinis V. MAP3K8 regulates Cox-2–mediated prostaglandin E 2 production in the lung and suppresses pulmonary inflammation and fibrosis. J Immunol. 2020;206(3):i2000862. doi:10.4049/JIMMUNOL.2000862
23. Yu J, Hui AY, Chu ESH. Expression of a cyclo-oxygenase-2 transgene in murine liver causes hepatitis. Gut. 2007;56(7):991–999. doi:10.1136/gut.2006.097923
24. Silva A, Weber A, Bain M, Reding T, Graf R. COX-2 is not required for the development of murine chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2011;300(6):G968. doi:10.1152/ajpgi.00497.2010
25. Espinosa E, Murad JP, Khasawneh FT. Aspirin: pharmacology and clinical applications. Thrombosis. 2011;2012(5):173124. doi:10.1155/2012/173124
26. Kleeff J, Whitcomb DC, Shimosegawa T, et al. Chronic pancreatitis. Nat Rev Dis Primers. 2017;3(1):17060. doi:10.1038/nrdp.2017.60
27. Yadav Dhiraj OMPG, O’Connell M, Papachristou GI. Natural history following the first attack of acute pancreatitis. Am J Gastroenterol. 2012;107(7):1096–1103. doi:10.1038/ajg.2012.126
28. Geisz A, Sahin-Tóth M. Sentinel acute pancreatitis event (SAPE) increases severity of subsequent episodes in mice. Gastroenterology. 2021;161(5):1692–1694. doi:10.1053/j.gastro.2021.06.013
29. Li-Fang D, Fan X, et al. Dachaihu decoction ameliorates pancreatic fibrosis by inhibiting macrophage infiltration in chronic pancreatitis. World J Gastroentero. 2017;23(40):7242. doi:10.3748/wjg.v23.i40.7242
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.