")
Back to Archived Journals » Hypoxia » Volume 4
Ascorbate availability affects tumor implantation-take rate and increases tumor rejection in Gulo–/– mice
Authors Campbell EJ, Vissers MC , Dachs G
Received 24 December 2015
Accepted for publication 3 February 2016
Published 8 April 2016 Volume 2016:4 Pages 41—52
DOI https://doi.org/10.2147/HP.S103088
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Elizabeth J Campbell,1 Margreet CM Vissers,2 Gabi U Dachs1
1Mackenzie Cancer Research Group, 2Centre for Free Radical Research, Department of Pathology, University of Otago, Christchurch, New Zealand
Abstract: In solid tumors, HIF1 upregulates the expression of hundreds of genes involved in cell survival, tumor growth, and adaptation to the hypoxic microenvironment. HIF1 stabilization and activity are suppressed by prolyl and asparagine hydroxylases, which require oxygen as a substrate and ascorbate as a cofactor. This has led us to hypothesize that intracellular ascorbate availability could modify the hypoxic HIF1 response and influence tumor growth. In this study, we investigated the effect of variable intracellular ascorbate levels on HIF1 induction in cancer cells in vitro, and on tumor-take rate and growth in the Gulo–/– mouse. These mice depend on dietary ascorbate, and were supplemented with 3,300 mg/L, 330 mg/L, or 33 mg/L ascorbate in their drinking water, resulting in saturating, medium, or low plasma and tissue ascorbate levels, respectively. In Lewis lung carcinoma cells (LL/2) in culture, optimal ascorbate supplementation reduced HIF1 accumulation under physiological but not pathological hypoxia. LL/2, B16-F10 melanoma, or CMT-93 colorectal cancer cells were implanted subcutaneously into Gulo–/– mice at a range of cell inocula. Establishment of B16-F10 tumors in mice supplemented with 3,300 mg/L ascorbate required an increased number of cancer cells to initiate tumor growth compared with the number of cells required in mice on suboptimal ascorbate intake. Elevated ascorbate intake was also associated with decreased tumor ascorbate levels and a reduction in HIF1α expression and transcriptional activity. Following initial growth, all CMT-93 tumors regressed spontaneously, but mice supplemented with 33 mg/L ascorbate had lower plasma ascorbate levels and grew larger tumors than optimally supplemented mice. The data from this study indicate that improved ascorbate intake is consistent with increased intracellular ascorbate levels, reduced HIF1 activity and reduced tumor initiation and growth, and this may be advantageous in the management of cancer.
Keywords: vitamin C, hypoxia inducible factor 1, C57BL/6 mice, B16-F10 melanoma, CMT-93 colorectal cancer
Introduction
In a rapidly expanding tumor mass, the availability of oxygen and nutrients is compromised, and this is known to drive the activation of the transcription factor HIF1, resulting in the upregulation of hundreds of genes that support cell survival in the stressful tumor microenvironment.1–3 HIF1 activity is regulated via posttranslational modification of the α-subunit, and involves the enzymatic hydroxylation of specific proline and asparagine residues by the HIF hydroxylases, which target the structurally modified HIF1α for proteasomal degradation4,5 and reduce interaction with the transcriptional coactivator p300/CBP.6 The hydroxylases are iron-containing dioxygenases that have a substrate requirement for molecular oxygen and 2-oxoglutarate,7,8 and Fe2+ and ascorbate are essential cofactors for this reaction. The HIF hydroxylases require millimolar concentrations of ascorbate in order to achieve optimal function in vitro, and these concentrations are within the normal range for intracellular ascorbate.8–11 However, very few studies have measured the effect of varied intracellular ascorbate availability on HIF1 activity.
The substrate and cofactor requirements of the HIF hydroxylases allow them to function as metabolic sensors and ensure that HIF1 responds to metabolic stress. This activity is advantageous for normal tissues, but in a growing tumor results in a more aggressive phenotype that is associated with enhanced tumor growth and resistance to therapy.12–15 We and others have shown that ascorbate availability influences HIF1 activation,9,16–18 and this is thought to be due to its role as an essential cofactor for the HIF hydroxylases. Low intracellular ascorbate levels decrease the activity of the HIF hydroxylases, resulting in increased HIF1 protein levels and gene expression in vitro.9,16,17 These observations led to the hypothesis that ascorbate availability could influence HIF1 activity in vivo, with consequent effects on tumor initiation and growth.
Recently, we have used the Gulo–/– mouse, a model of the human vitamin C-dependency condition, to investigate the effect of ascorbate availability on the growth characteristics of subcutaneous tumors.19 We have shown that optimal dietary intake levels of ascorbate resulted in tissue ascorbate levels equivalent to those seen in wild-type animals that synthesize their own ascorbate, and that are thus assumed to be at saturation levels. Restoration of these optimal ascorbate levels in Gulo–/– mice was associated with reduced HIF1 levels and slower tumor growth.19 These studies were focused on the growth of established tumors, and no information has previously been published on the potential for ascorbate to influence tumor initiation and rejection.
In this study, we used the Gulo–/– mouse to investigate dietary ascorbate availability on the implantation take and rejection rate, tumor-growth rate, and HIF1-pathway activity of lung, melanoma, and colorectal tumors in Gulo–/– mice. We show for the first time a significant association between tumor ascorbate availability and tumor initiation, with higher ascorbate resulting in decreased HIF1 activation and both preventing and slowing tumor growth.
Materials and methods
Cells and cell culture
Lewis lung carcinoma (LL/2), B16-F10 melanoma, and CMT-93 colon carcinoma cells are syngeneic tumor cell lines for the C57BL/6 mouse strain, and were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle’s Medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal calf serum (Thermo Fisher Scientific) in a humidified incubator at 37°C, 5% CO2. Cells were passaged using TrypLE Express (Thermo Fisher Scientific) and used for in vitro experiments and tumor implantation when they reached 60%–80% confluence. Only cells within the first six to eight passages from ATCC were used.
Ascorbate uptake in vitro
Solutions of sodium l-ascorbate (Sigma-Aldrich, St Louis, MO, USA) were made up fresh in phosphate-buffered saline, filter-sterilized with 0.22 μm filters, and added to the media. To investigate accumulation in LL/2, B16-F10, and CMT-93 cells over time, ascorbate was added to a final concentration of 50 or 500 μM and samples taken over a 24-hour period. After incubation with ascorbate, cells were washed with phosphate buffered saline to remove any extracellular ascorbate, and pelleted (1,000 rpm, 5 minutes) before being extracted in a 1:1 solution of 0.54 M perchloric acid (containing 50 mM diethylenetriaminepentaacetic acid) and H2O.9
Hypoxic conditioning
LL/2 cells were incubated for 8 hours in a HypoxyStation (H35; Don Whitley Scientific, Shipley, UK) under conditions of 0.1%–10% O2, with 5% CO2, balanced with N2. Cells were preloaded with 0, 50, or 500 μM of ascorbate for 16 hours before and during hypoxic exposure. Media, plastics, and supplements were left in the chamber for >3 hours to equilibrate prior to experiments. For Western blot analysis, cells were lysed with Tris-HCl lysis buffer (60 mM of 0.5 M Tris-HCl, pH 6.8, 20% glycerol, 2% w/v sodium dodecyl sulfate, 0.1 M of 1 M dithiothreitol, protease inhibitors), in the chamber to reduce loss of HIF1.
Animal ethics
Ethical approval for the study was obtained from the University of Otago Animal Ethics Committee (C04/11), and animal welfare was monitored and maintained following international guidelines.20,21
Mouse model
C57BL6/J B6.129P2-Gulotm1Unc/Ucd mice, originally obtained from the Mutant Mouse Resource Center, University of California, Davis, CA, USA, were bred in the Christchurch animal facility from homozygous Gulo–/– adults and genotyped by polymerase chain reaction after weaning.21 For colony maintenance, the animals were supplemented daily with 1 g/L ascorbate in their drinking water; the mouse chow did not contain any ascorbate. For these experiments, groups of female mice were maintained on water supplemented with 33 mg/L, 330 mg/L, or 3,300 mg/L ascorbate for 1 month prior to and following tumor inoculation.19 Ascorbate solutions (Sigma-Aldrich) were made fresh twice weekly and stabilized with 10 μM ethylenediaminetetraacetic acid (EDTA). Tumors were implanted when mice were 6–10 weeks of age.
Tumor models
LL/2, B16-F10, and CMT-93 cells were injected subcutaneously into the flanks of Gulo–/– mice at variable initial cell numbers (104–107 cells/mouse). Tumor size was measured using calipers every second day. The animals were killed by isoflurane (Baxter, Deerfield, IL, USA) overdose and cervical dislocation once tumors reached end point, i.e. a maximum tumor volume of 1,000 mm3 (tumor volume = width2 × length × π/6) or after 60 days in the absence of harvestable tumors. Tumors were excised, organs harvested, and blood collected. Tumors and organs were weighed, immediately flash-frozen, and were stored together with separated plasma at −80°C.
Tissue lysates
Frozen tissues were homogenized to a fine powder in liquid nitrogen, weighed, and suspended in phosphate buffer, followed by measurement of DNA concentration to standardize for cell content, as described previously.19,22,23 The homogenate was extracted with perchloric acid containing EDTA for ascorbate measurements.
Measurement of ascorbate
Ascorbate concentrations in cell lysates, tissues, and plasma were measured using reverse-phase high-performance liquid chromatography (HPLC; Waters 600 HPLC; Waters, MA, USA) with an ESA Coulochem II detector (Thermo Fisher Scientific), as described before.19,22,23 A fresh standard curve of 0–40 μM ascorbate was prepared for each HPLC run.
Western blot analysis
Western blot analysis was carried out on extracted cell-culture and tumor samples,19 and membranes were probed with primary antibodies against mouse HIF1α (1/800, AF1935), CAIX (1/800, AF2344), and β-actin (1/10,000) as loading control; all antibodies were from R&D Systems (Minneapolis, MN, USA). Secondary horseradish peroxidase-conjugated antibodies were from Agilent Technologies (Santa Clara, CA, USA). Protein bands were quantified using Alliance 2.7 software (Uvitec, Cambridge, UK) and the protein of interest normalized against a positive control (hypoxia-treated LL/2 whole-cell lysate) following confirmation of equal loading using β-actin.
VEGF ELISA
Mouse tumor VEGF-protein levels were measured using a Quantikine enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems), according to the manufacturer’s instructions.
Statistical analyses
Data were analyzed using SPSS 19.0 and GraphPad Prism version 5.08, with significance assumed at P<0.05. All data were analyzed by analysis of variance, followed by the appropriate post hoc tests. The Kolmogorov–Smirnov test determined the distribution of each variable, linear regression analysis was used for tumor growth and HIF1-pathway score analyses, and Student’s t-test determined differences in ascorbate levels.
Results
Uptake of ascorbate in murine cancer cells in vitro
Intracellular accumulation of ascorbate uptake into LL/2, B16-F10, and CMT-93 cells was investigated (Figure 1, A–C). Incubation of LL/2 cells for 8 hours with 10–1,000 μM ascorbate in the culture medium resulted in a concentration-dependent intracellular accumulation, saturating at 4–5 nmol/106 cells with a supply of 100 μM ascorbate or above (Figure S1). Subsequently, uptake was evaluated in all three cell lines over a 24-hour period, following incubation with either 50 μM or 500 μM ascorbate (Figure 1, A–C). Maximum intracellular concentrations of ascorbate were reached after 8 hours of incubation, with levels reaching 4.3±0.8 nmol/106 LL/2 cells, 0.4±0.1 nmol/106 B16-F10 cells, and 0.5±0.1 nmol/106 CMT-93 cells, following loading with 500 μM ascorbate. Significantly lower levels were reached with 50 μM ascorbate loading. After 16 hours of incubation, the intracellular ascorbate content reduced, and no difference was seen between 50 μM and 500 μM ascorbate availability in the medium. This may reflect a doubling of cancer cells during this period (Figure 1, D–F) or changes in transporter status in response to cell division or external stimuli. We did not determine the intracellular water volume for these cells, and thus ascorbate concentrations have not been determined, but we have previously noted that intracellular saturation results in low (0.5–10 mM) ascorbate concentrations in a number of cancer cell lines, with the extent of uptake varying with cell types.9
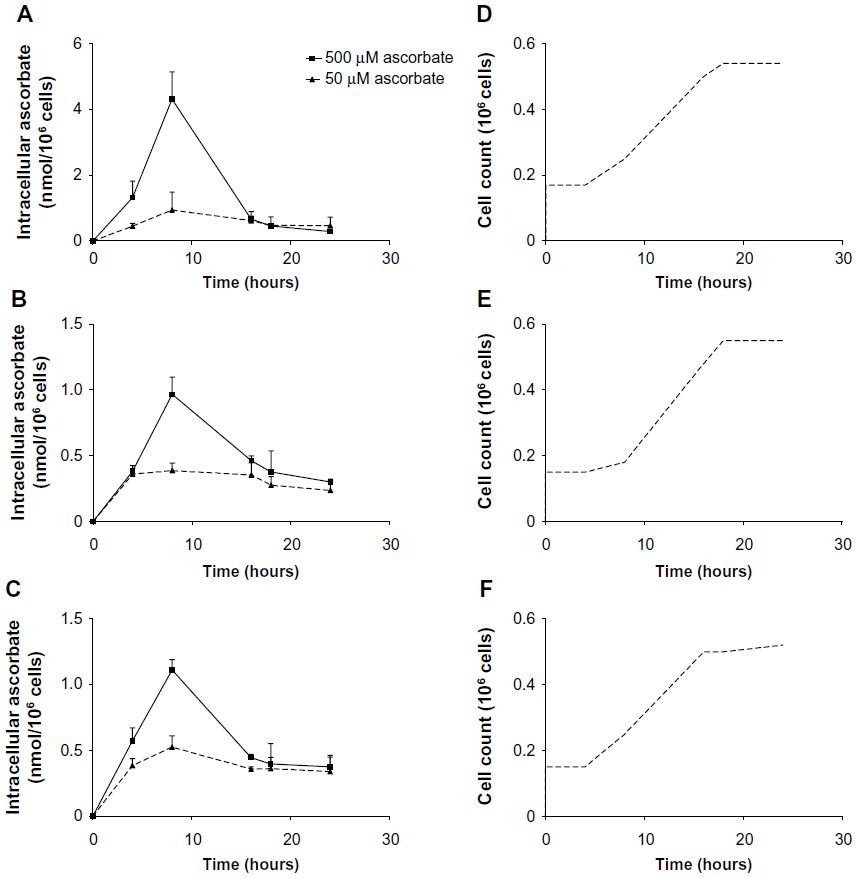 | Figure 1 Uptake of ascorbate in murine cancer cells. |
Effect of ascorbate on HIF1α accumulation in response to varied oxygen gradients
Preloading with either 50 μM or 500 μM ascorbate affected the stabilization of HIF1α in LL/2 cells in response to decreasing oxygen tension, with higher concentrations decreasing sensitivity to mild hypoxia (Figure 2). In the absence of ascorbate, HIF1 stabilization was seen when O2 availability was 5% or below. In contrast, LL/2 cells preloaded with 500 μM ascorbate showed a significant reduction in HIF1α accumulation under moderate physiological hypoxia (1%–5% O2) compared to cells lacking ascorbate (P<0.001). Preloading with 50 μM ascorbate was also able to reduce HIF1α levels significantly at 5% O2 (P<0.05), but not at 1% O2 (Figure 2). When O2 became limiting, as under severe, pathological hypoxia (0.1% O2), ascorbate had no effect on HIF1α accumulation. At 10% O2 or normoxia, HIF1α did not accumulate. Overall, increasing intracellular ascorbate concentrations were able to shift the HIF1α activation in response to hypoxia to the left (Figure 2), with more severe hypoxia being required before HIF1α protein was stabilized.
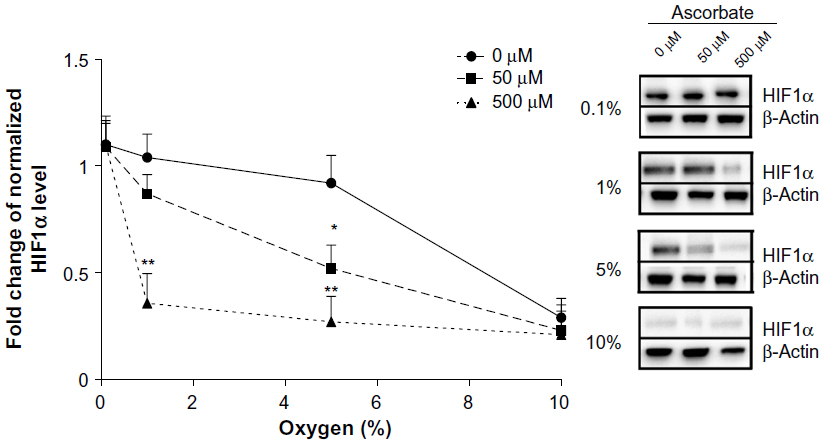 | Figure 2 The effect of intracellular ascorbate on the hypoxic response in LL/2 cells. |
Tissue ascorbate levels following dietary supplementation in mice
Varying the dietary intake of ascorbate in Gulo–/– mice resulted in achieving a range of plasma and tissue ascorbate levels from fully saturated (oral supplementation with 3,300 mg/L of ascorbate) to almost deficient (with water containing 33 mg/L) (Table 1). The levels reported in this study are in agreement with those reported previously.19,22
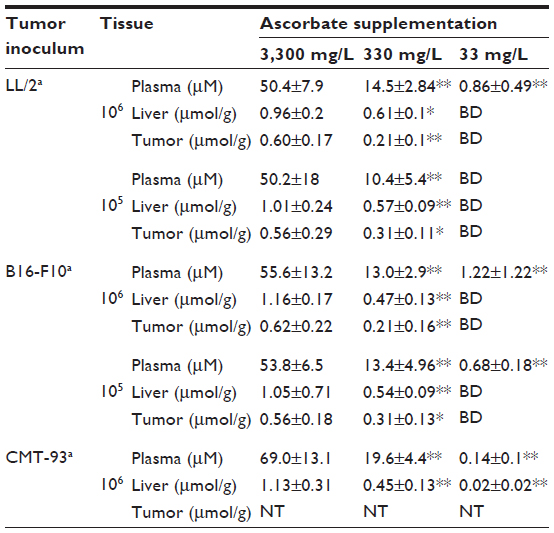 | Table 1 Plasma and tissue ascorbate concentrations in Gulo–/– mice inoculated with different starting numbers of LL/2 lung, B16-F10 melanoma, and CMT-93 colon carcinoma cells |
Tumor-take rate of LL/2, B16-F10, and CMT-93 tumors in Gulo–/– mice
Gulo–/– mice inoculated with 106 or 105 LL/2 cells grew harvestable tumors, but further reductions in the cell inoculum to 104 cells/mouse showed no tumor growth (Table 2). At the lowest cell concentration, there was no histological evidence of tumor growth up to 60 days postimplantation, when mice were killed (Table 2). With B16-F10 cells, however, six animals (of 30) grew tumors when injected with 104 cells, with mice inoculated at either 106 or 105cells all growing harvestable tumors (Table 2). In the B16-F10 low-implantation group, more tumors grew in mice supplemented with 33 mg/L than in animals kept on 330 mg/L, and more than on 3,300 mg/L, but low numbers precluded statistical assessment. For CMT-93, tumor growth was initiated in all mice inoculated at 106 and 107 cells/mouse, but all tumors regressed, resulting in a final take rate of zero (Table 2).
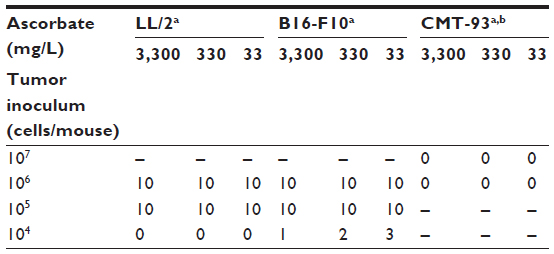 | Table 2 The effect of varying dietary ascorbate on tumor-take rate with different cell inocula |
CMT-93 tumor rejection in ascorbate-proficient mice
In mice implanted with CMT-93 cells, plasma and organ concentrations of ascorbate were significantly increased with increasing dietary supplementation (Table 1), reaching levels we have previously reported in non-tumor-bearing Gulo–/– mice.22 In mice injected with 106 CMT-93 cells, prior to complete regression, peak tumor growth was seen on day 13, with mice supplemented with 33 mg/L of ascorbate having greater tumor volumes (39±26 mm3) than those supplemented with either 330 mg/L (7.6±6.1 mm3, P<0.05) or 3,300 mg/L (4.2±2 mm3, P<0.01) ascorbate (Figure 3A). In mice injected with 107 CMT-93 cells, tumor development was accompanied by an apparent localized inflammation at the site of implantation, visible as redness and heat 3–4 days prior to complete regression. With this inoculum, peak tumor growth occurred on day 10 (Figure 3B), with mice supplemented with optimal levels of dietary ascorbate having smaller palpable tumors (7.9±8.6 mm3) compared to mice on 330 mg/L (24.8±21.3 mm3) or 33 mg/L (32±23 mm3) (P=0.06) ascorbate supplementation. No tumors were evident at 60 days post implantation (Figure 3C).
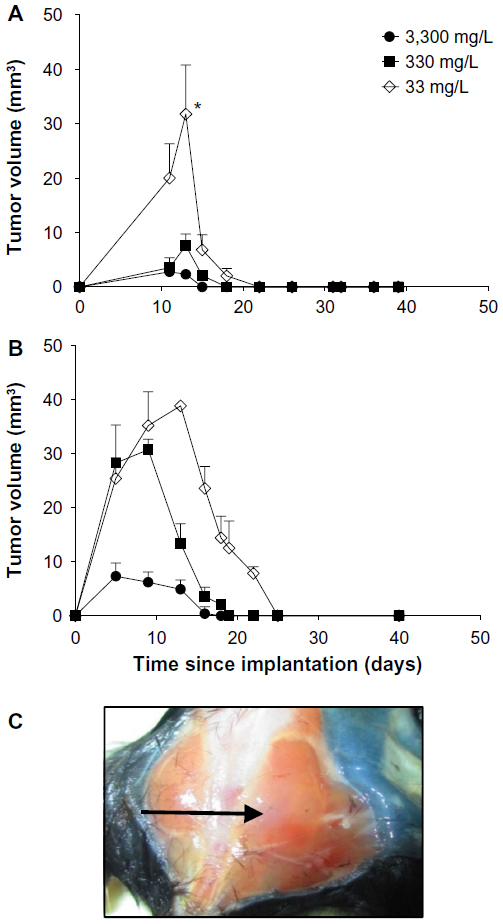 | Figure 3 CMT-93 tumor growth and regression in Gulo–/– mice. |
Tumor-growth rates according to ascorbate supplementation
Tumor-growth rates of LL/2 and B16-F10 tumors varied with ascorbate supplementation, similar to our previous report,19 and showed some dependency on initial inoculum. LL/2 tumors took significantly longer to grow to a palpable mass (106, P<0.001; 105, P<0.05) and to reach four times their size (106, P<0.05; 105, P<0.001) in 3,300 mg/L-supplemented mice, compared to 33 mg/L ascorbate-supplemented mice.
In mice inoculated with B16-F10 cells, very few tumors were initiated when mice were inoculated with 104 cells, and increasing levels of ascorbate supplementation prevented tumor initiation more effectively (Table 2). Those tumors that did establish also exhibited slower growth (tumors taking longer to reach 200 mm3 and to grow from 200 to 800 mm3), but low numbers precluded statistical analysis (data for individual mice shown in Table 3). With higher numbers of cells implanted, tumors also took significantly longer to start growing (106, P>0.05; 105, P<0.001) and to quadruple tumor size (106, P<0.001; 105, P<0.001) in 3,300 mg/L- vs 33 mg/L-supplemented mice (Table 3).
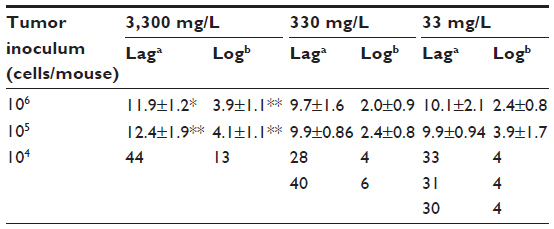 | Table 3 Growth characteristics of B16-F10 tumors in Gulo–/– mice at different initial cell inocula and varying dietary ascorbate supplementation |
It appeared that plasma ascorbate levels, and therefore tissue saturation, influenced tumor initiation: mice that produced tumors following implantation with 104 B16-F10 cells had plasma ascorbate levels that were notably lower and outside the 95% confidence interval of the group mean than those animals that did not produce tumors (Table 4). However, due to the low number of animals growing tumors in this group, the statistical significance of this could not be confirmed.
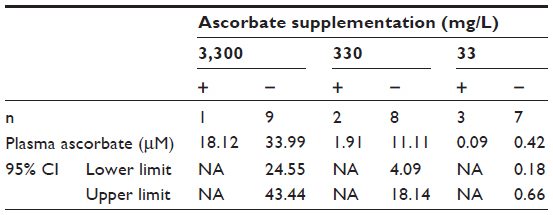 | Table 4 Plasma ascorbate levels of Gulo–/– mice implanted with 104 B16-F10 cells and maintained on varying dietary ascorbate supplementations |
Tumor-growth rates according to tumor ascorbate levels
Tumor ascorbate content was associated with both tumor initiation and growth rate. When measured levels of tumor ascorbate were compared with the tumor lag and log-growth phases of LL/2 and B16-F10 tumors, we found that there was a significant correlation between tumor ascorbate levels and increased lag- and log-phase growth, with the strength of the association varying by cell inoculum (Table 5).
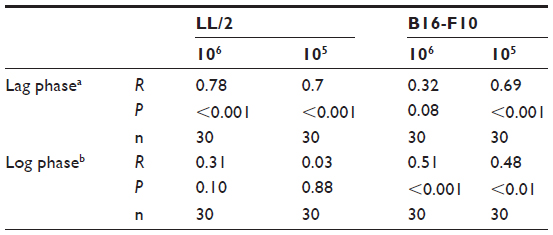 | Table 5 Pearson’s correlation between individual tumor ascorbate concentrations and tumor growth during lag and log phase of LL/2 and B16-F10 tumors |
Even in Gulo–/– mice supplemented with 3,300 mg/L of ascorbate, B16-F10 tumor-growth rates were highly variable (Figure 4A). This was not dependent on whether 106 or 105 B16-F10 cells were inoculated (Table 3) or on plasma ascorbate concentrations (Figure 4B), but was dependent on the level of tumor ascorbate. Tumors with higher than the median ascorbate concentration (>0.60 μmol/g) had significantly longer lag (P<0.05, t-test) and log phases (P=0.029, t-test) compared to tumors whose ascorbate levels were less than the median (<0.60 μmol/g) (Figure 4B).
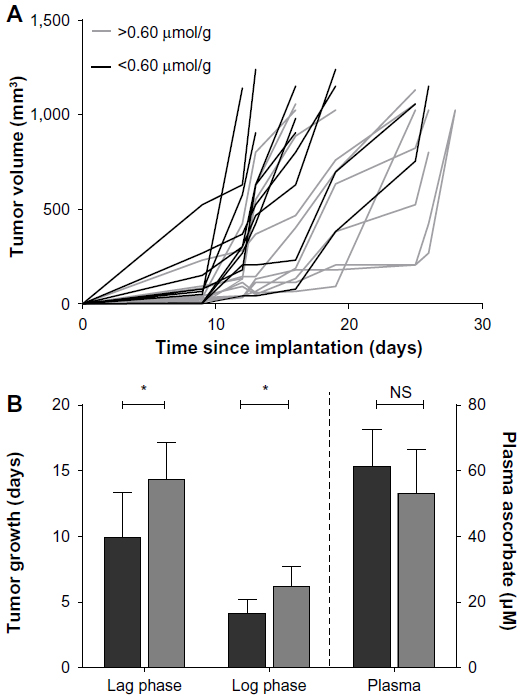 | Figure 4 Relationship between tumor ascorbate and B16-F10 tumors grown in mice supplemented with 3,300 mg/L ascorbate. |
HIF1α levels and HIF1 transcriptional activity in B16-F10 tumors
Levels of HIF1α, CAIX, and VEGF proteins detected in B16-F10 tumors grown from 105 cells increased as dietary ascorbate supplementation decreased (P<0.001; Figure 5, A and B). This is similar to our data in LL/2 tumors grown from 105 cells (data not shown), and supports our previous findings with LL/2 and B16-F10 tumors generated in mice inoculated with 106 cells/mouse.19 There was a significant inverse correlation between tumor ascorbate and each individual HIF1α-pathway protein in B16-F10 tumors, irrespective of cell inoculum, indicating that the tumor ascorbate content was the driving factor (Figure 5C). A comparison of VEGF levels revealed that higher dietary ascorbate intake resulted in significantly decreased tumor VEGF compared to those measured following lower levels of supplementation (P<0.05, Table 6).
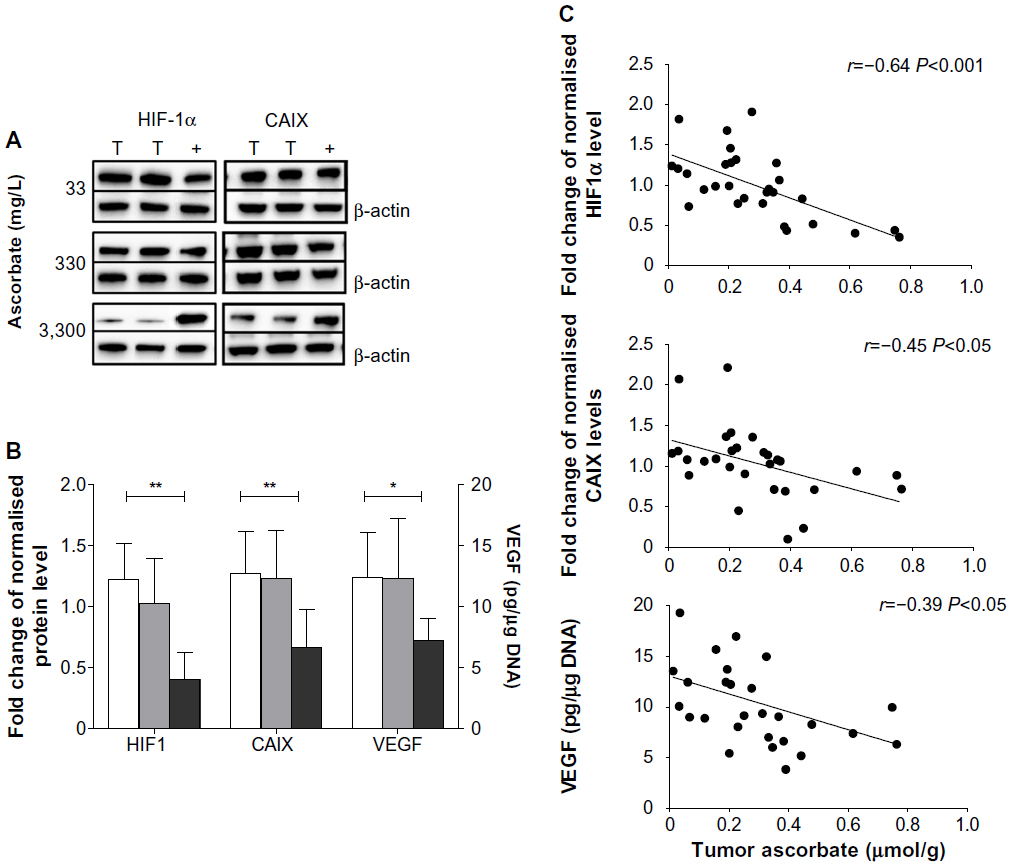 | Figure 5 Expression of HIF1-pathway proteins in B16-F10 tumors. |
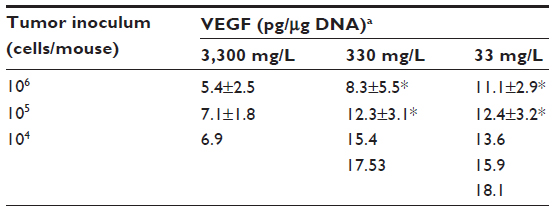 | Table 6 VEGF levels in tumors grown from different initial inocula and with variable ascorbate supplementation |
Discussion
Ascorbate has numerous potential roles in biological systems, but is particularly known to be an important cofactor for metalloenzymes with diverse functions.8–11 In this study, we investigated its role as a cofactor for the 2OG-dependent dioxygenases, which are involved in vital cellular processes ranging from the hypoxic response to collagen deposition and the regulation of epigenetic processes.10 This study has confirmed the fundamental importance of ascorbate in the regulation of the hypoxic response in cancer cells and tumors, and its ability to affect tumor initiation and growth.
Ascorbate is routinely absent from cell-culture media, and hence most cells in culture are maintained under ascorbate-deficient conditions. We showed that the three cell lines readily accumulated intracellular ascorbate from culture media, and that intracellular availability affected the HIF1 response of LL/2 cells to moderate levels of hypoxia (1%–5% O2), with more extreme hypoxia being required for HIF1 stabilization in the presence of physiological intracellular ascorbate. These levels of oxygenation are frequently measured in normal tissues (equivalent of 5% O2) and solid tumors (equivalent to 1% O2).2,24 These results extend previous findings by us and others in which physiological intracellular concentrations of ascorbate were able to suppress the HIF1 response in human fibroblasts and endothelial cells under conditions of moderate hypoxia (1% O2).16,17 Our current results are the first demonstration of the efficacy of ascorbate in the moderation of the hypoxic response over a range of physiological oxygen tensions. The ability of ascorbate to modulate the hypoxic response thus occurs at the margins of hypoxic sensitivity, and this varies with different cell lines.9 Ascorbate did not affect HIF1α stabilization in response to severe hypoxia (0.1% O2), and this likely reflects the absolute substrate requirement of prolyl hydroxylases for molecular oxygen.25 Accordingly, prolyl hydroxylase activity is almost completely suppressed at 0.1% oxygen.26 Our data indicate that ascorbate can have a marked impact on HIF1α stabilization under physiological hypoxia, and studies that have used only extreme hypoxia and low levels of ascorbate supplementation may have underestimated the importance of ascorbate as a regulator of the hydroxylases.27
We have identified an important relationship between tumor initiation and the level of dietary ascorbate supplementation. When tissue ascorbate loading was saturating or optimal, more B16-F10 cancer cells were required to drive tumor initiation, and initiation was significantly delayed compared to that seen in animals with lower ascorbate intake. Tumor ascorbate levels were inversely correlated with the extent of HIF1 activation, as measured by both HIF1α protein and downstream gene-expression levels. Our findings with the CMT-93 tumor model, which has previously been chosen for antitumor immunity studies,28 also indicate that increased ascorbate supplementation could support effective tumor rejection. Taken together, our results indicate that ascorbate uptake into tumor cells may influence HIF1 regulation, downregulating HIF1 activation and inhibiting tumor initiation and subsequent growth.
These results are consistent with our previous findings in Gulo–/– and wild-type C57BL/6 mice, where we showed that increased levels of ascorbate supplementation significantly reduced LL/2 HIF1 activation and tumor growth.19 Others have also reported reduced tumor growth in ascorbate-supplemented Gulo–/– mice29,30 and similarly deficient Sfx mice,31 although in these studies supplemented mice were compared with complete ascorbate withdrawal, resulting in signs of scurvy. Decreased tumor growth associated with lower ascorbate has also been reported, but once again only in conditions of complete ascorbate deficiency.32,33 Our experiments provide information on the effects of more physiologically relevant ascorbate intake, reflecting an optimal or suboptimal but still adequate dietary intake that does not result in scurvy-like conditions.
In addition, our data are the only measures available that document tumor ascorbate levels and HIF1 activation, and we were therefore able to determine whether there is an association between inhibition of HIF1 activity, tumor ascorbate, and tumor growth. Our observed association between ascorbate and HIF1-pathway activity is supported by a previous report in a human B-cell lymphoma model (grown in ascorbate-generating wild-type mice).34 Ascorbate inhibited lymphogenesis in unmodified tumors, whereas cells overexpressing a hydroxylase-resistant HIF1α were no longer sensitive to the effect of ascorbate.34 These results support the hypothesis that regulation of the HIF hydroxylases underpins the antitumor activity of ascorbate, but in this study neither tumor ascorbate levels nor tumor HIF1α levels were measured.
We have previously also measured ascorbate and HIF1 activation in human endometrial and colorectal cancer clinical samples.23,35 These studies employed retrospective analyses of tumor tissue taken at surgery, and ascorbate levels in the patients and their tumors were not manipulated. Our observations that tumor ascorbate levels were associated with HIF1 activity, and in the case of colorectal cancer patient disease-free survival,23,35 together with the results of our current studies with the Gulo–/– mouse model, suggest that human clinical intervention studies are warranted to determine whether changing tumor ascorbate levels would influence the HIF1 pathway in cancer patients.
The determining factor in the ability of ascorbate to influence tumor HIF1 activation appears to be the achievable concentration of ascorbate in the tissues rather than plasma levels. This would appear to be the case for normal physiological plasma concentrations, and this is well demonstrated by our analysis of B16-F10 tumors from mice supplemented with 3,300 mg/L ascorbate, which had saturating plasma ascorbate levels. In these mice, measured tumor ascorbate levels varied and were significantly correlated with HIF1 activity and tumor growth. Therefore, even when the highest physiological levels of ascorbate were supplied, unknown factors in mice or the tumors modulated tumor ascorbate accumulation and subsequently affected tumor growth. Similar patterns were observed in our analysis of patient clinical samples: ascorbate uptake into tumor tissue showed marked differences when compared with adjacent normal tissue, suggesting impaired transport in higher-grade tumors.23,35
In normal tissue, SVCT2 is the major facilitative transporter,36 but this may not be true for tumor tissue. A recent report has suggested that facilitated transport via upregulated GLUT1 in some colorectal tumors may influence tumor ascorbate, although this would only involve uptake of dehydroascorbate, the oxidized form of ascorbate.37 Previous reports have identified single-nucleotide polymorphisms in both SVCT1 and SVCT2 in humans that were associated with increased risk of cancer,38,39 suggesting that ascorbate delivery may be compromised in some cancer patients. In addition, tumors may differentially express these transporters, due to environmental changes.40 Therefore, differences in ascorbate-transporter status may explain some of the variation we have seen in the uptake of ascorbate in B16-F10 tumors.
The delayed tumor initiation in LL/2 and B16-F10 tumors and the spontaneous regressions of CMT-93 tumors also suggest that ascorbate may play a role in antitumor immunity. The potential of ascorbate to support the immune system is multiple: as a potent antioxidant, it protects immune cells from oxidative stress41 and promotes normal neutrophil apoptosis,17 thus supporting both the innate and adaptive immune response. A study comparing fully supplemented with completely deficient Gulo–/– mice given an ovarian cancer challenge showed a reduction in natural killer-cell activity in deficient mice, which was accompanied by reduced survival, compared to supplemented animals.30 These results, together with our own observations, suggest a role for ascorbate in anticancer immunity, and this warrants further analysis.
Recent meta-analyses have shown an inverse correlation between ascorbate intake and risk of head and neck42,43 and gastric cancer,44 and there is substantial public interest in dietary ascorbate as a cancer-preventive agent.45 In our study, the 330 mg/L dose produced plasma ascorbate levels halfway between saturation and deficiency, which is similar to levels in human populations,46 with mean ascorbate plasma levels in the general population being 40–50 μM, substantially below human saturation levels of ~80 μM.46,47 Tissue ascorbate status is dependent on plasma supply, and in humans this depends on dietary intake and metabolic turnover.46,48,49 Low plasma ascorbate levels have been reported in cancer patients, with levels of 15 μM in lung cancer and 18 μM in oral squamous cell carcinoma patients.50,51 The notable differences in tumor take and rejection, and in growth rates seen in our study between mice supplemented at 330 mg/L and 3,300 mg/L, is therefore of interest for further investigation, and supports the maintenance of body ascorbate status at saturating levels for the prevention of chronic diseases, including cancer.45–49
We have provided both in vitro and in vivo data that indicate that saturating levels of ascorbate dampen the HIF1 response under physiological hypoxia and optimal tumor ascorbate levels lead to a reduction and delay in tumor initiation and subsequent tumor growth. This coincided with a reduction in HIF1 activity, and most likely reflects the requirement for ascorbate as a cofactor for regulatory HIF hydroxylases, providing direct evidence for a plausible mechanism by which ascorbate can contribute to an anticancer effect.
Acknowledgments
The authors gratefully acknowledge financial support from the Health Research Council of New Zealand (MCMV, GUD), the Mackenzie Charitable Foundation (GUD), and the University of Otago (PhD scholarship to EJC).
Disclosure
The authors report no conflicts of interest in this work.
References
Semenza GL. Hypoxia and cancer. Cancer Metastasis Rev. 2007;26(2):223–224. | |
Semenza GL. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664–3671. | |
Ratcliffe PJ. Oxygen sensing and hypoxia signalling pathways in animals: the implications of physiology for cancer. J Physiol. 2013;591(8):2027–2042. | |
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22(16):4082–4090. | |
Maxwell PH, Wiesener MS, Chang GW, et al. The tumor suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. | |
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16(12):1466–71. | |
Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343–354. | |
Ozer A, Bruick RK. Non-heme dioxygenases: cellular sensors and regulators jelly rolled into one? Nat Chem Biol. 2007;3(3):144–153. | |
Kuiper C, Dachs GU, Currie MJ, Vissers MC. Intracellular ascorbate enhances hypoxia-inducible factor (HIF)-hydroxylase activity and preferentially suppresses the HIF-1 transcriptional response. Free Radic Biol Med. 2014;69:308–317. | |
Kuiper C, Vissers MC. Ascorbate as a co-factor for Fe- and 2-oxoglutarate dependent dioxygenases: physiological activity in tumor growth and progression. Front Oncol. 2014;4:359. | |
Myllylä R, Kuutti-Savolainen ER, Kivirikko KI. The role of ascorbate in the prolyl hydroxylase reaction. Biochem Biophys Res Commun. 1978;83(2):441–448. | |
Vaupel P, Thews O, Hoeckel M. Treatment resistance of solid tumors: role of hypoxia and anemia. Med Oncol. 2001;18(4):243–259. | |
Vaupel P, Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 2007;26(2):225–239. | |
Baba Y, Nosho K, Shima K, et al. HIF1A overexpression is associated with poor prognosis in a cohort of 731 colorectal cancers. Am J Pathol. 2010;176(5):2292–2301. | |
Zhong H, De Marzo AM, Laughner E, et al. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999;59(22):5830–5835. | |
Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ. Effect of ascorbate on the activity of hypoxia-inducible factor in cancer cells. Cancer Res. 2003;63(8):1764–1768. | |
Vissers MC, Gunningham SP, Morrison MJ, Dachs GU, Currie MJ. Modulation of hypoxia-inducible factor-1 alpha in cultured primary cells by intracellular ascorbate. Free Radic Biol Med. 2007;42(6):765–772. | |
Pihlajaniemi T, Myllylä R, Kivirikko KI. Prolyl 4-hydroxylase and its role in collagen synthesis. J Hepatol. 1991;13 Suppl 3:S2–S7. | |
Campbell EJ, Vissers MC, Bozonet S, Dyer A, Robinson BA, Dachs GU. Restoring physiological levels of ascorbate slows tumor growth and moderates HIF-1 pathway activity in Gulo(-/-) mice. Cancer Med. 2015;4(2):303–314. | |
Workman P, Aboagye EO, Balkwill F, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010; 102(11):1555–1577. | |
Balls M, Goldberg AM, Fentem JH, et al. The three Rs: the way forward – the report and recommendations of ECVAM Workshop 11. Altern Lab Anim. 1995;23(6):838–866. | |
Vissers MC, Carr AC, Pullar JM, Bozonet SM. The bioavailability of vitamin C from kiwifruit. Adv Food Nutr Res. 2013;68:125–147. | |
Kuiper C, Molenaar IG, Dachs GU, Currie MJ, Sykes PH, Vissers MC. Low ascorbate levels are associated with increased hypoxia-inducible factor-1 activity and an aggressive tumor phenotype in endometrial cancer. Cancer Res. 2010;70(14):5749–5758. | |
Carreau A, El Hafny-Rahbi B, Matejuk A, Grillon C, Kieda C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J Cell Mol Med. 2011;15(6):1239–1253. | |
Epstein AC, Gleadle JM, McNeill LA, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43–54. | |
Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286(15):13041–13051. | |
Nytko KJ, Maeda N, Schläfli P, Spielmann P, Wenger RH, Stiehl DP. Vitamin C is dispensable for oxygen sensing in vivo. Blood. 2011; 117(20):5485–5493. | |
Hu JY, Wang S, Zhu JG, Zhou GH, Sun QB. Expression of B7 costimulation molecules by colorectal cancer cells reduces tumorigenicity and induces anti-tumor immunity. World J Gastroenterol. 1999;5(2):147–151. | |
Cha J, Roomi MW, Ivanov V, Kalinovsky T, Niedzwiecki A, Rath M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int J Oncol. 2013;42(1):55–64. | |
Kim JE, Cho HS, Yang HS, et al. Depletion of ascorbic acid impairs NK cell activity against ovarian cancer in a mouse model. Immunobiology. 2012;217(9):873–881. | |
Amano A, Aigaki T, Maruyama N, Ishigami A. Ascorbic acid depletion enhances expression of the sodium-dependent vitamin C transporters, SVCT1 and SVCT2, and uptake of ascorbic acid in livers of SMP30/GNL knockout mice. Arch Biochem Biophys. 2010;496(1):38–44. | |
Telang S, Clem AL, Eaton JW, Chesney J. Depletion of ascorbic acid restricts angiogenesis and retards tumor growth in a mouse model. Neoplasia. 2007;9(1):47–56. | |
Parsons KK, Maeda N, Yamauchi M, Banes AJ, Koller BH. Ascorbic acid independent synthesis of collagen in mice. Am J Physiol Endocrinol Metab. 2006;290(6):1131–1139. | |
Gao P, Zhang H, Dinavahi R, et al. HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell. 2007;12(3):230–238. | |
Kuiper C, Dachs GU, Munn D, et al. Increased tumor ascorbate is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in human colorectal cancer. Front Oncol. 2014;4:10. | |
Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010;500(2):107–115. | |
Jun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–1396. | |
Chen AA, Marsit CJ, Christensen BC, et al. Genetic variation in the vitamin C transporter, SLC23A2, modifies the risk of HPV16-associated head and neck cancer. Carcinogenesis. 2009;30(6):977–981. | |
Skibola CF, Bracci PM, Halperin E, et al. Polymorphisms in the estrogen receptor 1 and vitamin C and matrix metalloproteinase gene families are associated with susceptibility to lymphoma. PLoS One. 2008;3(7):e2816. | |
Hong SW, Lee SH, Moon JH, et al. SVCT-2 in breast cancer acts as an indicator for L-ascorbate treatment. Oncogene. 2013;32(12):1508–1517. | |
Ströhle A, Wolters M, Hahn A. Micronutrients at the interface between inflammation and infection – ascorbic acid and calciferol: part 1, general overview with a focus on ascorbic acid. Inflamm Allergy Drug Targets. 2011;10(1):54–63. | |
Edefonti V, Hashibe M, Parpinel M, et al. Natural vitamin C intake and the risk of head and neck cancer: a pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. Int J Cancer. 2015;137(2):448–462. | |
de Munter L, Maasland DH, van den Brandt PA, Kremer B, Schouten LJ. Vitamin and carotenoid intake and risk of head-neck cancer subtypes in the Netherlands Cohort Study. Am J Clin Nutr. 2015;102(2):420–432. | |
Mazda J, Elio R, Pietro F, et al. Plasma and dietary vitamin C levels and risk of gastric cancer in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST). Carcinogenesis. 2006;27(11):2250–2257. | |
New Zealand Ministry of Health. Nutrient Reference Values for Australia and New Zealand, 2014. Available from: https://www.nrv.gov.au/chronic-disease/summary. Accessed March 8, 2016. | |
Levine M, Wang Y, Padayatty SJ, Morrow J. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98(17):9842–9846. | |
Levine M, Conry-Cantilena C, Wang Y, et al. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93(8):3704–3709. | |
Harrison FE, Green RJ, Dawes SM, May JM. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010;1348:181–186. | |
Padayatty SJ, Sun H, Wang Y, et al. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004; 140(7):533–537. | |
Fiaschi AI, Cozzolino A, Ruggiero G, Giorgi G. Glutathione, ascorbic acid and antioxidant enzymes in the tumor tissue and blood of patients with oral squamous cell carcinoma. Eur Rev Med Pharmacol Sci. 2005;9(6):361–367. | |
Calişkan-Can E, Firat H, Ardiç S, Simşek B, Torun M, Yardim-Akaydin S. Increased levels of 8-hydroxydeoxyguanosine and its relationship with lipid peroxidation and antioxidant vitamins in lung cancer. Clin Chem Lab Med. 2008;46(1):107–112. |
Supplementary materials
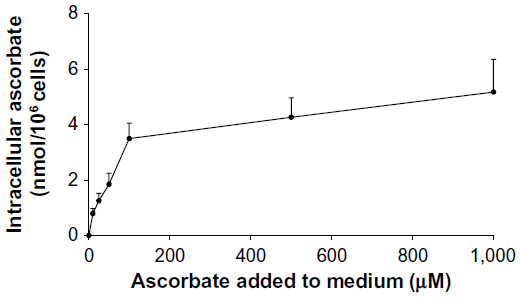 | Figure S1 Intracellular ascorbate levels in LL/2 cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.