")
Back to Journals » OncoTargets and Therapy » Volume 12
ASAP3 is a downstream target of HIF-1α and is critical for progression of lung adenocarcinoma
Authors Zhang P, Sun J, Kai J, Peng Y, Liu X, Zhou F, Wu J
Received 26 December 2018
Accepted for publication 8 May 2019
Published 17 July 2019 Volume 2019:12 Pages 5793—5803
DOI https://doi.org/10.2147/OTT.S199603
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gaetano Romano
Pingping Zhang,1,2,* Junwei Sun,2,* Jindan Kai,2 Yi Peng,2 Xiyou Liu,2 Fuxiang Zhou,1 Jianping Wu2
1Department of Radiation Oncology and Medical Oncology, Zhongnan Hospital, Wuhan University, Wuhan 430079, People’s Republic of China; 2Department of Oncology, Hubei Cancer Hospital, Affiliated Hubei Cancer Hospital of Huazhong University of Science and Technology, Wuhan 430079, People’s Republic of China
*These authors contributed equally to this work
Background: ASAP3 was first identified as a protein that promotes cell proliferation in hepatocellular carcinoma and later reported to be an Arf6-specific Arf GTPase-activating protein that regulates cell migration associated with cancer cell invasion.
Materials and methods: Patients and tissue samples were from Hubei Cancer Hospital, human lung adenocarcinoma cell lines were obtained from the cell bank of the Chinese Academy of Science, nude mice (BALB/c nu/nu) were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. Our methods contained immunohistochemistry, Western blotting, reverse-transcription polymerase chain reaction (RT-PCR), immunofluorescence staining, stable transfection of lung adenocarcinoma cells, chromatin immunoprecipitation (CHIP) and luciferase assay, wound healing and cell migration assay.
Results: In this study, we show that ASAP3 overexpression promotes migration and invasiveness in human lung adenocarcinoma cells and accelerates tumor progression in a xenograft mouse model. In patient tumor samples, ASAP3 overexpression was significantly associated with lymph node metastasis and reduced overall survival. We also show that ASAP3 expression is induced under hypoxic conditions through hypoxia-inducible factor 1α (HIF-1α), which binds directly to HER1 or/and HER2 (hypoxia response element) in the ASAP3 promoter. ASAP3 overexpression counteracts the inhibition of lung adenocarcinoma progression caused by HIF-1α knockdown both in vitro and in vivo.
Conclusion: Our results identify ASAP3 as a downstream target of HIF-1α that is critical for metastatic progression in lung adenocarcinoma.
Keywords: lung adenocarcinoma, ASAP-3, hypoxia-inducible factor-1α, metastasis
Introduction
Lung cancer is the leading cause of cancer-related deaths, lung adenocarcinoma comprises 60% of the lung cancer and is the predominant subtype of cancer cases affecting women, non-smokers, and young adults.1,2 Survival rates for lung adenocarcinoma are low, and the rapid invasion and metastasis greatly limit treatment options and accounts for 90% of the cancer-related deaths.3 The underlying mechanisms that trigger adenocarcinoma cell invasion and metastasis remain largely unknown, although it is believed that the hypoxic microenvironment and hypoxia-induced factors are critical drivers of metastasis and progression.4 In particular, HIF-1α expression levels are known to be associated with tumor progression, poorly differentiated cells, lymph node metastasis, and poor survival.4
Migration requires temporally and spatially coordinated changes in the cellular membrane and cytoskeleton.5 ADP-ribosylation factors are small G proteins that control cytoskeletal remodeling and function by cycling between GTP-bound and GDP-bound states.6,7 The Arf family of GTP-binding proteins and Arf GTPase-activating proteins (GAPs) regulate cytoskeletal remodeling by regulating hydrolysis of GTP bound to Arf proteins.8,9 ASAP3, previously known as DDEFL1 and ACAP4, is a member of the ASAP subfamily (Arf GAP, SH3, Ank repeat, and PH domains) of Arf GAPs, which possess Ankyrin repeats, Arf GAP, Bin/amphiphysin/Rvs (BAR), and PH domains. ASAP3 expression is low or negligible in normal epithelia, but has been found to be dramatically increased in many human carcinomas, including lung carcinomas.10,11 Several studies have reported that ASAP3 expression promotes metastasis in multiple cancers.5,11,12 and may contribute to poor clinical outcome in non-small cell lung carcinoma.10 These effects may be linked to the role of ASAP3 in regulating cell migration and thus cancer cell invasion. In support of this, a recent study reported that ASAP3 promotes migration of embryonic fibroblast cells in vitro.5,13 However, the molecular mechanisms underlying ASAP3 overexpression and the role of ASAP3 in lung adenocarcinoma progression are still not fully understood.
In this study, we aimed to investigate the role of ASAP3 in lung adenocarcinoma progression. We show that ASAP3 is overexpressed in lung adenocarcinoma, and that ASAP3 overexpression promotes lung adenocarcinoma migration and invasion in vitro and in xenograft mouse models in vivo. ASAP3 overexpression in lung adenocarcinoma is mediated by HIF-1α, which directly binds to and activates the ASAP3 promoter. Our findings indicate that ASAP3 is a novel direct downstream target of HIF-1α that promotes lung adenocarcinoma metastasis and progression.
Materials and methods
Patients and tissue samples
While some patients were dead or not convenient to return to the hospital, we would inform their relatives, therefore, all patients or their relatives gave their informed consent for this study. The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Hubei Cancer Hospital (LLHBCH2018KY-009).
Tumor specimens and paired normal non-neoplastic tissues were obtained from 101 patients (65 males and 36 females) with lung adenocarcinoma who underwent surgical resection at Hubei Cancer Hospital between January 2009 and December 2012. Normal tissues were taken from the same patient at >5 cm from the edge of the primary tumor. Survival was defined as the time from the day of surgery to the end of follow-up or the day of death due to recurrence or metastasis. All specimens were reevaluated to confirm lung cancer diagnosis based on the 2015 criteria of the World Health Organization14 and the revised criteria of the Union for International Cancer Control.15
Immunohistochemistry (IHC)
IHC was performed on patient tissue samples using antibodies against HIF-1α (ab203848, Abcam, USA, dilution 1:200) and ASAP3 (ab208169, Abcam, USA, dilution 1:200) and the DAB Substrate Kit (USEN Biotechnology, Shanghai, China) according to manufacturer protocols. Sections were scored for the proportion of immunopositive cells and intensity of immunoreactivity. The final score (ranging from 0 to 9) was determined by multiplying the score for immunopositive proportion and the score for relative staining intensity as described previously.16
Cell culture and hypoxic treatment
Human lung adenocarcinoma cell lines human bronchial epithelial (HBE), CALU3, DMS53, A549, PC9, H1299, and CALU6 were obtained from the cell bank of the Chinese Academy of Science (Shanghai, China) and grown at 37°C in a humidified atmosphere of 5% CO2 using Dulbecco’s modified Eagle medium (DMEM; Solarbio, Shanghai, China) with 10% fetal bovine serum (FBS; Gibco, USA). To induce hypoxia, cells were placed in a modulator incubator (Thermo Electron, Forma, MA, USA) in an atmosphere of 93.5% N2/5% CO2/1.5% O2 or 85% N2/5% CO2/10% O2.
Western blotting
Cells were harvested and lysed in 10 volumes of PIPA lysis buffer with a proteinase inhibitor cocktail (Sigma). After centrifugation, the supernatant was collected and quantified. An equal amount of total protein from each sample was then loaded onto a 10% gel for SDS-PAGE separation and transferred to a PVDF membrane. The membrane was then incubated at 4°C overnight with antibodies against ASAP3 (dilution 1:1,000) or HIF-1α (dilution 1:1,000) and GAPDH (ab8245, Abcam, USA, dilution 1:1,000). After incubation with a horseradish peroxidase-conjugated secondary antibody (Santa Cruze, CA, USA) at room temperature for 2 hrs, protein bands were visualized using enhanced chemiluminescence and detected using the BioImaging System (GE, USA).
Reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from transfected cells with TRIzol Reagent (Takara, Dalian, China) and used for first-strand cDNA synthesis using the First-Strand Synthesis System for RT-PCR (Takara). The primers are listed in Table S1. Each sample was processed in triplicate, and β-actin mRNA was quantified as an internal control. Each experiment was performed at least three times.
Immunofluorescence staining
Cells were fixed with 4% paraformaldehyde, blocked with 1% bovine serum albumin (Gibco, USA) and incubated overnight at 4°C with anti-ASAP3 polyclonal antibody (ab208169, dilution 1:50). The samples were then incubated with secondary antibodies conjugated to rhodamine/fluorescein isothiocyanate (FITC; Genesil Biotechnology). Nuclei were counterstained with Hoechst 33258 (Genesil Biotechnology).
Stable transfection of lung adenocarcinoma cells
Human ASAP3 cDNA was cloned into the pcDNA3.1 vector (Genesil Biotechnology), while a pcDNA3.1-HIF-1α plasmid was prepared as described.17 For stable transfections, the pLV-cDNA lentivirus-mediated expression system was used (Genesil Biotechnology) according to the manufacturer’s protocol. Lentiviral DNA was packaged as described.18 Following transfection, the medium containing lentiviruses was collected, filtered, and transferred onto CALU3 and DMS53 lung adenocarcinoma cells. Infected cells were selected with puromycin (1 μg/mL) for 7 days.
Chromatin immunoprecipitation (CHIP) and luciferase assay
Chromatin was immunoprecipitated using the ChIP-IT® Express Enzymatic Shearing kit (Active Motif Biotechnology, Shanghai, China) according to the manufacturer’s instructions. Briefly, A549 cells were treated with or without hypoxia and then immunoprecipitated with anti-HIF1a antibody, the immunoprecipitated products were detected by RT-PCR assays. Primers flanking the HRE of the promoter of the VEGF gene were used as a positive control.16
Luciferase analysis was performed as described.19 When cells were 80% confluent, they were transfected for 48 hrs with pGL3-ASAP3 using Lipofectamine 2000 (Invitrogen). Cells transfected with pcDNA-HIF1a or control vector (pcDNA-vector) were transfected with pGL3-ASAP3-promoter, pGL3-ASAP3-promoter mutation (MUT), or pGL3-empty vector (pGL3). Forty-eight hours later, cells were then collected and analyzed for luciferase activity using the Dual-Luciferase Reporter Assay System (Promega) as described.17 Results were expressed as a fold induction relative to the cells transfected with the control vector (pcDNA3.1) after normalization to Renilla activity.
ASAP3 knockdown in lung adenocarcinoma cells
For stable gene silencing, short hairpin (sh)RNA sequences against ASAP3 and HIF-1α were designed and inserted into the pLKO.1 vector as described.20 In parallel, a negative control pLKO.1 vector containing scrambled shRNA was constructed. Each of these three constructs was co-transfected into A549 and H1299 cells with packaging plasmids using Lipofectamine 2000 (Invitrogen). One set of cultures was transfected with empty pLKO.1 vector under the same conditions. Transfected cells were selected with 2 mg/mL puromycin for 10 days, and knockdown efficiency was confirmed by quantitative RT-PCR.
Wound healing and cell migration assay
A wound-healing assay was performed as described.16 Migration and invasion assays were performed in 24-well Transwell plates with inserts containing 8.0-μm pores. Cells (1×105) in DMEM were plated in the upper chamber. DMEM with 10% FBS was added to the lower chamber, and the plates were incubated for 24 hrs. For invasion assays, matrigel-coated filters were inserted into the wells (BD Bioscience, diluted 1:6). Cells that migrated to the bottom of the filter were stained with a three-step stain kit (Thermo Scientific). Data were collected from at least three independent experiments, each carried out in triplicate. Data are presented as percentages calculated by normalizing against the values obtained for cells transfected with scrambled shRNA vector.
Growth and metastasis of xenogaft lung adenocarcinoma cancer in mice
All animal experiments were approved by the Animal Research Ethics Committee of Hubei Cancer Hospital and performed in accordance with the guidelines for animal care of the hospital’s Institutional Animal Care and Use Committee. Nude mice (BALB/c nu/nu) (Shanghai SLAC Laboratory Animal Co. Ltd) were housed in the animal facility of the Experimental Animal Center of Hubei Cancer Hospital.
A549 adenocarcinoma cells were transfected with pLKO.1, pLKO.1-ASAP3, pLKO.1-HIF-1α, or the combination of pLKO.1-HIF-1α and pcDNA3.1-ASAP3. The four sets of cultures were harvested by trypsinization, washed in phosphate-buffered saline (PBS), resuspended at 107 cells/mL in a 1:1 solution of PBS/Matrigel, and injected subcutaneously into the right flank of five-week-old immunodeficient female nude mice (n=6 mice per group). After 42 days, mice were sacrificed by CO2 asphyxiation. Primary tumors were harvested from the mice, measured in 3 dimensions (a, b, c), and volume was calculated as a×b×c×0.52.21 Part of the tumor was fixed in 4% formalin and embedded in paraffin, while the rest of the tumor was used for protein extraction and analysis.
Statistical analysis
Statistical analyses were performed using SPSS 18.0 (IBM, Chicago, IL, USA) with p<0.05 defined as significant. Spearman’s rank correlation coefficient was utilized to test the correlation of ordinal variables. Student's t-test was used to compare the mean. The log-rank test was conducted to obtain a P value for Kaplan–Meier survival curves’ divergence. Experiments were conducted at least three times, and values are presented as mean ± SD, unless otherwise stated.
Results
ASAP3 mRNA and protein expression in lung adenocarcinoma tissues
To investigate the role of ASAP3 in lung adenocarcinoma progression, we used RT-PCR to determine ASAP3 mRNA levels in 60 lung adenocarcinoma surgical specimens and adjacent normal lung tissues. ASAP3 mRNA expression was detected in 60% (60/101) of adenocarcinoma samples and control tissues (Figure 1A), and mRNA levels were significantly higher in adenocarcinoma than in normal tissue. Significantly higher expression in lung adenocarcinoma tissue and located mainly in the cytoplasm was confirmed by Western blot and immunohistochemistry (Figure 1B–D and G). These results suggest that ASAP3 transcription was up-regulated in lung adenocarcinoma cells.
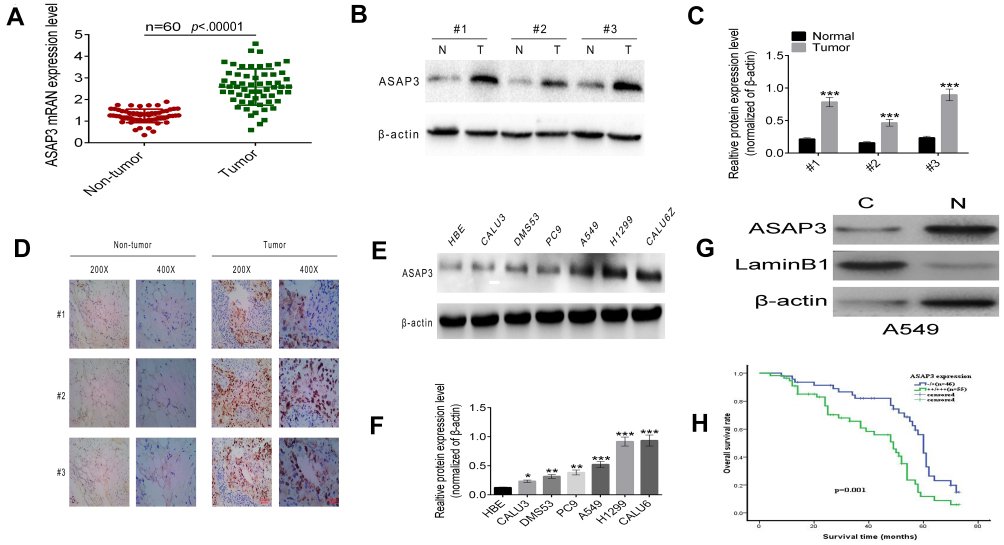 |
Figure 1 Up-regulation of ASAP3 is associated with lung adenocarcinoma. (A) Levels of ASAP3 mRNA in human lung adenocarcinoma tissues and matched adjacent non-neoplastic tissues. (B and C) Western blot analysis of ASAP3 protein levels in three pairs of human lung adenocarcinoma tissues and matched adjacent non-tumorous tissues. ASAP3 expression levels were normalized with β-actin. (D) Representative immunohistochemistry images of ASAP3 protein expression in human lung adenocarcinoma specimens compared with matched adjacent normal lung tissues. (E and F) ASAP3 expression in lung adenocarcinoma cell lines as detected by Western blot. (G) ASAP3 is located mainly in the cytoplasm. (H) Kaplan–Meier analysis of overall survival of the 101 lung adenocarcinoma patients according to different ASAP3 levels by IHC. * p<0.05, ** p<0.01, *** p<0.001.Abbreviation: IHC, immunohistochemistry. |
Western blotting showed that ASAP3 protein levels were significantly lower in HBE cells than in the lung adenocarcinoma cell lines CALU3, DMS53, PC9, A549, H1299, and CALU6 (Figure 1E and F). ASAP3 expression was significantly higher in the more invasive A549 and H1299 cell lines than in the less invasive CALU3 and DMS53 lines.
We further investigated the correlation between ASAP3 mRNA expression and clinicopathologic parameters in patients (Table S2). High ASAP3 expression, which was defined as being at least the median expression level, correlated significantly with larger tumors (p=0.005), lymph node metastasis (p=0.018), poor differentiation (p=0.011), and advanced tumor/node/metastasis (TNM) stage (p=0.013). The overall survival times of lung adenocarcinoma patients with low ASAP3 were significantly longer than those with high ASAP3 level (42.15±2.74 vs 54.71±3.02 months, p=0.001, Figure 1H). Taken together, these data indicate that high ASAP3 expression in lung adenocarcinoma correlates with lymph node metastasis and poorer patient prognosis.
ASAP3 overexpression promotes migration and invasiveness of human lung adenocarcinoma cells in vitro
To determine whether ASAP3 plays a role in lung adenocarcinoma cell invasion and migration in vitro, we performed wound-healing and invasion assays. We used the pLKO.1-ASAP3 vector containing shRNA against ASAP3 to knockdown ASAP3 expression in the lung adenocarcinoma cell lines A549 and H1299, which show high endogenous ASAP3 levels (Figure 2A and B). Of the two shRNA sequences that we tested, pLKO.1#2 efficiently knocked down ASAP3 expression by more than 50% at the mRNA level (Figure 2A, right) and protein level (Figure 2B, right). Therefore, this sequence was used in all subsequent studies. To determine whether ASAP3 overexpression is sufficient to promote lung adenocarcinoma cell migration, we also used the pcDNA3.1-ASAP3 vector to overexpress ASAP3 in the lung adenocarcinoma cell lines CALU3 and DMS53, which show low endogenous ASAP3 levels (Figure 2A and B, left). Immunofluorescence was performed to confirm ASAP3 knockdown and overexpression. Labeling with antibodies against ASAP3 confirmed the cytoplasmic localization of the protein (Figure 2E).
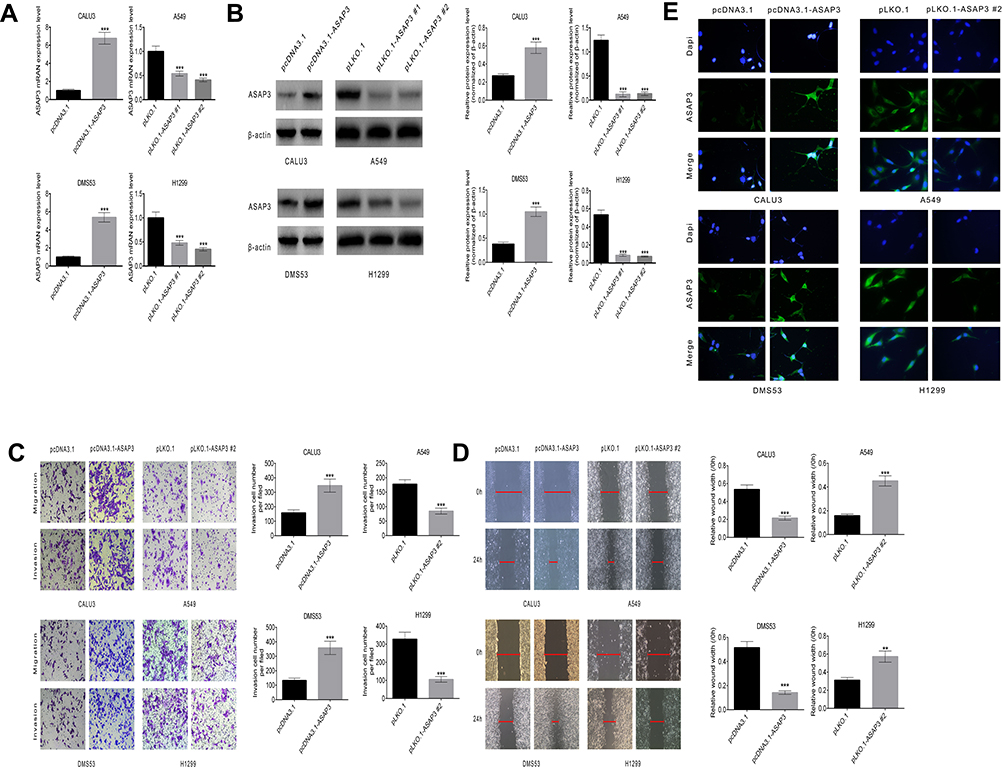 |
Figure 2 ASAP3 regulates migration and invasion of human lung adenocarcinoma cells. (A and B) Levels of ASAP3 mRNA and protein in CALU3, DMS53, A549, and H1299 cells transfected with pLKO.1, pLKO.1-ASAP3, or pcDNA3.1-ASAP3 vectors. (C) Number of cells that penetrated into the lower chamber within 24 hrs in the transwell assay. Top panels, migration assay; lower panels, invasion assay. (D) Representative images of wound-healing assays with CALU3, DMS53, A549, and H1299 cells after 24-hr treatment. Scale bar, 200 nm. (E) Immunofluorescence staining against ASAP3 in A549 and H1299 cells transfected with pLKO.1 or pLKO.1-ASAP3, and in CALU3 and DMS53 cells transfected with pcDNA3.1 or pcDNA3.1-ASAP3. Cells were immunostained against ASAP3 (green) and stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Magnification, 600×. **p<0.01, ***p<0.001. |
Up-regulation of ASAP3 increased migration and invasiveness in CALU3 and DMS53 cells, as demonstrated in both the transwell and invasion assay (Figure 2C, left) as well as the wound-healing assay (Figure 2D, left). In contrast, suppression of ASAP3 in A549 and H1299 cells significantly decreased the number of invasive cells in these assays (Figure 2C and D). These data indicate that ASAP3 expression is sufficient to promote the migration and invasion of lung adenocarcinoma cells.
HIF-1α regulates the expression of ASAP3 in lung adenocarcinoma cells
The microenvironment in solid tumors is typically hypoxic. To determine whether hypoxia promotes the expression of ASAP3 in lung adenocarcinoma cells, we examined whether HIF-1α regulates the expression of ASAP3 in lung adenocarcinoma in vitro.
We established a lentivirus construct expressing HIF-1α (pcDNA3.1-HIF-1α). CALU3 and DMS53 cells transfected with pcDNA3.1-HIF-1α demonstrated at least two-fold higher levels of ASAP3 mRNA and protein than cells transfected with control vector (p<0.001; Figure 3A and B). We also transfected A549 and H1299 cells with the pLKO.1-HIF-1α vector to knock down HIF-1α expression, and found that the corresponding levels of ASAP3 mRNA and protein were diminished by >40% compared with mock-transfected controls (p<0.001; Figure 3A and B).
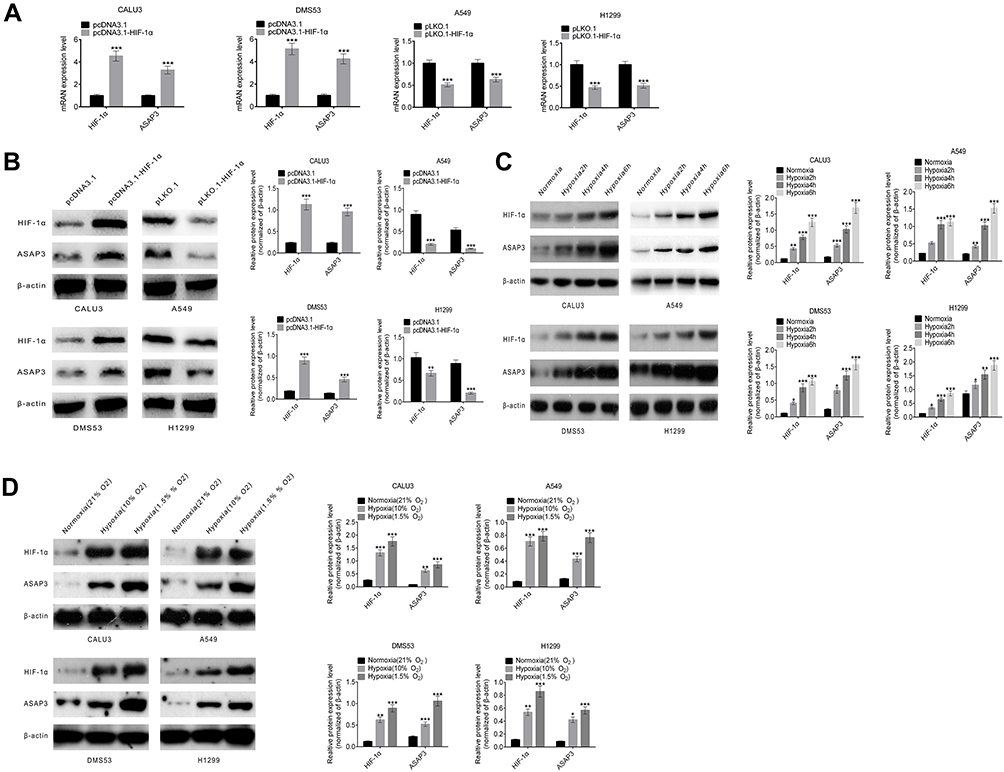 |
Figure 3 HIF-1α directly regulates the expression of ASAP3. (A) Quantitative RT-PCR and (B) Western blot analysis of HIF-1α and ASAP3 expression in CALU3 and DMS53 cells transfected with pcDNA3.1-HIF1α, and in A549 and H1299 cells transfected with pLKO.1-HIF-1α. (C) HIF-1α and ASAP3 expression in adenocarcinoma cells cultured under normoxia (21% O2) and hypoxia (1.5% O2) for different time (2, 4, or 6 hrs) as determined by Western blot. (D) HIF-1α and ASAP3 expression in adenocarcinoma cells cultured under normoxia (21% O2) and different hypoxia concentration (10% O2 and 1.5% O2) as determined by Western blot analysis. *p<0.05, **p<0.01, ***p<0.001. |
To determine whether ASAP3 expression changes over time with hypoxic exposure, we investigated HIF-1α and ASAP3 expression in CALU3, DMS53, A549, and H1299 cells cultured under normoxia (21% O2) and hypoxia (10% O2) for 2, 4, and 6 hrs. Expression of both proteins gradually increased with the duration of hypoxia, as assessed by Western blot (Figure 3C). In fact, expression of both proteins was higher under severe hypoxia (1.5% O2) than under mild hypoxia (10% O2) (p<0.001, Figure 3D). These results suggest that a hypoxic microenvironment may induce ASAP3 overexpression via HIF-1α in lung adenocarcinoma.
HIF-1α and ASAP3 expression are correlated in lung adenocarcinoma tissues
To investigate whether HIF-1α regulates the expression of ASAP3 in lung adenocarcinoma patients, we performed IHC to measure HIF-1α and ASAP3 levels in tumor samples from lung adenocarcinoma patients. ASAP3 was found to colocalize with HIF-1α in consecutive sections of lung adenocarcinoma tissues (Figure S1 and Table S3 and S4). HIF-1α expression levels positively and significantly correlated with ASAP3 levels (p<0.01), suggesting that tumors in a hypoxic microenvironment also overexpress ASAP3 (Table S1, p<0.01). These data indicate that HIF-1α may play a critical role in regulating ASAP3 overexpression in lung adenocarcinoma in human patients.
ASAP3 overexpression is able to restore lung adenocarcinoma progression following HIF-1α knockdown
To examine whether HIF-1α regulates ASAP3 expression in vivo, we created an orthotopic mouse model of lung adenocarcinoma by transfecting A549 adenocarcinoma cells with pLKO.1, pLKO.1-ASAP3, pLKO.1-HIF1α, or the combination of pLKO.1-HIF1α + pcDNA3.1-ASAP3. Transfected cells were injected into nude mice to create orthotopic tumors. We evaluated the association between HIF-1α and ASAP3 by Western blot (Figure 4A and B) and IHC (Figure 4C), and the results suggest that expression of ASAP3 was decreased as a result of the HIF-1α level reduction. However, tumor cells transfected pLKO.1-HIF1α showed reduced ASAP3 expression, which was rescued by co-transfection of pLKO.1-HIF1α + pcDNA3.1-ASAP3, as assessed by Western blot (Figure 4A and B). Mice injected with cells from the pLKO.1-ASAP3 and pLKO.1-HIF1α groups developed tumors with significantly smaller volume and weight than mice injected with cells transfected with the empty pLKO.1 vector (p<0.001; Figure 4C and E). Mice in the pLKO.1-HIF1α + pcDNA3.1-ASAP3 group developed significantly larger primary tumors than mice in the pLKO.1-HIF1α group (p<0.001; Figure 4D and F). These results suggest that ASAP3 overexpression is critical for lung adenocarcinoma progression induced by HIF-1α.
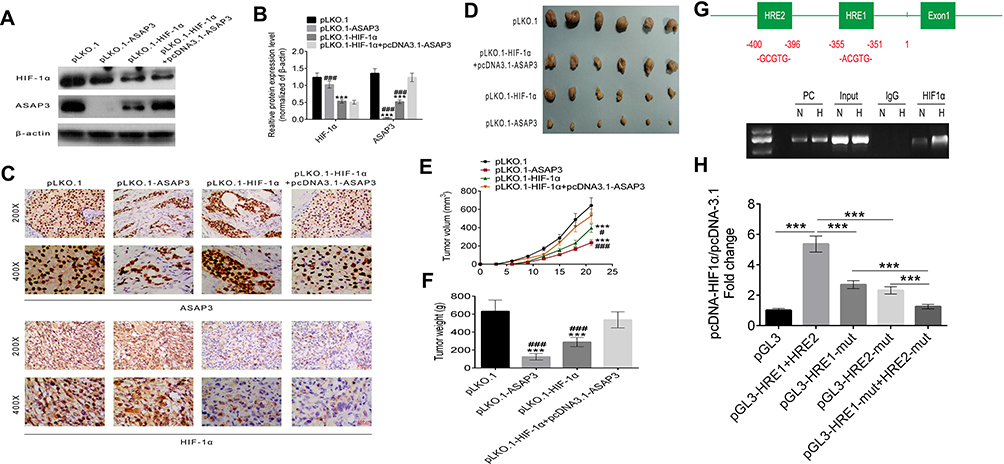 |
Figure 4 (A and B) Western blotting of ASAP3 and HIF-1α in A549 cells transfected with pLKO.1-ASAP3, pLKO.1-HIF1α or the combination of pLKO.1-HIF1α + pcDNA3.1-ASAP3. (C) Representative immunohistochemical staining against HIF-1α and ASAP3 in orthotopic tumors in nude mice. Magnification, 200 and 400×. (D) Representative images of tumors formed in nude mice. Volume (E) and weight (F) of primary tumors in nude mice expressed as mean ± SD. (G) Above, DNA sequence of the ASAP3 promoter. Two candidate HRE sites are indicated. Below, HIF1α chromatin immunoprecipitation analysis. The VEGF promoter was used as a positive control (PC). (F) Luciferase expression assay in A549 cells transfected with pcDNA-HIF1α or pcDNA3.1 (control vector) as well as pGL3-ASAP3-HRE1 + HRE2, pGL3-ASAP3-HRE1-mutated, pGL3-ASAP3-HRE2-mutated, pGL3-ASAP3-HRE1-mutated + HRE2-mutated, or empty pGL3. (H) Results are expressed as fold induction relative to cells transfected with empty pcDNA3.1, after normalization to Renilla activity. ***p<0.001; #p<0.05; ###p<0.001.Abbreviations: H, hypoxia; N, normoxia; IgG, negative control. |
HIF-1α directly regulates ASAP3 expression by binding the HRE in its promoter
To understand how HIF-1α regulates ASAP3 expression, we identified two potential HIF-1α–binding sites (HRE1-2) within the promoter and upstream of the transcription start site (Figure 4G). To determine whether HIF-1α binds these HREs, we performed a CHIP assay using anti-HIF-1α antibodies in A549 cells cultured under hypoxic (1.5% O2) or normoxic (21% O2) conditions. DNA fragments containing the HRE1 and/or HRE2 binding sites were significantly more abundant under hypoxic conditions (Figure 4G), suggesting that HIF-1α regulates ASAP3 expression by binding to these regions.
To determine whether binding of HIF-1α to the HREs activates the ASAP3 promoter, we constructed a full-length ASAP3 luciferase promoter vector containing the HREs (−400 to −351 bp) and cotransfected this reporter construct with or without HIF-1α cDNA into A549 cells. In addition, we generated a reporter construct in which HRE1 and/or HRE2 was mutated to abolish HIF-1α binding. Indeed, mutation in either HRE almost completely abolished activation of the ASAP3 promoter by HIF-1α (Figure 4H). These data suggest that HIF-1α activates the ASAP3 promoter by binding to the HRE1 and/or HRE2 regions.
Discussion
ASAP3 was first identified as a protein that contributes to cell proliferation in hepatocellular carcinoma and was later identified as an ArfGAP that may regulate cell migration associated with the invasion of normal tissue by cancer cells.5 ASAP3 promotes migration, invasion and indicates poor survival outcome in hepatocellular cancer tissues.10,12,13 Our data indicate that ASAP3 may play a causal role in lung adenocarcinoma progression and metastasis. ASAP3 expression levels were higher in lung adenocarcinoma than in adjacent non-neoplastic tissues, and they positively correlated with lymph node metastasis and poor clinical prognosis. Furthermore, we show that ASAP3 overexpression promotes migration and invasiveness of human lung adenocarcinoma cells and accelerates tumor progression in a xenograft mouse model of human lung adenocarcinoma. Taken together, our results from in vitro and in vivo experiments indicate that ASAP3 overexpression is a critical driver of lung adenocarcinoma cell progression and metastasis.
In the hypoxic microenvironment typical of solid tumors, HIF-1α expression provides a selective advantage by promoting angiogenesis and the epithelial-to-mesenchymal transition, which ultimately increases tumor aggressiveness.22,23 HIF-1α overexpression in lung tumor cells is associated with increased invasiveness and metastasis, and HIF-1α serves as a biomarker of poor prognosis in human lung cancer.24 In response to hypoxia, HIF-1α and a broad array of its downstream targets are synthesized de novo, helping promote cancer cell invasion and migration. We investigated HIF-1α and ASAP3 expression in four human lung adenocarcinoma cell lines and found that ASAP3 expression increased in a step-wise manner with increasing time of exposure to hypoxia and decreasing O2 concentration. Our mechanistic studies suggest that HIF-1α influences ASAP3 overexpression by binding to HER1 or/and HER2 in the ASAP3 promoter. ASAP3 overexpression restores lung adenocarcinoma progression caused by HIF-1 knockdown, both in vitro and in a xenograft mouse model. IHC staining indicated a strong correlation between HIF-1α and ASAP3 levels. These observations provide strong evidence that ASAP3 is a novel downstream target of HIF-1α in lung adenocarcinoma.
In summary, we demonstrate that ASAP3 is up-regulated in lung adenocarcinoma and correlates with poor clinical prognosis. ASAP3 overexpression promotes invasive phenotypes in vitro and in vivo. HIF-1α regulates ASAP3 expression by binding to HRE sites in the ASAP3 promoter. Targeted down-regulation of ASAP3 may be an effective strategy for the treatment of metastatic lung adenocarcinoma.
Acknowledgments
This work was supported by grants from Chen Xiao-ping Foundation for the Development of Science and Technology of Hubei Province (grant numbers: CXPJJH11800001-2018329).
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest related to this work.
References
1. Johnson DH, Schiller JH, Bunn PA. Recent clinical advances in lung cancer management. J Clin Oncol. 2014;32(10):973–982. doi:10.1200/JCO.2013.54.6911
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. Cancer J Clin. 2015;65(1):05–29. doi:10.3322/caac.21254
3. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Natural Rev Cancer. 2002;2(8):563–572. doi:10.1038/nrc865
4. Ren W, Mi D, Yang K, et al. The expression of hypoxia-inducible factor-1α and its clinical significance in lung cancer: a systematic review and meta-analysis. Swiss Med Wkly. 2013;143:w13855.
5. Vi LH, Sanita B, Hiroki I, i ASAP3 is a focal adhesion-associated Arf GAP that functions in cell migration and invasion. J Biol Chem. 2008;283(22):14915–14926. doi:10.1074/jbc.M709717200
6. Donaldson JG. Multiple roles for Arf6: sorting, structuring, and signaling at the plasma membrane. J Biol Chem. 2003;278(43):41573–41576. doi:10.1074/jbc.R300026200
7. Kahn RA, Cherfils J, Elias M, et al. Nomenclature for the human Arf family of GTP-binding proteins: ARF, ARL, and SAR proteins. J Cell Biol. 2006;172(5):645–650. doi:10.1083/jcb.200512057
8. Randazzo PA, Hirsch DS. Arf GAPs: multifunctional proteins that regulate membrane traffic and actin remodelling. Cell Signal. 2004;16(4):401–413. doi:10.1016/j.cellsig.2003.09.012
9. Sabe H, Onodera Y, Mazaki Y, Hashimoto S. ArfGAP family proteins in cell adhesion, migration and tumor invasion. Curr Opin Cell Biol. 2006;18(5):558–564. doi:10.1016/j.ceb.2006.08.002
10. Fan C, Tian Y, Miao Y, et al. ASAP3 expression in non-small cell lung cancer: association with cancer development and patients’ clinical outcome. Tumour Biol. 2014;35(2):1489–1494.
11. Luo Y, Kong F, Wang Z, et al. Loss of ASAP3 destabilizes cytoskeletal protein ACTG1 to suppress cancer cell migration. Mol Med Rep. 2014;9(2):387–394. doi:10.3892/mmr.2013.1812
12. Tian H, Qian J, Ai L, et al. Upregulation of ASAP3 contributes to colorectal carcinogenesis and indicates poorsurvival outcome. Cancer Sci. 2017;108(8):1544–1555. doi:10.1111/cas.13281
13. Okabe H, Furukawa Y, Kato T, et al. Isolation of development and differentiation enhancing factor-like 1 (DDEFL1) as a drug target for hepatocellular carcinomas. Int J Oncol. 2004;24(1):43–48.
14. Travis WD, Brambilla E, Nicholson AG, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thoracic Oncol. 2015;10(9):1243–1260. doi:10.1097/JTO.0000000000000630
15. Yang L, Wang S, Zhou Y, et al. Evaluation of the 7th and 8th editions of the AJCC/UICC TNM staging systems for lung cancer in a large North American cohort. Oncotarget. 2017;8(40):66784–66795. doi:10.18632/oncotarget.18158
16. Zhao X, Gao S, Ren H, et al. Hypoxia-inducible factor-1 promotes pancreatic ductal adenocarcinoma invasion and metastasis by activating transcription of the actin-bundling protein fascin. Cancer Res. 2014;74(9):2455–2464. doi:10.1158/0008-5472.CAN-13-3009
17. Zhao T, Gao S, Wang X, et al. Hypoxia-inducible factor-1alpha regulates chemotactic migration of pancreatic ductal adenocarcinoma cells through directly transactivating the CX3CR1 gene. PLoS One. 2012;7(8):e43399. doi:10.1371/journal.pone.0043399
18. Yang S, Zhang JJ, Huang XY. Mouse models for tumor metastasis. Methods Mol Biol. 2012;928:221–228.
19. N. Li, Li Y, Li Z, et al. Hypoxia inducible factor 1 (HIF-1) recruits macrophage to activate pancreatic stellate cells in pancreatic ductal adenocarcinoma. Int J Mol Sci. 2016;17(6):e799. doi:10.3390/ijms17010039
20. Zhang L, Wang Q, Wang F, et al. LncRNA LINC01446 promotes glioblastoma progression by modulating miR-489-3p/TPT1 axis. Biochem Biophys Res Commun. 2008;503(3):1484–1490. doi:10.1016/j.bbrc.2018.07.067
21. Chaturvedi P, Gilkes DM, Wong CC, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123(1):189–205. doi:10.1172/JCI69244
22. Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102(4):639–644. doi:10.1038/sj.bjc.6605530
23. Cheng CW, Chen PM, Hsieh YH, et al. Foxo3a-mediated overexpression of microRNA-622 suppresses tumor metastasis by repressing hypoxia-inducible factor-1α in ERK-responsive lung cancer. Oncotarget. 2015;6(42):44222–44238. doi:10.18632/oncotarget.5826
24. Lou Y, McDonald PC, Oloumi A, et al. Targeting tumor hypoxia: suppression of breast tumor growth and metastasis by novel carbonic anhydrase IX inhibitors. Cancer Res. 2011;71(9):3364–3376. doi:10.1158/0008-5472.CAN-10-4261
Supplementary materials
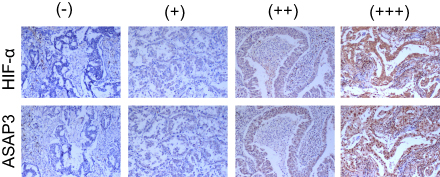 |
Figure S1 Representative immunohistochemical staining against HIF-1α and ASAP3 in human lung adenocarcinoma tissue samples. Upper, HIF-1α; lower, ASAP3. Magnification, 200×. |
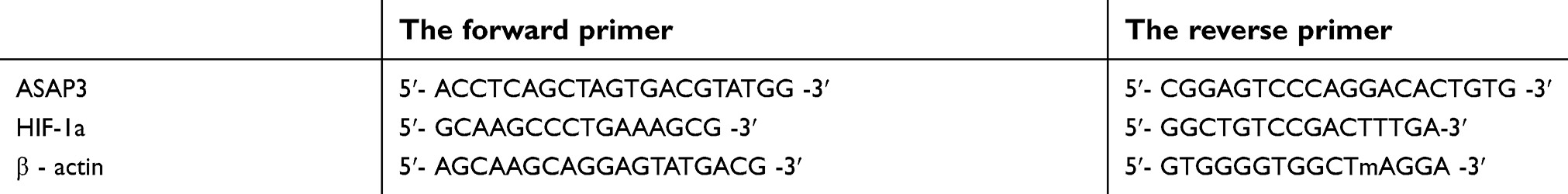 |
Table S1 The primer sequences for ASAP3, HIF-1a andβ- actin. |
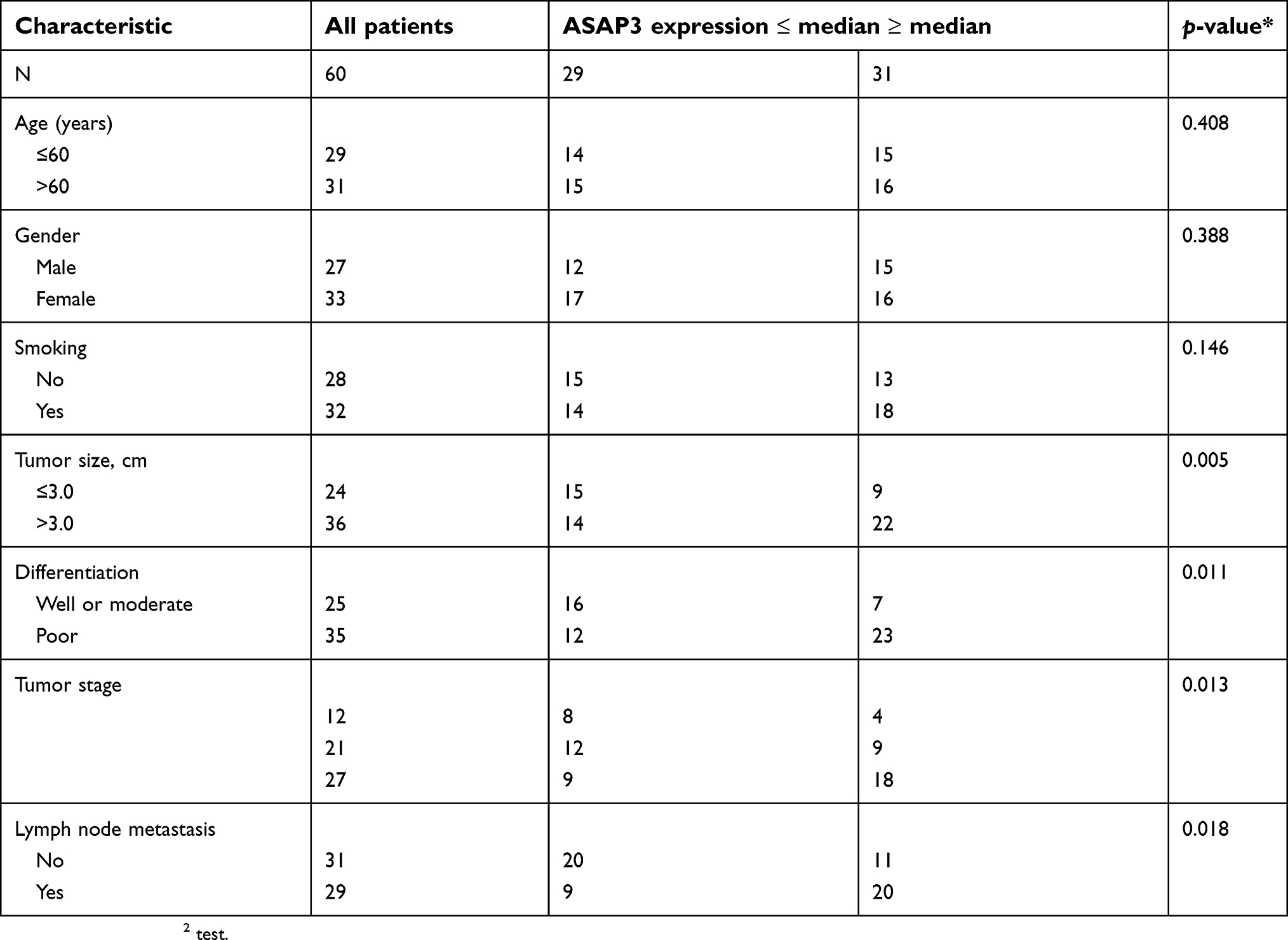 |
Table S2 Clinicopathological characteristics of patients with lung adenocarcinoma, stratified by level of ASAP3 mRNA |
 |
Table S3 Chi-square analysis of immunohistochemical analysis of HIF-1α and ASAP3 expression in human lung adenocarcinoma surgical samples |
 |
Table S4 Association between ASAP3 expression levels and regional lymph node (LN) metastasis in patients with lung adenocarcinoma. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.