")
Back to Journals » ClinicoEconomics and Outcomes Research » Volume 11
Untangling the clinical and economic burden of hospitalization for cardiac amyloidosis in the United States
Authors Quock TP , Yan T, Tieu R, D'Souza A, Broder MS
Received 28 February 2019
Accepted for publication 23 June 2019
Published 15 July 2019 Volume 2019:11 Pages 431—439
DOI https://doi.org/10.2147/CEOR.S207127
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Samer Hamidi
Tiffany P Quock,1 Tingjian Yan,2 Ryan Tieu,2 Anita D’Souza,3 Michael S Broder2
1Medical Affairs, Prothena Biosciences Inc, South San Francisco, CA, USA; 2Health Services Research, Partnership for Health Analytic Research, LLC, Beverly Hills, CA, USA; 3Division of Hematology and Oncology, Medical College of Wisconsin, Milwaukee, WI, USA
Purpose: Cardiac dysfunction is common in amyloid light-chain (AL) amyloidosis, a rare disease caused by extracellular deposition of misfolded immunoglobulin light chains. This study aimed to examine economic/clinical disease burden in hospitalized cardiac amyloidosis patients.
Patients and methods: Cardiac amyloidosis patients ≥18 years old hospitalized between 2014 and 2016 were identified in claims if they had ≥1 inpatient claim consistent with amyloidosis and evidence of cardiac dysfunction. Descriptive statistics were reported.
Results: 3239 cardiac amyloidosis patients [1795 (55.4%) with concurrent renal disease] were identified. Mean (SD) length of stay was 8.3 (11.1) days. 25.2% were admitted to the intensive care unit. Mean overall hospitalization costs were USD$20,584. In-hospital mortality was 9.0% overall. 16.8% were readmitted within 30 days, with 11.2% dying in-hospital and a mean readmission cost of USD$18,536.
Conclusion: Hospitalization for cardiac amyloidosis is costly, with high rates of readmission and mortality. Opportunities exist to improve care.
Keywords: cardiac amyloidosis, hospitalization, burden of illness, cost
Plain language summary
This is the first study of the real-world economic and clinical burden of disease in hospitalized patients with cardiac amyloidosis. Using data from a large electronic healthcare database, we identified hospitalized adult patients with cardiac amyloidosis in order to provide an estimate of the burden associated with this rare disease. Patient demographics and clinical characteristics, as well as admitting hospital characteristics, are also reported. We found that hospitalization for cardiac amyloidosis is costly, with high rates of readmission and mortality. These results indicate a potential area of cost savings through programs to increase awareness of readmission risks and the need for better outpatient disease management.
Introduction
Amyloidoses refer to a group of rare disorders of protein folding characterized by extracellular tissue deposition of misfolded and aggregated autologous proteins as β-pleated sheet fibrils.1 The incidence of amyloidosis has been estimated to be 8–14 cases per million persons per year.2–6 The heart is one of the most frequently affected organs, and amyloid deposits can be found in the atria, ventricles, perivascular space, valves, and conduction system.7,8 Cardiac amyloidosis usually manifests as right-sided heart failure, commonly associated with dyspnea, edema, hepatomegaly, and ascites.9 Pericardial disease from amyloid deposits and arrhythmias is less common but also observed. During the diagnostic workup for a patient with unexplained heart failure, increased wall thickness and non-dilated left ventricle suggest cardiac amyloidosis, as do hepatomegaly, and neuropathy. Concomitant renal disease is also common, as low cardiac output contributes to renal dysfunction resulting from amyloid deposition.
Cardiac dysfunction is most common in three types of amyloidosis: light chain (AL), hereditary transthyretin (TTR) (or ATTRv; v for variant10), and ATTR wild type (wt) amyloidosis.11 Advances in identification (introduction of serum-free light-chain assay) and treatment (addition of bortezomib to melphalan and dexamethasone for the treatment of transplant ineligible patients) of AL amyloidosis over the last decade have improved outcomes, but survival is still poor.9,12–14 When symptomatic cardiac dysfunction is present (as it is in 50–70% of AL amyloidosis15), the median overall survival without treatment is only 6 months.16 There are two therapies that are currently available, tafamidis (Vyndaqel®, Pfizer) and patisiran (Onpattro®, Alnylam), which have reported data on treating amyloid cardiomyopathy in ATTRv and ATTRwt amyloidosis patients.17–19 However, typical treatments for heart failure are of limited use in amyloidosis patients because of disease-related contraindications.20
A search of PubMed identified no articles reporting on the cost of cardiac amyloidosis in the real-world setting. No doubt because of its rarity, most of what is known about health care utilization and costs in amyloidosis, in general, come from data collected on patients with other diseases, such as multiple myeloma,21 and extrapolated to amyloidosis. In a recent study using commercial insurance claims, we found that inpatient hospitalization accounted for more than a third of costs for prevalent patients with AL amyloidosis and that nearly a third of patients had evidence of congestive heart failure. To examine the clinical and economic outcomes for patients with cardiac amyloidosis in more detail, we conducted the current retrospective cohort study using hospital discharge data.
Materials and methods
This was a retrospective, cohort study using 2014–2016 data from the Premier Healthcare Database (Premier, Inc., Charlotte, NC, USA). The Premier database covers over 45 million hospital discharges and includes data on complete clinical coding, hospital cost, and patient billing data from close to 600 hospitals throughout the US out of 5500 in total.22 Participating hospitals submit data on patient demographic and payer information, primarily derived from hospital billing records. Available data include medications; laboratory, diagnostic, and therapeutic services; and primary and secondary diagnoses. Hospital geographic location, bed size, and teaching status are also included. Before the information is added to the database, a series of quality assurance and validation checks are completed. The data are de-identified, contain no protected health information, and are compliant with the Health Insurance Portability and Accountability Act (HIPAA) of 1996. As such, institutional review board approval was not required for this study.
Hospitalized patients ≥18 years old with cardiac amyloidosis were identified for inclusion if, during calendar years 2014–2016, they had ≥1 inpatient claim consistent with amyloidosis in any diagnosis field and evidence of cardiac dysfunction. Amyloidosis claims were identified using International Classification of Diseases, 9th Revision, Clinical Modification (ICD-9-CM) codes 277.3 (amyloidosis); except 277.31, which codes for familial Mediterranean fever and the equivalent ICD-10-CM codes: E85.4 (other amyloidosis), E85.8 (organ-limited amyloidosis), and E85.9 (amyloidosis, unspecified). In patients with multiple qualifying hospitalizations, only the first was included, so no patients appear in the study twice. All measures were based on data from the index hospitalization only. Cardiac dysfunction was defined as having an ICD-9/10 code for at least one of: heart failure, syncope, postural/orthostatic hypotension, tricuspid/mitral regurgitation murmur, sudden cardiac death, dyspnea, edema, or ascites. We excluded patients with claims for types of amyloidoses not on the list above (ie, E85.0-E85.3) and those with evidence of conditions associated with inflammatory amyloidosis (ie, rheumatoid arthritis, inflammatory bowel diseases, bronchiectasis, chronic osteomyelitis).
We described patient demographics, payment source, concomitant conditions, hospital characteristics, cost, and utilization. Patient characteristics included age, gender, and race. Payment source included Medicare, Medicaid, commercial, or other. We reported Charlson Comorbidity Index (CCI) as an overall synthetic measure of comorbidity burden, as well as the proportion with ICD-9/10 codes for hypotension, multiple myeloma, and monoclonal gammopathy of undetermined significance (MGUS). Admitting hospital characteristics included admission type (urgent/emergent or elective), hospital geographic region, bed size (over 500 beds vs 500 beds or less), and whether the admission took place in an urban location (yes/no) or teaching hospital (yes/no). Costs (2016$), length of stay (LOS), intensive care unit (ICU) use, in-hospital mortality and readmissions were also reported. Because the presence of renal dysfunction is a potential indicator of more severe illness, all outcomes were stratified by whether the individual had evidence of renal disease (defined by an ICD-9/10 code for renal failure, nephropathy, nephrotic syndrome, dialysis, or renal transplantation during the same admission).
Descriptive statistics including mean, SD, and relative frequencies for continuous data, and percentages for categorical data were reported. As the intent of the study was to describe patients with cardiac amyloidosis, no statistical confidence testing was done to compare the groups studied. All data transformations and analyses were performed using SAS© version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
We identified 7727 unique patients admitted to a hospital in the database in calendar years 2014–2016 with a qualifying amyloidosis diagnosis. Of these, 174 were excluded because they were <18 years old on admission, had a code for hereditary types of amyloidosis, or had evidence of an inflammatory cause for their disease (eg, concomitant rheumatoid arthritis). Of the 7553 remaining, 3239 had evidence consistent with cardiac disease (eg, ICD-9/10 code for heart failure, syncope, dyspnea), and these patients comprised the final study cohort. This final cohort comprises 1795 (55.4%) patients with cardiac dysfunction plus evidence of renal disease and 1444 without such evidence (Figure 1).
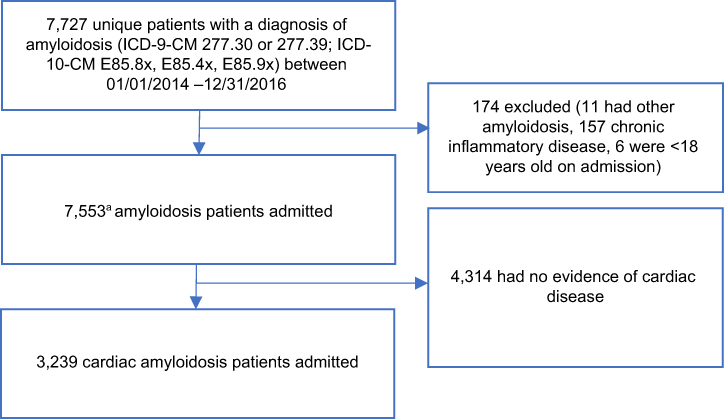 |
Figure 1 Attrition table for identifying hospitalized patients with cardiac amyloidosis. The final cohort comprised 3239 amyloidosis patients with cardiac disease. Note: aOnly the first qualified hospitalization for each patient was included in the study. |
The population mean (SD) age was 72.6 (11.8) years, 59.3% were male, and 63.6% were White. The cardiac without renal disease group was slightly older (74.3 vs 71.3 years), had a higher proportion of females (43.8% vs 38.3%), and a higher proportion of Whites (70.1% vs 58.4%) than the cardiac and renal disease group. Medicare was the primary payer for 77.5% of patients overall (Table 1). Patients in both groups were identified in equal proportions from the three study years. Patients without evidence of renal disease had fewer comorbid conditions, both as measured by CCI (3.0 vs 4.7) and by the proportion with a code for hypotension (18.8% vs 24.7%), multiple myeloma (11.7% vs 16.9%), and MGUS (2.4% vs 5.1%). During the admission, purpura (12.6%), stroke (11.4%), and claudication (5.9%) were frequently coded, while peripheral neuropathy (2.8%), hepatomegaly (0.8%) and carpal tunnel syndrome (0.3%) were coded less commonly. The distribution of conditions differed between the cardiac only and cardiac plus renal groups (Table 2).
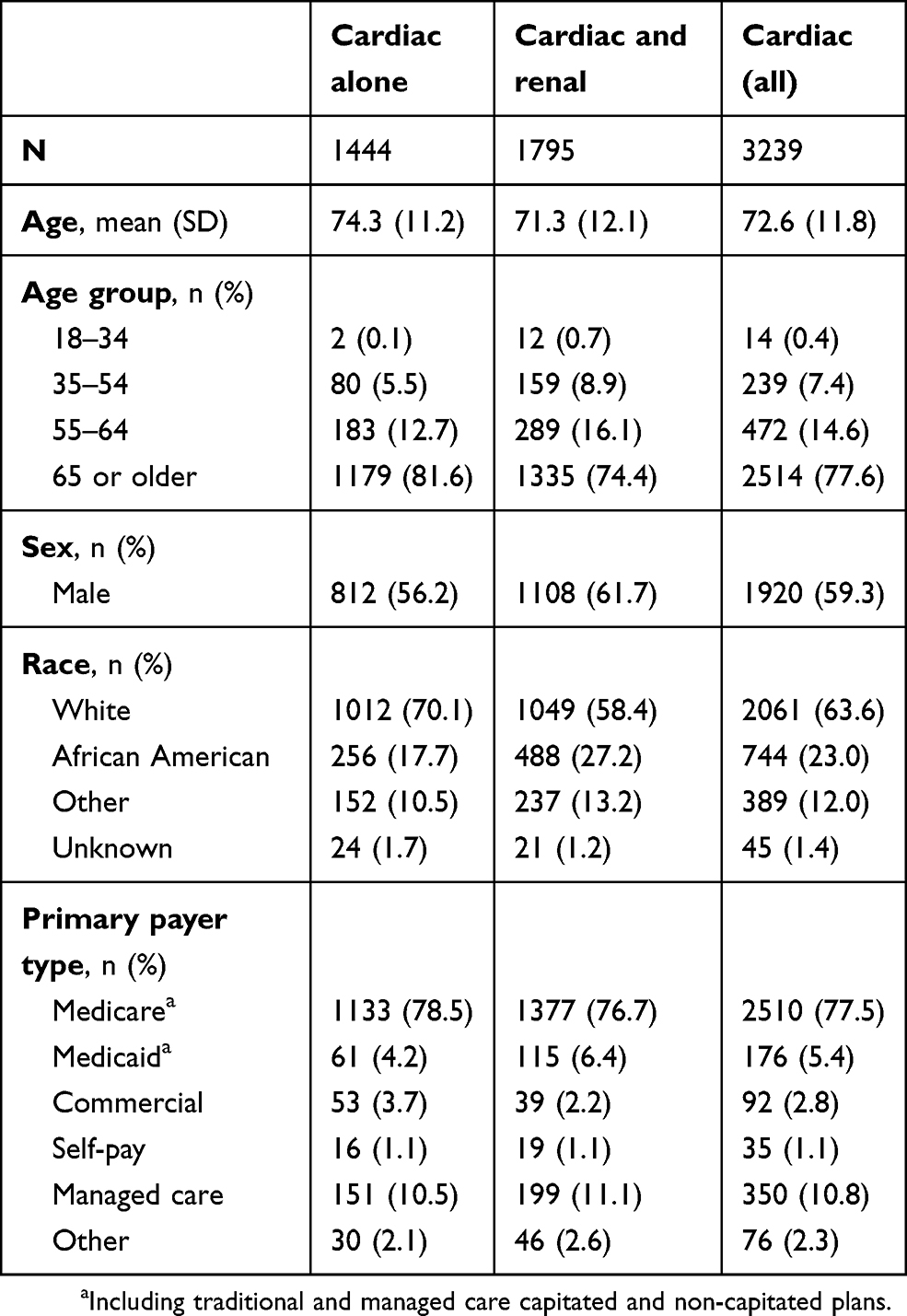 |
Table 1 Demographics of hospitalized cardiac amyloidosis patients |
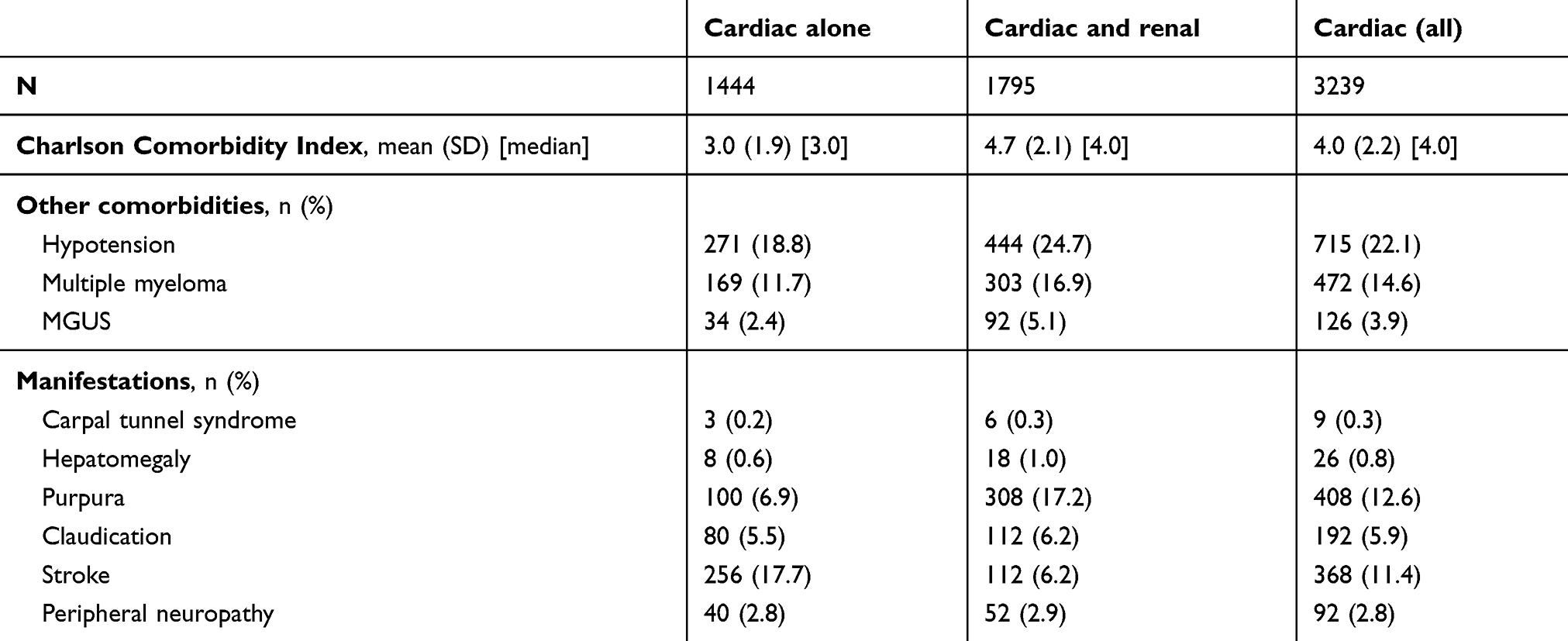 |
Table 2 Comorbidities and disease manifestations |
Most (90.9%) admissions were considered non-elective (eg, urgent or emergent), and admissions to hospitals from all regions of the US were represented in the database. Teaching hospitals were the site of the majority (51.3%) of admissions and 93.3% of admissions were to hospitals in urban settings (Table 3). The mean (SD) LOS was 8.3 (11.1) days overall: 6.7 (8.1) days among patients with cardiac dysfunction only and 9.7 (12.8) for the group with cardiac and renal disease. Overall, 25.2% of patients were admitted to the ICU. Mean (SD) hospitalization costs in USD were $20,584 ($30,030) overall, $16,041 ($19,746) for cardiac only and $24,238 ($35,834) for patients with cardiac dysfunction plus renal disease. Among selected hospital procedures, mean (SD) costs were highest for stent placement: $5370 ($6954) overall, $5929 ($9,149) for patients with cardiac dysfunction only, and $4903 ($4505) cardiac dysfunction plus renal disease. The in-hospital mortality rate was 9.0% overall: 4.5% for patients with cardiac dysfunction alone and 12.5% for patients with cardiac and renal disease. Just over half (56.2%) of patients were discharged home, and 30.5% were transferred to other facilities (including rehab and skilled nursing facility) (Table 4). Among 2,860 patients discharged alive and for whom 30-day follow up was available, 481 (16.8%) were readmitted to the hospital within 30 days. The mean (SD) readmission cost was $18,536 (39,415) and 11.2% died in the hospital (Table 5).
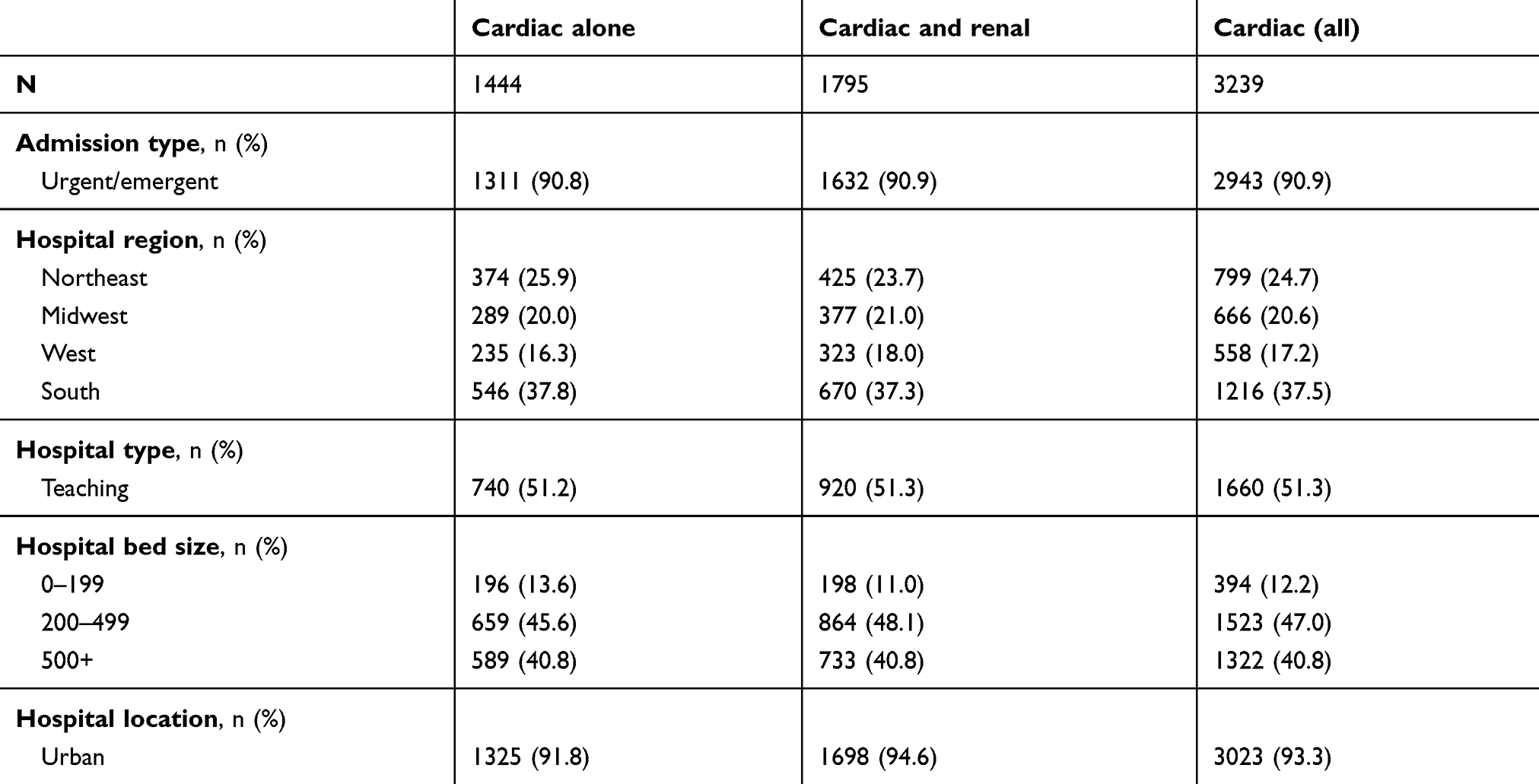 |
Table 3 Hospital and admission characteristics |
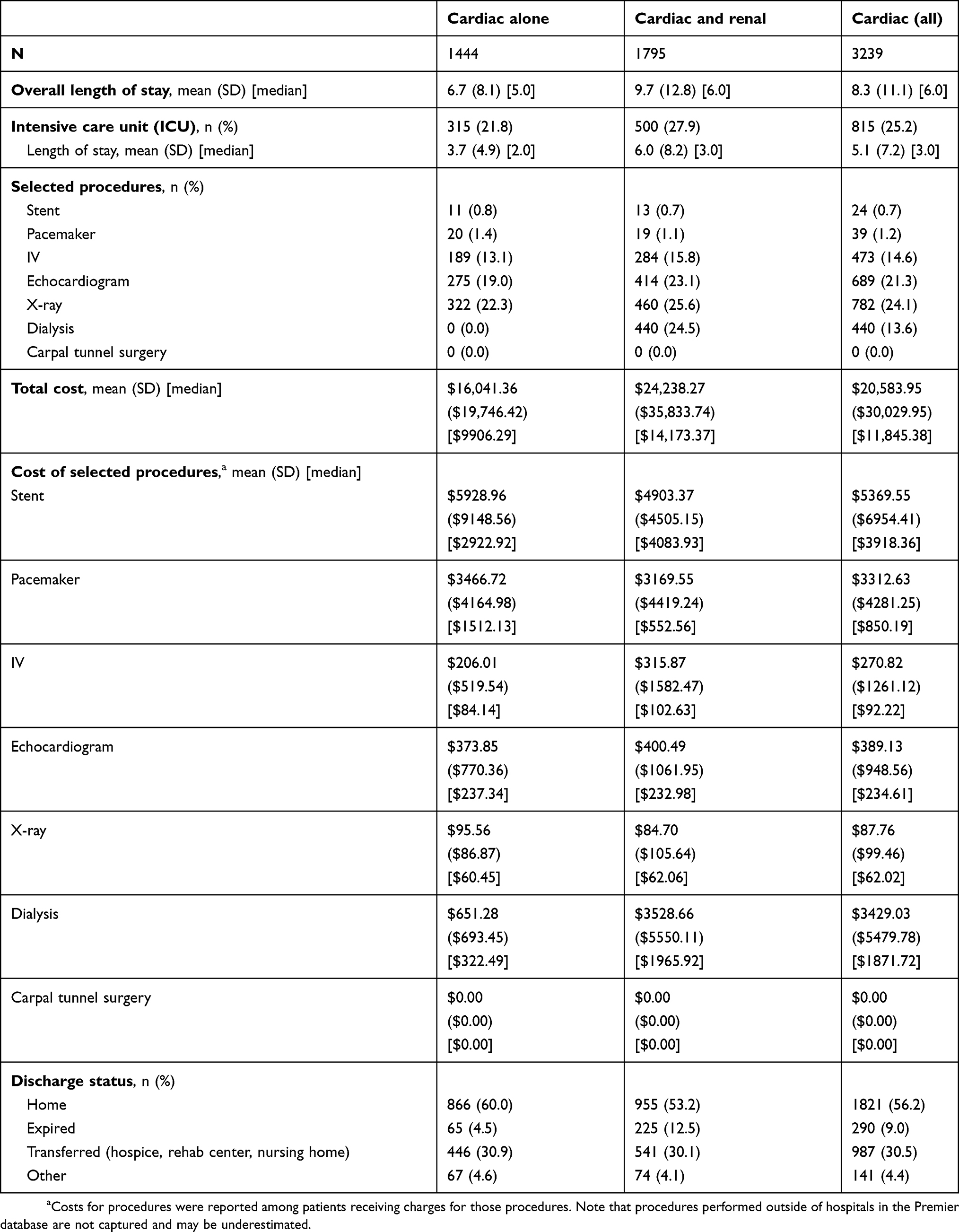 |
Table 4 Hospital utilization and cost |
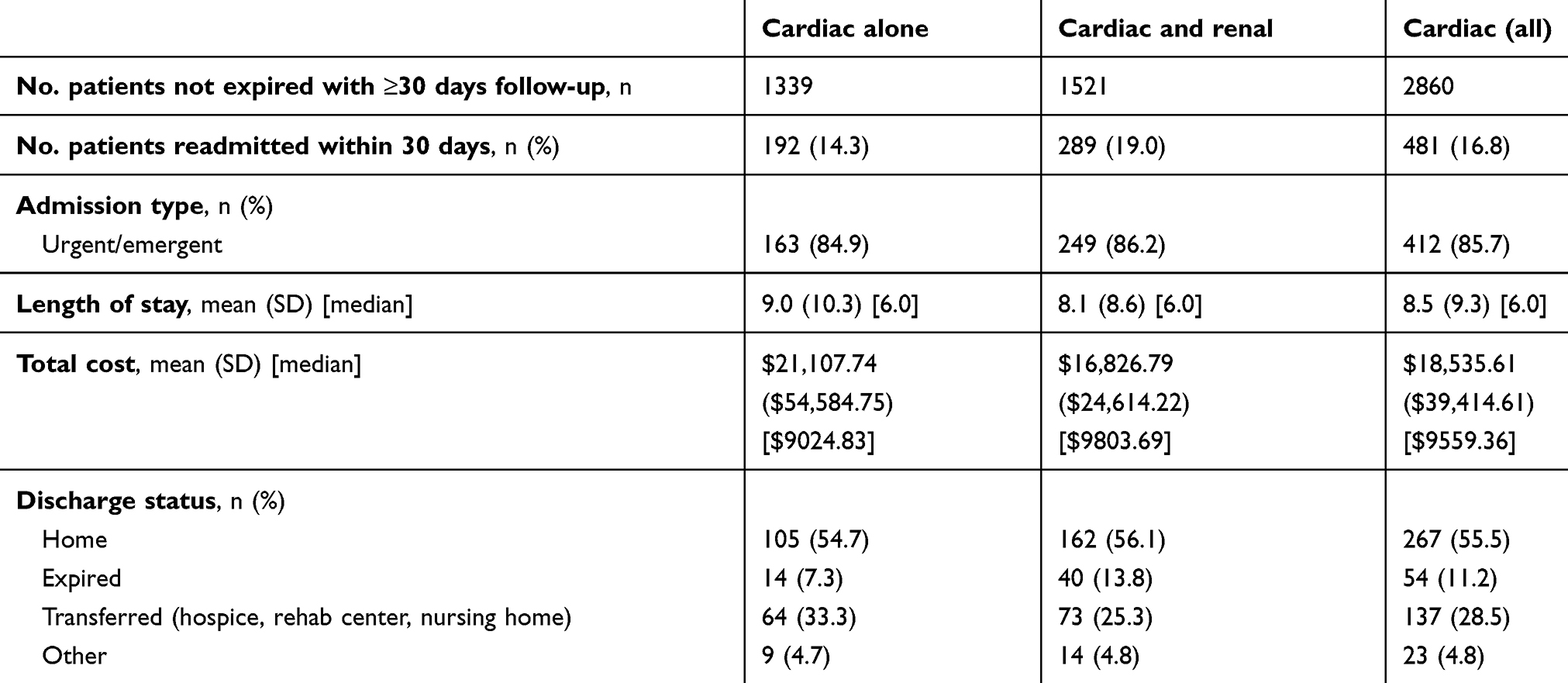 |
Table 5 Readmission characteristics, utilization, and cost |
Discussion
Cardiac amyloidosis, one of the most frequent types of solid organ disease in AL and ATTR amyloidosis, is known to be a severe, progressive, and often fatal disease manifestation, but in a search of Pubmed, we found no quantitative data on cost and very limited information on outcomes.11,22,23 Examining more than 3000 hospital admissions of patients with cardiac amyloidosis, we found an overall LOS of more than 8 days at an average cost of more than $20,000 per admission. During the admission, one-quarter of patients entered the ICU, spending an average of 5 days in intensive care. Nine percent of patients died in hospital, and just over half were discharged home, with the remainder transferred to other hospitals, skilled nursing facilities, or rehabilitation centers. Among patients who were discharged, 1 in 6 was readmitted within 30 days with an 11% mortality rate during that subsequent admission.
In the current study, more than half of cardiac amyloidosis patients had evidence of concomitant renal disease (specifically, a code for renal failure, nephropathy, nephrotic syndrome, dialysis, or kidney transplant) on admission, roughly consistent with smaller studies that used clinical data.24,25 Lacking clinical data, we could not determine the specific type of cardiac amyloidosis or which amyloidosis type produced the cardiac and renal disease in the patients we studied, although we hope to do so in future studies. A large case series suggests two-thirds of cardiac amyloidosis patients have AL, most of the remainder have ATTRv, and under 10% have ATTRwt,25 but these proportions could not be evaluated in the current study. Patients with both organ systems involved appear to be sicker than the group as a whole with an average LOS of nearly 10 days, compared to 7 days in patients without renal dysfunction, and costs were 50% higher than for those without renal disease. Nearly three times as many (12.5% vs 4.5%) of these patients died in hospital compared to those without renal disease.
Survival among patients with cardiac amyloidosis specifically has not been well studied, and what has been reported is not directly comparable to our study. In a single-center study of an AL amyloidosis cohort, Muchtar et al found 60% survived 2 years in the most recent period (2010–2014) compared with 42% ten years earlier.12 The authors reported 76% of patients had cardiac dysfunction, 61% renal, and 20% three or more organ systems (cardiac, renal, hepatic, or neurologic) involved. More recently, the same group reported 10-year survival rates in cardiac amyloidosis, varying from 68% in those with the deepest organ response (as measured by reduction in N-terminal pro b natriuretic peptide [NT-proBNP]) to 24% in those with the least response.23 An Italian series following 233 patients with cardiac amyloidosis found 2-year survival of 63% in AL, 98% in ATTRv, and 100% in ATTRwt. They reported 39% of cardiac amyloidosis patients had renal dysfunction, with nearly all patients with renal disease having AL amyloidosis.25
The 17% 30-day readmission rate in cardiac amyloidosis has not been previously reported. Hospital readmissions have been widely studied in recent years, and beginning in 2013, hospitals have been penalized by Medicare for exceeding national averages. Previous quality improvement programs have been credited for reducing readmission rates after a first hospitalization for heart failure from 25% in 2005–2008 to 22% in 2011–2014.26 While the current study included a mixture of patients admitted for the first time and those with one or more prior admissions, the relatively high readmission rate suggests that opportunities exist to optimize in-hospital cardiac amyloid care and that management of the disease is still poor. Even the early awareness of the likelihood of readmission and more straightforward methods to identify patients at high-risk for readmission may offer opportunities to improve care.
Although the hospitals in the Premier database are not a random sample of all US hospitals, the data represent close to 600 out of 5500 total US hospitals and have a similar geographic distribution.22 Teaching hospitals are slightly underrepresented in the database, making up about a quarter of the sample compared to more than a third of all hospitals. The finding that more than half of admissions in the current study were to teaching hospitals is consistent with the higher degree of specialty care needed to treat this rare disease.27
This study had limitations. Perhaps most importantly, cardiac disease, and amyloidosis, in general, were identified using coded data, not clinical records. Codes are primarily applied to support billing, not research. Miscoding does occur, and the extent of coding errors cannot be directly ascertained. We are currently planning a study to validate the codes using medical records. Clinical information, such as results of echocardiograms, cardiac troponin, or other biomarkers, were not available, so patients could not be staged by severity. Privacy restrictions that permit the use of coded data explicitly prevented us from seeking additional data on patients identified in the database, so pathology, laboratory, or other clinical notes could not be used. Our data set did not contain longitudinal information on the patients we identified; all data pertained only to the hospital admission of interest. As a result, we could not investigate how long prior to admission the diagnosis of amyloidosis was made, nor could we estimate the impact of early or late diagnosis on outcomes. The data source represents more than 10% of US acute-care hospitals, geographically dispersed across the country, but does not include federally funded (eg, U.S. Department of Veterans Affairs) or closed panel health maintenance orgranization facilities. Finally, the study was descriptive in nature; we did not attempt to control for differences between groups, nor to make statistical comparisons between them. Despite these limitations, information on a large, US population derived from across the country is not readily available without the use of databases like the one used here, making this as useful starting point from which to monitor trends and outcomes.
Conclusions
To our knowledge, this is the first study of the real-world economic and clinical burden of disease in hospitalized patients with cardiac amyloidosis. Our findings show that hospitalizations for cardiac amyloidosis are costly, with high rates of readmission and mortality. Quality improvement programs, along with increased awareness of readmission risks and better outpatient disease management, may improve care while reducing costs.
Abbreviations list
AL, light chain; ATTR, amyloid transthyretin; CCI, Charlson Comorbidity Index; HIPAA, Health Insurance Portability and Accountability Act; ICD-9-CM, International Classification of Diseases, 9th Revision, Clinical Modification; ICD-10-CM, International Classification of Diseases, 10th Revision, Clinical Modification; ICU, intensive care unit; LOS, length of stay; MGUS, monoclonal gammopathy of undetermined significance; NT-proBNP, N-terminal pro b natriuretic peptide; TTR or ATTRv, hereditary transthyretin; wt, wild type.
Acknowledgments
Sohum Gokhale, MPH, an employee of PHAR, LLC, provided technical assistance and proofread the manuscript.
Author contributions
All authors contributed to study design, interpretation of the data, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Quock is an employee of Prothena Biosciences Inc, which funded the research described in this manuscript. D’Souza is an employee of the Medical College of Wisconsin and was paid by Prothena Biosciences Inc to consult as a subject matter expert. Yan, Tieu, and Broder are employees of Partnership for Health Analytic Research, LLC, which received funding from Prothena Biosciences Inc to conduct the research described in this manuscript. The authors report no other conflicts of interest in this work.
References
1. Rajkumar SV. Pathogenesis of immunoglobulin light chain (AL) amyloidosis and light and heavy chain deposition diseases [Internet]. UpToDate; 2018 [
2. Kyle RA, Linos A, Beard CM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817–1822.
3. Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012;26(11):2317–2325. doi:10.1038/leu.2012.100
4. Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood. 2013;121(26):5124–5130. doi:10.1182/blood-2013-01-453001
5. Lousada I, Comenzo RL, Landau H, Guthrie S, Merlini G. Light chain amyloidosis: patient experience survey from the amyloidosis research consortium. Adv Ther. 2015;32(10):920–928. doi:10.1007/s12325-015-0250-0
6. Quock TP, Yan T, Chang E, Guthrie S, Broder MS. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018;2(10):1046–1053. doi:10.1182/bloodadvances.2017010645
7. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047–2060. doi:10.1161/CIRCULATIONAHA.104.489187
8. Koike H, Misu K, Sugiura M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63(1):129–138. doi:10.1212/01.WNL.0000132966.36437.12
9. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32(1):45–59.
10. Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–219. doi:10.1080/13506129.2018.1549825
11. Banypersad SM, Moon JC, Whelan C, Hawkins PN, Wechalekar AD. Updates in cardiac amyloidosis: a review. J Am Heart Assoc. 2012;1(2):e000364–e000364. doi:10.1161/JAHA.111.000364
12. Muchtar E, Gertz MA, Kumar SK, et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood. 2017;129(15):2111–2119. doi:10.1182/blood-2016-11-751628
13. Reich G, Held T, Siegert W, Kampf D, Dörken B, Maschmeyer G. Four patients with AL amyloidosis treated with high-dose chemotherapy and autologous stem cell transplantation. Bone Marrow Transplant. 2001;27(3):341–343. doi:10.1038/sj.bmt.1702766
14. Weiss BM, Lund SH, Bjorkholm M, et al. Improved survival in AL amyloidosis: a population-based study on 1,430 patients diagnosed in Sweden 1995–2013. Blood. 2016;128(22):4448. doi:10.1182/blood-2016-06-724161
15. Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med. 2015;278(2):126–144. doi:10.1111/joim.2015.278.issue-2
16. Rosenzweig M, Giralt S, Landau H. Light-chain amyloidosis: SCT, novel agents and beyond. Bone Marrow Transplant. 2013;48(8):1022–1027. doi:10.1038/bmt.2012.199
17. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
18. Quarta CC, Solomon SD. Stabilizing transthyretin to treat ATTR cardiomyopathy. N Engl J Med. 2018;379(11):1083–1084. doi:10.1056/NEJMe1810074
19. European Medicines Agency. Onpattro Summary of Product Characteristics.Amsterdam: European Medicines Agency; 2019.
20. Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–540. doi:10.1016/j.acvd.2013.06.051
21. Lin HM, Gao X, Cooke CE, et al. Disease burden of systemic light-chain amyloidosis: a systematic literature review. Curr Med Res Opin. 2017;33(6):1017–1031. doi:10.1080/03007995.2017.1301903
22. Health Forum, LLC. Fast facts on U.S. hospitals, 2018 [Internet]. American Hospital Association – Data Insights – Statistics; 2018 [
23. Muchtar E, Dispenzieri A, Leung N, et al. Depth of organ response in AL amyloidosis is associated with improved survival: grading the organ response criteria. Leukemia [Internet]; 2018. Available from http://www.nature.com/articles/s41375-018-0060-x. Accessed April 12, 2018. doi:10.1038/s41375-018-0060-x
24. Lee M-H, Lee S-P, Kim Y-J, Sohn D-W. Incidence, diagnosis and prognosis of cardiac amyloidosis. Korean Circ J. 2013;43(11):752. doi:10.4070/kcj.2013.43.11.752
25. Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212. doi:10.1161/CIRCULATIONAHA.108.843334
26. Boccuti C, Casillas G. Aiming for Fewer Hospital U-turns: The Medicare Hospital Readmission Reduction Program [internet]. Washington, DC: The Henry J. Kaiser Family Foundation; 2017. March. Available from: https://www.kff.org/report-section/aiming-for-fewer-hospital-u-turns-the-medicare-hospital-readmission-reduction-program-issue-brief/#endnote_link_211172-5.
27. Premier Applied Sciences. Premier Healthcare Database: Data that Informs and Performs. Charlotte, NC: Premier Inc.; 2018. February.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.