")
Back to Journals » Patient Related Outcome Measures » Volume 8
The health and life path of rare disease patients: results of the 2015 French barometer
Authors Heuyer T, Pavan S, Vicard C
Received 24 December 2016
Accepted for publication 22 May 2017
Published 13 September 2017 Volume 2017:8 Pages 97—110
DOI https://doi.org/10.2147/PROM.S131033
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Liana Bruce
Thomas Heuyer,1 Sonia Pavan,2 Christine Vicard1
1Maladies Rares Info Services, 2SPScienceCom, Paris, France
Purpose: A barometer has been set up to provide better knowledge about the daily situation of French rare disease (RD) patients, their families and relatives, in order to contribute to the elaboration of improvement measures. This report focuses on the care and life path of RD patients.
Patients and methods: A preliminary survey was carried out with three patients, five parents and three RD experts to identify the main hurdles and disruptions in the life path of RD patients. It was used to design a larger survey comprising 60 questions as well as open fields allowing free expression. Respondents (448) comprised patients, parents of RD children and close relatives of patients. The Percentage of Maximum Deviation, Yates’ correction for continuity and Fisher’s test were employed to compare the responses between groups.
Results: Large disparities in the delays to obtain a diagnosis were identified (<1 year to >20 years), and longer delays were associated with negative perception of care conditions. While good interactions with education teams were reported (59% of respondents), the professional situation of both patients and parents was strongly and negatively impacted by the disease (51% did not work or stopped working). Three hundred respondents expressed various needs and psychological and personal issues were reported by 62% and 75% of respondents, respectively. Interestingly, the medical care path and daily life of RD patients were positively impacted by the follow-up in a specialized consultation, as reflected by changes in scores measured by our barometer (Fisher’s test, p<0.05).
Conclusion: Some of the main hurdles and sources of disruption in the life path of RD patients were identified, as well as some positive outcomes. These data could serve not only as a background for further studies, but also to better adapt the support to real needs and to improve the synergies between the many people involved in the life path of RD patients.
Keywords: rare disease, survey, patients, life path, diagnosis, care
Introduction
Rare diseases (RDs) are estimated to affect around 30 million people in Europe1 and 3 million in France. Yet, there are about 7,000 RDs, some of them affecting no more than a few individuals in the world. In Europe, a disease is considered as rare when it affects <1 in 2,000 people. RDs are often chronic and associated with very diverse clinical and mental manifestations. The rarity of each disease leads to a number of specific issues compared to more common diseases, such as the scarcity of information, unavailability of treatments, delays in diagnosis, difficulty to find a specialist, disability issues, social care access and isolation.2
Maladies Rares Info Services is the French RD information and support service.3 It comprises telephone, email, chat and forum services managed by a professional team (scientists and medical doctors [MDs]). About 5,500 requests were addressed to the service in 2015: 50% from people directly affected by an RD and 40% from family members, relatives or people close to an RD patient. The service has not only provided information about diseases (38% of the requests), but has also helped to find specialists, patient associations, global and social support.4 Although the issues specific to RDs are known, the impact on patients and their caregivers is not well-documented and quantitative data are scarce or incomplete. Cost of illness is sometimes documented, especially for diseases with available specific therapies. Such studies aim at estimating the economic burden on society, but the patient’s point of view is not addressed.5 Unmet needs for health care or social support and life situations have been investigated to various extents for some specific diseases.6–9 While they provide very valuable information to improve the care and support of given subpopulations of patients, global approaches to quantitatively estimate the specific issues faced by a majority of RD patients are limited. Yet, these data would be useful to more efficiently implement policy recommendations for RD quality of care management. In 2009, the European Organization for Rare Diseases carried out two surveys to describe and compare patients’ experiences and expectations about diagnosis and access to health services. These surveys provide useful information about 16 diseases from 12,000 patients over 17 countries, and aim to contribute to shaping patient-centered public health policies.2 In 2013, Shire published an impact report to help quantify the health, psychosocial and economic impact of RDs based on online surveys conducted on US and UK patients and caregivers.10 In 2011, an RD barometer was set up by Maladies Rares Info Services, in order to increase knowledge about the situation of French patients and close people. The barometer collects quantitative data about the patient’s daily life with the aim to take an inventory and to evaluate the extent of the hurdles faced. The participants are patients, parents, relatives and, occasionally, health professionals using either the telephone helpline or online services of Maladies Rares Info Services. The first survey aimed to better characterize the delay in diagnosis and its consequences for RD patients, the access to information and the financial support for care, products and services.11 In 2012, a second survey was carried out to clarify the conditions and consequences of diagnosis announcement, the practical difficulties related to drugs and other health products and the coordination between health and care professionals.12
Based on the experience from the 2011 and 2012 studies, a third survey was performed in 2015 to further analyze the health and care path of RD patients. A preliminary study was carried out with a small number of participants to identify the disruptions in this path. It was used to specify the details of a larger quantitative survey. The covered topics were related to the provided medical and social care and the consequences of the disease on daily life, school, work and private life. This article presents the results of the 2015 survey.
Methods
Preliminary survey
The goals of this survey were to outline the main steps of the health and life path of RD patients, to identify potential disruptions and difficulties in daily life and to better prepare statistical analysis of the larger quantitative survey: choice of the topics to be developed, validation of preliminary observations, collection and treatment of data about situations not identified prior to the setup of the survey. Eleven participants were included in the preliminary survey. They were all volunteers and have given their informed consent. Criteria for their selection were chosen so as to include people with diverse profiles. Three RD patients from 20 to 55 years of age were included. They lived in rural areas or in cities, were diagnosed with Stiff man syndrome, neurofibromatosis and pulmonary arterial hypertension, and had either stopped working due to their disease or were looking for a job. Five parents of RD patients from 21 months of age to 26 years of age (four mothers and one father) were included. They lived in rural areas or in cities, had a professional activity or stopped working to take care of their child. The RD children were diagnosed with Smith–Magenis syndrome, Cornelia de Lange syndrome, 17p11p12 chromosomal deletion, or were incompletely diagnosed (ataxia). Three health professionals were included: an MD from a reference center (rare anorectal and pelvic malformations), a pediatric coordinator (MD) from a regional center for disabled people and a social worker from a patient association on neurodegenerative disorders. These health professionals were involved in daily care and/or social support of RD and disabled patients. Interviews were conducted by telephone by a professional used to carrying out interviews with patients. They lasted for 2 hours on average, and participants were all volunteers. They were asked to talk about the key steps, including obstacles and disruptions, during their life (or their patient’s life) with the disease, from the first signs up to their current situation. They could spontaneously express themselves or be guided with more specific and adapted questions if needed. All the participants were contacted again on the next day to ensure they were not unsettled following the evocation of painful situations. The interviews were recorded and subsequently transcribed. Then, manual, textual analysis was carried out. The steps were identified based on the recurrence of testimonies among all participants.
Quantitative survey
A questionnaire was constructed based on the outcome of the preliminary study and on the experience acquired from the previous barometers. The questionnaire comprised 60 questions about the respondents’ demographic and general characteristics, the medical care situation, daily life, the use of an adapted medical or social infrastructure, school or work and private life. Open comments and suggestion fields were also included. Three different questionnaires were developed with slight variations in the wording, depending on the type of participant: patient, parent or relative. The quantitative survey took place from 17 November 2014 to 9 January 2015, with a 2-day interruption during the French Telethon 2014. During this period, each person contacting Maladies Rares Info Services (by telephone or email) was asked whether he/she would like to participate in the survey, provided the following inclusion criteria were met: 1) to be a patient, a parent of a child patient between 0 and 18 years old, or a close relative of a patient (partner, sibling, child); 2) to have an RD according to the French definition based on prevalence, or to have an undiagnosed pathology likely to be an RD, and 3) to be emotionally able to answer the survey (not crying, not excessively stressed by the evocation of diagnosis and so on). In addition, the questionnaire was distributed through social networks (the discussion forum of Maladies Rares Info Services, Facebook and Twitter). The questionnaire could be filled either online or on paper sent to the participants by Maladies Rares Info Services. All participants were volunteers. They were informed about the aim of the barometer, that is, to improve knowledge about the daily life of RD patients, and that the collected data could be used to contribute to the elaboration of improvement measures. Moreover, the questionnaires were anonymous and in conformity with the requirements from the Commission Nationale de l’Informatique et des Libertés, the French Authority for the control of protection of personal data. No individual data were presented in this study and the commission did not require written informed consent. Both the steering committee (composed of patient association representatives) and the board of directors (patient association representatives and qualified health professionals) of Maladies Rares Info Services have approved the barometer and this study.
Five hundred and forty questionnaires were returned to Maladies Rares Info Services. After quality control, 448 questionnaires were retained for the analyses and 92 were excluded due to inconsistency of the answers, a majority of lacking answers and diagnoses of non-RDs. Analyses including answers from all three types of respondents (patient, parent, relative) were performed to characterize the respondents, the provided medical and social care, daily life, private life, school and work. Then, specific analyses were performed to identify potential differences between subgroups (by type of respondent, date of diagnosis, follow-up in a specialized consultation [SC], and so on). The qualitative and ordinal variables were described by the number of answers (n) and by the frequency (%) of each modality. The quantitative variables were described by the number of answers, mean, standard deviation, minimum, maximum and median. Statistical analyses were performed using Modalisa v7 or v8 (Kynos Sarl, Paris, France). The Percentage of Maximum Deviation and Pearson’s chi-squared test were used to compare qualitative variables, and when the calculated theoretical populations were <5, a Yates’ correction for continuity was applied. Fisher’s test was employed to compare quantitative variables. Differences were considered statistically significant for p<0.05. The terms “respondent” and “participant” were employed to encompass responses from all types of respondents. Keeping in mind that their responses were not related to their own situation but to the patient’s situation, the term “patient” was used to describe patient-related results (whether they responded for themselves or not). When differences were observed depending on the type of respondent, this was specified in the text, as well as when non-patient participants responded for themselves (e.g., for parents of underage patients responding for themselves for work-related questions).
The surveys were in French. The questions have been translated into English for inclusion in this paper.
Results
Identification of the main steps of the health and life path of RD patients
From the preliminary study, the testimony of 11 participants including RD patients, parents of RD patients and RD health professionals allowed identifying the main steps of the health and life path of RD patients, as well as some of the difficulties/hurdles associated with each step (Figure 1). In spite of the heterogeneity of conditions involved, some common hurdles were reported. The first steps before a diagnosis is made, especially the first signs of the disease and the feeling of being poorly understood, may last for years. Depending on the symptoms, the initial recognition that “something is wrong” is sometimes attributed to the family context or the psychological condition of the person, and direct orientation toward a specialist is rare. Diagnosis announcement plays an important role in the way people react. While some patients are relieved and start fighting, others totally collapse or even conceal the issue. Various consequences may occur during this phase: depression, divorce, distance kept by the relatives, uneasiness of RD children. Acceptance of the disease also appears to be a key step that determines the future plans and life of patients and their ability to adapt to the disease progression. Medical and psychological care is often perceived as unsuitable to RDs, and sometimes useless. Living daily with an RD is an important source of disruptions: professional, social, emotional, familial and medical.
 | Figure 1 Summary of the main steps of the health and life path of rare disease patients and examples of associated characteristics or difficulties. |
All these answers were taken into account to build the quantitative survey. An additional input of this preliminary survey was the possibility for the participants to freely express themselves outside the frame of medical and social care, which is an uncommon feature in this type of study.
Analysis of the situation of RD patients at different steps of their life
Fifty-five percent of the respondents were RD patients, 28% were parents of underage children and 17% were close parents or relatives (parents of adult patients, partners, siblings, patients’ children) (Table 1). A majority of women responded to the survey (87%), which reflects the higher proportion of women using Maladies Rares Info Services and, more generally, using social and medical helplines. The questionnaires contained 180 free comments.
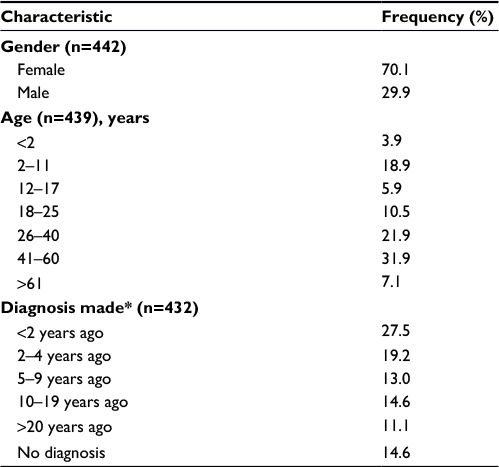 | Table 1 Characteristics of the patients Note: *At the time of the survey. Abbreviation: n, number of respondents. |
Characteristics of the RD patients
Characteristics of the patients are summarized in Table 1. Seventy percent of the patients were women, with variable female gender proportion depending on the type of respondent. Among the patients responding for themselves, 80% were women. However, when the respondents were relatives, the proportion of male and female patients for whom they filled in the questionnaire was more equally distributed (47% and 53%, respectively). The geographic origin followed the global population density of France (data not shown). A total of 195 RDs were represented. The groups of pathologies were diverse and representative of the global frequencies (data not shown). At the time of the survey, the patients had a diagnosis made 8 years ago on average and <5 years ago for 47% of them (Table 1).
First signs of the disease and delay in diagnosis
Two peaks were observed for the first signs of the disease: for one-third of the patients, they appeared before the age of 2 years (47% before 12 years old) and for about 40%, they appeared during adulthood (Figure 2A). A slightly lower response rate from the participants (80%) was obtained for the delay between the first signs and the diagnosis, because the respondents were not always able to accurately remember when the first signs appeared or when the diagnosis was made. In addition, 14.6% of the patients did not have a diagnosis at the time of the survey and, thus, could not answer this question. Nevertheless, a clear picture about the delay in diagnosis could be obtained from 359 participants (Figure 2B). Almost 45% of the patients with a diagnosis had obtained it within 1 year after appearance of the first signs (median =1 year). Very long delays in diagnosis were also revealed. For 22 patients, the delay was of 20 years or more. Among the very long delays in diagnosis, several participants mentioned a diagnosis made fortuitously, following a medical appointment for another reason or for another member of the family. Some patients were given psychological or psychiatric care in justification for their unexplained symptoms. The time between the appearance of the first signs and the diagnosis was sometimes perceived as a difficult period during which they were not heard and believed (observation from the free comments). Among the patients without a diagnosis, an average of 10 years was reported from appearance of the first symptoms. Twelve percent of them had been waiting >20 years. Seventy-six percent of them presented heavy symptoms, and 55% expressed a feeling of isolation (data not shown).
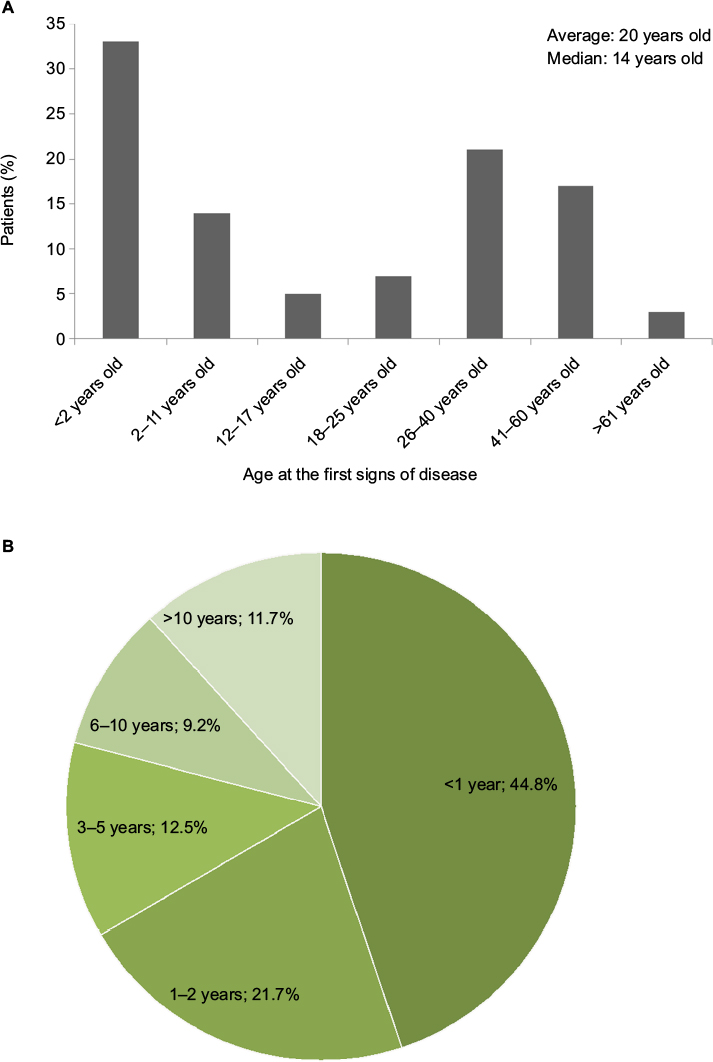 | Figure 2 Age at the first signs of the disease (A) and duration of delays in diagnosis (percentage of patients) (B). |
Medical care
Several questions were dedicated to a better characterization of the patient’s perception of medical care (Figure 3). Most respondents estimated that health professionals outside of the hospital did not have enough knowledge about the pathology (90%). This perception was significantly correlated with the delay in diagnosis: the average delay in diagnosis was 4.5 years among respondents who estimated that health professionals had insufficient knowledge of the disease vs 1.5 years among respondents who estimated that health professionals had sufficient knowledge of the disease. Fifty-seven percent of respondents considered they (or the patients for whom they answered) had undergone inappropriate examinations, tests or treatments, and 72% mentioned insufficient coordination between ambulatory health care professionals (family doctor, nurses, physiotherapists and so on). For 45% of respondents, unsatisfactory relay between the hospital and the family doctor (transfer of the patient’s hospital care to the follow-up by the family doctor) was reported. Some of these frequencies were significantly higher when the responses concerned only patients with longer delays in diagnosis (≥6 years): 70% considered they had undergone inappropriate examinations, tests or treatments, 89% mentioned insufficient coordination between ambulatory health care professionals and 60% reported unsatisfactory relay between the hospital and the family doctor (data not shown). Hospital stays were often planned for 55% of patients, while the other 45% experienced hospital stays in emergency situations (most often due to sudden disease aggravation). When the respondents were parents or relatives, the answers to subjective questions were those perceived by the respondents and, therefore, could differ from the real patients’ perceptions. However, statistical analyses revealed no differences between the answers from the patients themselves, parents and relatives. Finally, home return after hospital stay often or very often proceeded smoothly for 88% of the patients. For the 43 participants who expressed difficulties on returning home, financial issues (35%), insufficient home assistance (14%) and psychological issues (14%) were the most frequently mentioned reasons.
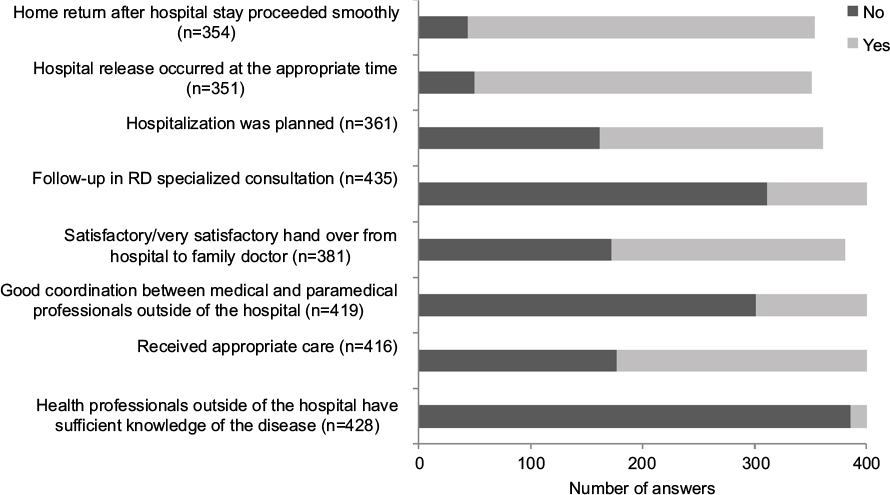 | Figure 3 Perception of medical care. Note: This figure shows an English translation of the original version of these questions, which were presented in French. Abbreviation: RD, rare disease. |
Follow-up in an RD specialized consultation (represented in France by the reference or competence centers for RD) was observed for 28% of the patients on average, with a higher proportion of children younger than 12 years of age (36% of the children <12 years old, 26% of the patients >12 years old, significant difference). A slightly higher proportion of patients living in Ile-de-France (Paris and the surrounding departments [administrative regions]) was followed in an SC (37.5%), compared to patients living in provinces (26%, significant difference; data not shown).
Daily life
Impact of the disorders on everyday life was perceived as heavy or very heavy by 73% of the respondents (n=436). Only 4% declared very minor troubles. For 77% of the participants, no evaluation was carried out to determine the patients’ needs (Table 2). This high proportion should be modulated by the fact that for some patients such evaluation was irrelevant, depending on their pathology. Of note, for children, needs were evaluated in a significantly higher proportion: 34% of the children <18 years old vs 23% on average. This proportion was even higher for children <2 years old (44%). When the evaluation was carried out, it was performed mostly in a regional center for disabled people, corresponding to the French Maisons Départementales des Personnes Handicapées [Disabled people department home] (for 65% of the evaluations). Interestingly, for 27% of the people who benefited from an evaluation (27/101 patients), the needs and life plan were estimated as not enough, or not at all considered. Several needs were expressed in the survey by 300 respondents (whether they had undergone an evaluation of their needs or not). The most frequently reported were financial, technical (wheelchair, bed, bath seat and so on) and home support. Subgroup analyses by type of respondent showed that parents and relatives significantly more frequently expressed needs than patients themselves (Table 2). Procedures to obtain support were considered as difficult or rather difficult for 71% of respondents, mostly for administrative reasons. The proportions of met and unmet needs among those expressed are not known. In addition, 33% of the total panel of participants did not express needs. It is not known whether the concerned patients really did not need help or needed help but did not answer the question.
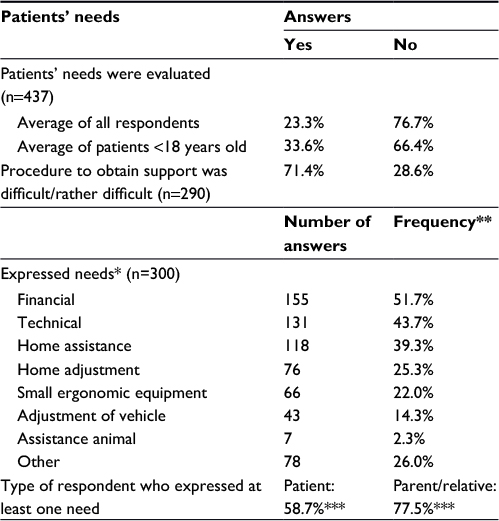 | Table 2 Patients’ needs Notes: *These needs are those expressed in the survey, independent of the needs that could have been expressed during the evaluation by a health care professional. **300 participants answered the question, but some declared several needs. The frequency was calculated based on the number of respondents (n=300) and not on the total number of answers. ***Percentages of the participating patients or parents/relatives. Abbreviation: n, number of respondents. |
Medicosocial center/service
In France, medicosocial centers encompass various establishments dedicated to social or medicosocial support of disabled or vulnerable people (centers for accommodation, education, psychological care, rehabilitation and so on). One-fifth of the patients (83/431 respondents) stayed or were
followed in a medicosocial center. Eighty-three percent of them were children, which represented half of the children involved in the survey, while adults represented less than one-tenth of the adult patients in the survey. Due to the small number of patients concerned by questions about the medicosocial center, and thus who answered them, no advanced analysis could be made. However, the answers provide interesting information about this particular step in the life of some RD patients. For 61 respondents, staying or being followed in a medicosocial center was anticipated, 44 indicated they could choose between several centers, 64 answered their expectations about the center were met, and 70 considered that the stay or follow-up in a medicosocial center had favorable or very favorable consequences (better quality of life, better care, increased social relationships). Yet, a small proportion of patients felt uprooted (23) or isolated from their family (21).
School and work
One hundred and seventy-eight respondents were concerned by school-related questions, with an average patient age of 21 at the time of the survey. Patients who had responded for themselves were 16–78 years old (average 38 years old). Responses provided by parents and relatives concerned patients from <2 to 50 years of age (average 13 years of age). Fifty-nine percent of the respondents estimated that the contact with education teams was good or very good and that they were listened to well. This proportion reached 82.5% for young and adolescent patients (Table 3). Among the respondents who negatively perceived the contact and communication with educational teams, the situation was described as difficult, with a feeling of isolation. Forty-two percent of respondents expressed the need of a special needs teaching assistant, whether it was actually provided or not, and 42% benefited from specific school adjustment/support. Thirteen patients from 16 to 23 years of age replied to this question by themselves. None of them mentioned the need for a school assistant.
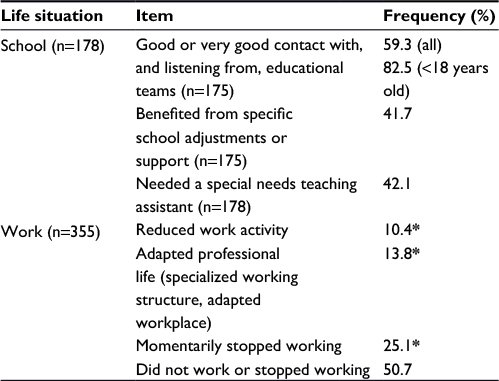 | Table 3 School and work situation Notes: *For work-related questions, several answers were possible. Percentages were calculated based on the 355 respondents. This table shows an English translation of the original version of these items, which were presented in French. Abbreviation: n, number of respondents. |
Half of the respondents to work-related questions declared they did not work or stopped working as a result of the disease (Table 3). Of note, for this topic, answers corresponded not only to the situation of patients themselves, but also to the situation of parents when they responded for <18-year-old patients. Consequences for both patients and families were income reductions, which added a hurdle to the daily life difficulties, as expressed in the free comments.
Consequences of the disease on personal life
More than one-third of the patients were advised to have psychological assistance (Table 4). Among patients between 2 and 17 years of age, this proportion was significantly higher (48%). About two-thirds of the participants in the survey reported depression, breakdowns, behavior troubles and other psychological issues. Among patients who were not advised to have psychological assistance (n=277), more than half declared having psychological troubles (whatever the extent), and each type of trouble (depression, breakdown and so on) was present in the same proportion as for the patients who were advised to have support (data not shown). The feeling of being isolated from friends and relatives was present for half of the participants, and was also often underlined in the free comments. The need to hide the disease (own disease or disease of a child) was expressed by 33% of the respondents. Subgroup analyses showed that 45% of the patients responding for themselves felt the need to hide their disease, while this feeling was significantly less present among parents and relatives (18%). Half of the participants mentioned an impact of the disease on sentimental or emotional life, and various degrees of couple issues were often reported (for both patients and parents), with the disease as a causative or triggering factor for a majority of them (75% and 70%, respectively). Family issues were also declared by about 60% of participants.
 | Table 4 Psychological, emotional and family aspects associated with the disease Notes: *Answers to this question concerned both patients and parents. This table shows an English translation of the original version of these issues, which were presented in French. Abbreviation: n, number of respondents. |
Focus on the specific situation of patients followed in specialized consultations
Statistical analyses of answers concerning 124 patients followed in SCs (28% of the patients from the survey, see the “Medical care” section) showed some differences compared to the answers concerning patients not followed in SCs (311). Answers with significant differences between the two populations are detailed in this section and reported in Figure 4. While a majority of respondents considered that health professionals outside of the hospital had insufficient knowledge of the disease, this proportion was reduced for responses concerning patients followed in an SC (85% for patients followed in an SC vs 92% for patients not followed in an SC). Fewer patients followed in an SC estimated (themselves or through the respondents’ answers) they had undergone inappropriate examinations, tests or treatments: 50% of patients followed in an SC vs 61% of patients not followed in an SC. Follow-up in an SC was correlated with a higher degree of satisfaction regarding the relay between the hospital and the family doctor (66% vs 51% for patients not followed in an SCs). Thirty-five percent of patients followed in an SC considered the coordination between medical and paramedical professionals outside of the hospital was good vs only 26% of patients not followed in an SC. Less hospitalizations in emergency situations were reported for patients followed in SCs (37% vs 48% for patients not followed in SCs), and hospital release was more frequently estimated to occur at the appropriate time (92% vs 82% for patients not followed in SCs). A higher rate of needs evaluation was reported for patients followed in an SC (31%) than for patients not followed in an SC (20%). Similarly, more patients followed in an SC were advised to have psychological support (44%) than patients not followed in an SC (34%).
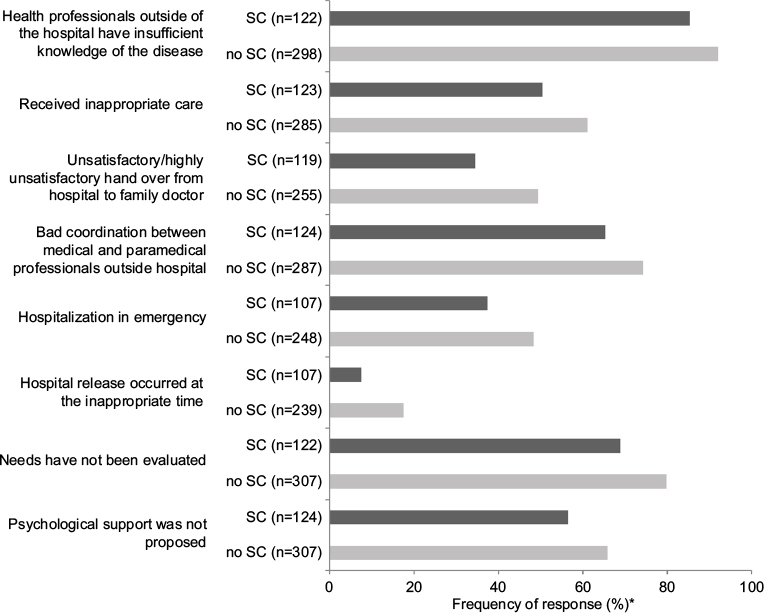 | Figure 4 Comparison between answers of patients followed in an SC (black bars) and patients not followed in an SC (gray bars). Notes: *Percentage of patients who answered ‘yes’. All answers are significantly different between the two populations. This figure shows an English translation of the original version of these questions, which were presented in French. Abbreviation: SC, specialized consultation. |
Discussion
This study allowed the collection of quantitative data about the situations to which patients, their parents, relatives and caregivers are confronted. Results from 448 questionnaires covered 195 different diseases. Respondents were represented by a majority of women, which is likely to reflect the higher proportion of women who generally participate in this type of survey rather than epidemiological data. This proportion has also been observed in the previous barometer12 and in an Australian survey carried out with self-selected respondents.13 The results presented here primarily aimed to quantify the responses of the participants, in order to describe the journey of the involved patients. These participants represented a panel of users of Maladies Rares Info Services distributed in all regions of metropolitan France and, therefore, may not be fully representative of all RD patients in France. Yet, although extrapolations to all RD patients should be done carefully, these results provide interesting data and trends about the patients’ situations.
Some of the key findings of the 2015 barometer are highlighted in Table 5. The sometimes extremely long delay in diagnosis confirms a well-known issue in the field of RDs. In the USA and UK, averages of 7.6 and 5.6 years have been reported, respectively.10 In a survey involving Australian adults living with an RD, delays in diagnosis close to those found in our study were reported, with about half of the respondents having waited for 1 year or more and one third waiting for five or more years.13 In France, a survey involving 22 different RDs and 844 patients has described a delay in diagnosis >5 years for about 25% of patients.14 In our study, no obvious correlation was found between the disease type or prevalence and the duration of the delay in diagnosis. However, among the diseases with reported delays of ≥20 years, some are well-known for being often diagnosed late. This is the case, for instance, for Ehlers–Danlos syndrome,2,15,16 Marfan disease2,17 or chronic intestinal pseudo-obstruction,18 which were also associated with very long delays in our survey. These diseases are highly representative of the long delay issue. They have a noncharacteristic presentation, symptoms overlapping with several other disorders including non-rare ones, intrafamilial and interfamilial variability and no known specific biomarkers.19,20 By contrast, diseases with specific biomarkers, pathognomonic signs, early (neonatal, infancy) and severe presentation are likely to be diagnosed quickly.
 | Table 5 Summary of the key conclusions from the 2015 barometer |
Moreover, the results of the barometer have highlighted the impact of the length of delay in diagnosis on the life path of RD patients. Longer delays (≥6 years) between the onset of the first symptoms and the diagnosis were correlated with a higher impression of having undergone inappropriate tests and treatments, higher impression of insufficient coordination between ambulatory health care professionals and higher dissatisfaction regarding the relay between the hospital and the family doctor. Very different pathologies were represented in this study, as well as different conditions of age, life and care. To better understand the factors involved in the length of diagnosis delays and the associated negative perceptions, it would be interesting to carry out studies on specific subgroups of respondents. This would help improve the dialogue between patients and professionals (better listening and understanding of the first symptoms, orientation toward appropriate expert centers, increased role of medicosocial professionals and better link with health professionals, patient associations and so on). In the European Organization for Rare Diseases survey, correlations were observed between longer time to diagnosis and prior misdiagnosis, and between shorter time to diagnosis and personal initiatives of RD patients, such as the suggestion that their disease might be a rare one, or the identification of a diagnostic structure by themselves.2 The feeling of being unheard and disbelieved, which was reported repeatedly through the free comments in our study, has also been reported for pathologies with known long delays in diagnosis and may play a role in the lengthening of this step.16 Three main levers could be used to reduce delays in diagnosis and the associated negative impact. First, the capacities of high-throughput sequencing should be developed, while ensuring its equity of access. Then, the initial and continuing training of physicians should be strengthened to improve the medical orientation of patients. Finally, the information and communication on RDs should be strengthened more generally. Although various resources are available in France, they are poorly known of by patients and professionals outside of specialized RD centers. The number of inappropriate tests or treatments could be reduced by developing the doubt-based culture in medical practice: “and if it was an RD?” It would prevent nonexpert physicians from proceeding by trial and error, and it would promote earlier patient orientation toward a more adapted resource center. French physicians from the ambulatory sector have poor awareness of the definition of RD and of the available resources that can be proposed for patients. Changing the game on this aspect is a major concern of the Third National Plan on RDs that is currently being set up in France.
School life and professional life were identified as important steps in the life path of RD patients. Interestingly, school needs seemed to be rather well-evaluated and taken into account. Good communication with educational teams was also globally reported, particularly among younger patients. It is possible that the measures taken during the last decade to better consider the needs and rights of disabled people have played a role in improving school-related issues for RD patients. Indeed, following the disability law from 11 February 2005 for equality of rights and chances, involvement and civic rights of disabled people,21 the reception conditions and recognition of disabled people’s needs and rights have improved,22 and may therefore be perceived differently by younger patients. However, the difference in the degree of satisfaction between younger and older patients could also be explained by differences between responses of patients themselves (the older ones) and responses of parents or relatives (for younger patients). Indeed, patients who are in direct and frequent contact with the educational teams may have perceived the relationships more negatively than the parents. By contrast with the school period, the professional situation of both patients and parents seemed strongly and negatively impacted by the disease. No quantitative data about the impact on families of having to stop working (or not working) were collected, but the affected people often reported financial, sometimes severe, deterioration of their situation. In addition, the absence of work activity may induce social and psychological consequences: social relationships may be reduced, or a feeling of isolation and social exclusion may appear.
Various needs, psychological and personal issues were reported and quantified. Psychological support seemed to be variably proposed, and needs were only partly evaluated or taken into consideration. Regarding the consequences of the disease on personal life, differences were observed depending on the type of respondent, especially between patients and parents. A higher proportion of patient respondents felt the need to hide their disease, than parent respondents. This difference could be correlated with the “risk” for an adult to reveal his/her pathology to the employer, while parents are likely to benefit from better assistance and acceptance from teachers, friends and children themselves when they better communicate about the disease. This difference could also be correlated with another difference: the higher proportion of parents/relatives who expressed needs compared to patients themselves. Needs may also be perceived more strongly among parents, especially parents of younger children. For instance, parents represented the majority of respondents who expressed the need for a school assistant, while very few patients and none of the adolescent participants responding for themselves expressed this need. It is possible that the latter did not express this need due to the difficulty of being accepted at school. However, these results should be considered with care due to the low number of adolescent respondents and because some of the respondents may actually not have needed assistance, depending on their pathology. Indeed, it is possible that participants who responded for themselves had less severe disabilities and needed less assistance than those for whom the parents responded.
A small proportion of patients were followed up or stayed in a medicosocial center. This particular situation was globally associated with better social and care conditions of life for a majority of patients and their families. However, one-third of these patients felt isolated and deserve higher vigilance from their caregivers to avoid more life path disruptions. Access to medicosocial centers is naturally limited by the fostering capacities of these establishments, which is not specific to RDs. However, better information and training of the different health care stakeholders would help in taking into account the specificities of RDs to improve access to the appropriate medicosocial services.
A significant impact of the follow-up in SCs on the life of RD patients was revealed in this study. Although some bias due to confounding factors related to the disparity of situations (age, disease, degree of disability, social and professional situation and so on) among the respondents should not be excluded, several aspects of the medical care path and daily life of RD patients seemed to be positively impacted by the follow-up in an SC.
These data about the role of RD SCs are particularly interesting with regard to the recent setup of the French RDs health care networks (“filières de santé maladies rares”),23 which aim to drive and coordinate the actions between all the stakeholders involved in RD care and also with regard to the creation of the European Reference Networks to improve the care for patients with low prevalence diseases or RDs throughout the European Union.24 The German National Action League for People with Rare Diseases founded in 2010 to improve medical care in the field of RDs has identified similar priorities for action: to introduce reference centers for RDs, to take measures to accelerate the diagnosis process, and for the promotion of research and information management.25 Enhanced networking of RD specialists is expected to help improve some of the current issues RD patients are facing. The French RDs health care networks are expected to improve the coordination between reference and competence centers specialized in the same groups of pathologies. However, for patients not yet included in these SCs, the role of the family doctor remains essential to direct them toward the appropriate center. Facilitation of the journey of RD patients would most probably involve, among others, increased communication among caregivers, increased interdisciplinary exchanges, better assistance of patients throughout their life projects by simplifying some administrative procedures, limiting the hurdles in professional life, providing adapted support to reduce psychological, social and familial difficulties, providing the opportunity to be listened to at any time, facilitating the contacts with social services or patient associations, and providing the means for parents and relatives to take some time off. It would be important for reference centers to have more financial resources to provide free care that tackles the psychological consequences of the disease (e.g., the family-related issues), not only for patients but also for their families. To prevent isolation, services or establishments where patients could have social interactions should be proposed. In addition, targeted and recurring communication about these resources should be made. This communication would also help in promoting recognition of patients who have an RD, and should become a goal, at the same level as for other severe, non-rare, pathologies that are now well-known and accepted by society.
Conclusion
This study raises awareness about the positive aspects as well as the many hurdles and sources of disruption in the life path of RD patients. Despite the diversity of pathologies, several issues were common to RD patients. The results, based on the direct perception of patients, their parents or relatives, provide a background not only for further in-depth studies, but also to better adapt the support to real needs and to improve the synergies between the many people involved in the life path of RD patients.
Acknowledgments
The 2015 barometer for rare diseases has received support from Genzyme and from the Groupama Foundation for Health. The funders had no role in the study design, data collection and analysis, decision to publish and preparation of the manuscript. We deeply acknowledge the participating patients, parents, relatives and health professionals, who have shared, for several hours, their life stories and experience of RDs, and also the referral listening and information staff from Maladies Rares Info Services. We thank Marieke Podevin from Argo Santé who carried out the qualitative study interviews and analysis, and contributed to the design and analysis of the quantitative survey questionnaire. We also thank Antoine Decaris, Emmanuel Bartholo and Violette Rouzet from Kynos who edited the questionnaires and analyzed the results of the quantitative study. Finally, we thank Lara Chappell for proofreading the English of the manuscript.
Author contributions
CV and TH: conception and design of the study, selection of participants, data collection, analysis and interpretation of the data, and literature search. SP: literature search, analysis and interpretation of the data, preparation of figures and tables, and writing of the manuscript. All authors contributed toward data analysis, drafting and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Council of the European Union. Council recommendation of 8 June 2009 on an action in the field of rare diseases (2009/C 151/02). Off J Eur Union 2009;151:7–10. | ||
EURORDIS. The Voice of 12,000 Patients. Experiences and Expectations of Rare Disease Patients on Diagnosis and Care in Europe; 2009. Available from: http://www.eurordis.org/publication/voice-12000-patients. Accessed December 15, 2016. | ||
Maladies Rares Info Services. Available from: http://www.maladiesraresinfo.org/. Accessed December 15, 2016. French. | ||
Maladies Rares Info Services [homepage]. Rapport d’activité 2015 [2015 activity report]. Available from: http://www.maladiesraresinfo.org/mris/en-savoir-plus-sur-maladies-rares-info-services/rapport-activite.html. Accessed December 15, 2016. French. | ||
Angelis A, Tordrup D, Kanavos P. Socio-economic burden of rare diseases: a systematic review of cost of illness evidence. Health Policy. 2015;119(7):964–979. | ||
van Walsem MR, Howe EI, Iversen K, Frich JC, Andelic N. Unmet needs for healthcare and social support services in patients with Huntington’s disease: a cross-sectional population-based study. Orphanet J Rare Dis. 2015;10:124. | ||
Dwyer AA, Quinton R, Morin D, Pitteloud N. Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support. Orphanet J Rare Dis. 2014; 9:83. | ||
Sjoberg L, Nilsagard Y, Fredriksson C. Life situation of adults with congenital limb reduction deficiency in Sweden. Disabil Rehabil. 2014;36(18):1562–1571. | ||
Velvin G, Bathen T, Rand-Hendriksen S, Geirdal AO. Systematic review of the psychosocial aspects of living with Marfan syndrome. Clin Genet. 2015;87(2):109–116. | ||
Shire. Rare disease impact report: insights from patients and the medical community. J Rare Disord. 2014:1–34. | ||
Maladies Rares Info Services. Les résultats 2011 de l’Observatoire des maladies rares [The 2011 results of the rare diseases barometer]. Available from: http://www.maladiesraresinfo.org/assets/pdf/Rapport_Observatoire_MR_2011_web.pdf. Accessed December 15, 2016. French. | ||
Maladies Rares Info Services. Résultats et analyses 2012 de l’Observatoire des maladies rares [The 2012 results and analyses of the rare diseases barometer]; 2012. Available from:http://www.maladiesraresinfo.org/assets/pdf/Rapport_Observatoire_maladies_rares_2012.pdf. 2012. Accessed December 15, 2016. French. | ||
Molster C, Urwin D, Di Pietro L, et al. Survey of healthcare experiences of Australian adults living with rare diseases. Orphanet J Rare Dis. 2016;11:30. | ||
Alliance maladies rares. ERRADIAG – L’errance diagnostique dans les maladies rares [ERRADIAG - diagnostic delay in rare diseases]; 2016. Available from: http://www.alliance-maladies-rares.org/erradiag-enquete-sur-lerrance-diagnostique-dans-les-maladies-rares/. Accessed Dec 15, 2016. French. | ||
Colombi M, Dordoni C, Chiarelli N, Ritelli M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am J Med Genet C Semin Med Genet. 2015;169C(1):6–22. | ||
Knight I. The role of narrative medicine in the management of joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type. Am J Med Genet C Semin Med Genet. 2015;169C(1):123–129. | ||
Roll K. The influence of regional health care structures on delay in diagnosis of rare diseases: the case of Marfan syndrome. Health Policy. 2012;105(2–3):119–127. | ||
Bernardi MP, Warrier S, Lynch AC, Heriot AG. Acute and chronic pseudo-obstruction: a current update. ANZ J Surg. 2015;85(10):709–714. | ||
Sobey G. Ehlers-Danlos syndrome: how to diagnose and when to perform genetic tests. Arch Dis Child. 2015;100(1):57–61. | ||
De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82(1):1–11. | ||
LOI n 2005-102 du 11 février 2005 pour l’égalité des droits et des chances, la participation et la citoyenneté des personnes handicapées [Law 2005-102 from February 11, 2005 on equal rights and opportunities, participation and citizenship of disabled people]. JORF n 36 du 12 février 2005 [Official Journal of the French Republic number 36, February 12, 2005]. Available from: http://www.legifrance.gouv.fr/affichTexte.do?cidTexte=JORFTEXT000000809647&categorieLien=id. 2005. Accessed December 15, 2016. French. | ||
Campion CL, Debré I. Loi Handicap: des avancées réelles, une application encore insuffisante. Rapport d’information n 635 (2011-2012) fait au nom de la commission pour le contrôle de l’application des lois [Disability Law: real progress, still insufficient application. Information report number 635 (2011-2012) written on behalf of the commission for the control of law enforcement]; 2012. Available from: http://www.senat.fr/rap/r11-635/r11-635_mono.html#toc147. Accessed April 6, 2016. French. | ||
Ministère des Affaires sociales, de la Santé et des Droits des femmes. Les filières de santé maladies rares [French rare diseases healthcare networks]; 2014. Available from: http://social-sante.gouv.fr/soins-et-maladies/prises-en-charge-specialisees/article/les-filieres-de-sante-maladies-rares. Accessed December 15, 2016. French. | ||
Hollak CE, Biegstraaten M, Baumgartner MR, et al. Position statement on the role of healthcare professionals, patient organizations and industry in European reference networks. Orphanet J Rare Dis. 2016;11(1):7. | ||
Frank M, Eidt-Koch D, Aumann I, Reimann A, Wagner TOF, Graf von der Schulenburg JM. Maßnahmen zur Verbesserung der gesundheitlichen Situation von Menschen mit seltenen Erkrankungen in Deutschland. Ein Vergleich mit dem Nationalen Aktionsplan [Measures to improve the health situation of patients with rare diseases in Germany. A comparison with the National Action Plan]. Bundesgesundheitsblatt Gesundheitsforschung Gesundheitsschutz. 2014;57(10):1216–1223. German. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.