")
Back to Journals » Journal of Pain Research » Volume 10
Opioid analgesics-related pharmacokinetic drug interactions: from the perspectives of evidence based on randomized controlled trials and clinical risk management
Authors Feng XQ, Zhu LL, Zhou Q
Received 3 April 2017
Accepted for publication 2 May 2017
Published 24 May 2017 Volume 2017:10 Pages 1225—1239
DOI https://doi.org/10.2147/JPR.S138698
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Michael Schatman
Xiu-qin Feng,1 Ling-ling Zhu,2 Quan Zhou3
1Nursing Administration Office, Division of Nursing, 2VIP Care Ward, Division of Nursing, 3Department of Pharmacy, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang, People’s Republic of China
Background: Multimorbidity results in complex polypharmacy which may bear a risk of drug interactions. A better understanding of opioid analgesics combination therapy used for pain management could help warrant medication safety, efficacy, and economic relevance. Until now there has been no review summarizing the opioid analgesics-related pharmacokinetic drug interactions from the perspective of evidence based on randomized controlled trials (RCTs).
Method: A literature search was performed using PubMed, MEDLINE, and the Cochrane Library, using a PRISMA flowchart.
Results: Fifty-two RCTs were included for data interpretation. Forty-two RCTs (80.8%) were conducted in healthy volunteers, whereas 10 RCTs (19.2%) enrolled true patients. None of the opioid–drug/herb pairs was listed as contraindications of opioids involved in this review. Circumstances in which opioid is comedicated as a precipitant drug include morphine–P2Y12 inhibitors, morphine–gabapentin, and methadone–zidovudine. Circumstances in which opioid is comedicated as an object drug include rifampin–opioids (morphine, tramadol, oxycodone, methadone), quinidine–opioids (morphine, fentanyl, oxycodone, codeine, dihydrocodeine, methadone), antimycotics–opioids (buprenorphine, fentanyl, morphine, oxycodone, methadone, tilidine, tramadol), protease inhibitors–opioids (ritonavir, ritonavir/lopinavir–oxycodone, ritonavir–fentanyl, ritonavir–tilidine), grapefruit juice–opioids (oxycodone, fentanyl, methadone), antidepressants–opioids (paroxetine–tramadol, paroxetine–hydrocodone, paroxetine–oxycodone, escitalopram–tramadol), metoclopramide–morphine, amantadine–morphine, sumatriptan–butorphanol nasal sprays, ticlopidine–tramadol, St John’s wort–oxycodone, macrolides/ketolides–oxycodone, and levomepromazine–codeine. RCTs investigating the same combination, almost unanimously, drew consistent conclusions, except two RCTs on amantadine–intravenous morphine combination where a different amantadine dose was used and two RCTs on morphine–ticagrelor combination where healthy volunteers and true patients were enrolled, respectively. RCTs investigating in true patients may reflect a realistic clinical scenario and overcome the limitation of RCTs performed in healthy volunteers under standardized conditions. Further research opportunities are also presented in this review.
Conclusion: Effective and safe combination therapy of opioids can be achieved by promoting the awareness of potential changes in therapeutic efficacy and toxicities, prescribing alternatives or changing administration strategy, tailoring dose, reviewing the appropriateness of orders, and paying attention to medication monitoring.
Keywords: adverse drug reaction, clinical efficacy, combination therapy, drug-drug interactions, drug metabolism, drug transporter, pain management, pharmacokinetics, polypharmacy
Introduction
Multimorbidity is the principal cause of complex polypharmacy. Polypharmacy is not a problem in itself, but there is a risk of drug–drug interactions (DDIs) or herb–drug interactions in the event of poor awareness or a lack of coordination among care providers and patients.
Opioid analgesics are commonly prescribed for acute and chronic pain. A better understanding of drug-metabolizing enzymes and transporters-involved interactions with opioids will provide greater insight into potential DDIs and could help warrant safety and efficacy of opioids.1 Moreover, opioid pain management involves economic issue. Summers et al observed that chronic pain patients exposed to a potential opioid-related DDI of major clinical significance had significantly higher monthly health care costs in the 90-day post-index period compared to those not exposed to an opioid-related DDI.2
A randomized controlled trial (RCT) is considered to provide the most reliable evidence on the effectiveness of interventions because the processes used during the conduct of an RCT minimize the risk of confounding factors influencing the results.3 There have been several reviews on DDIs associated with opioids, including a review by Kotlinska-Lemieszek et al which only included 32 case reports or case series published till January 2014,4 a review by Overholser and Foster published in 2011 which lacked literature search strategy in spite of describing potential DDIs associated with opioids that are frequently encountered in managed care,5 and a review by Gharavi et al published in 2015 which lacked literature search strategy in spite of reporting the literature related to opioids and their drug transporter interactions.6 Given that a review summarizing the latest literature on opioid analgesics-related pharmacokinetic DDIs from the perspective of evidence based on RCTs has not been available, we present an updated narrative review and propose relevant clinical risk management to enhance rational combination use of opioids.
Methods
Search strategy
The relevant literature was identified by searching PubMed, MEDLINE, and the Cochrane Library till Mar 16, 2017, using a query “opioids and drug interaction and pharmacokinetics and analgesics” with a filter of “language: English; article type: randomized controlled trials; species: humans”.
Selection criteria
Two reviewers (XQF and LLZ) independently searched the literature and screened the relevant studies. In the event of disagreement between the two reviewers, a third reviewer (QZ) was consulted. Studies on opioid analgesics-associated pharmacokinetic DDIs were included. The following studies were excluded: documents with actually irrelevant topics despite being retrieved using the search terms; studies investigating DDIs related to opioids indicated for anesthesia or alcohol-related studies; documents such as comments, letters, and editorials in spite of reporting RCT results; studies involving drugs which have been withdrawn from the market, studies involving neutral DDIs between “A” and “B”, while the similar drugs within the same therapeutic category of “A” did not have DDIs with “B” or the similar drugs within the same therapeutic category of “B” did not have DDIs with “A”. Fifty-two articles were finally included under this search strategy and inclusion/exclusion criteria. The PRISMA flowchart of study selection is illustrated in Figure 1. The full text of each included article was critically reviewed, and valuable information was summarized by data interpretation.
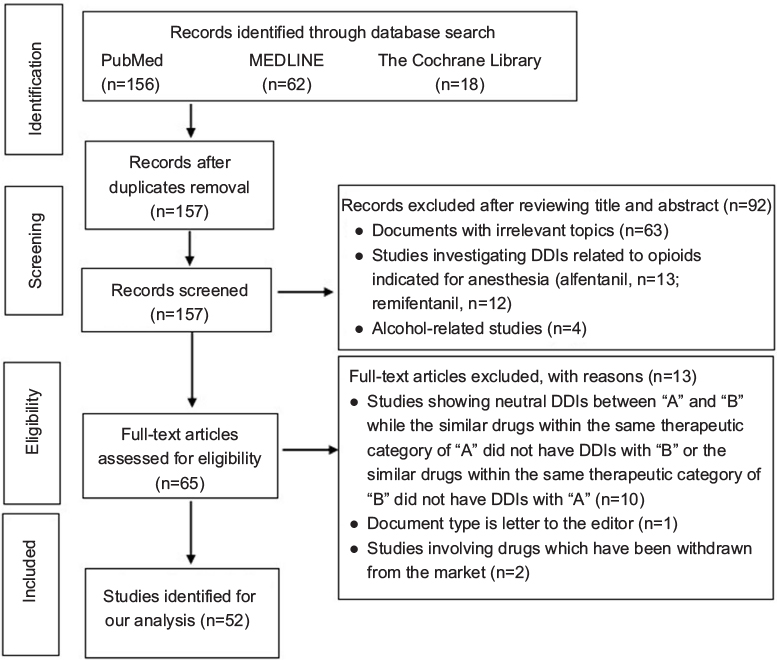 | Figure 1 Flowchart of literature selection. Abbreviation: DDIs, drug–drug interactions. |
Results and discussion
Metabolism and transporter profiles of common opioid analgesics
Opioids metabolized by the drug-metabolizing enzymes of the CYP system are associated with numerous DDIs that may result in either a reduction in opioid effect or excess opioid effects. In contrast, opioids that are not metabolized by that system tend to be involved in fewer CYP-associated pharmacokinetic DDIs.5,7
However, some opioids are UGT substrates, and evidence have shown the role of P-gp in the intestinal disposition and/or effects of morphine, oxycodone, methadone, fentanyl, and metabolites of some opioids.8–11 OCT1 plays an important role in the hepatocellular uptake of morphine, and polymorphisms in OCT1 gene significantly affect morphine pharmacokinetics after codeine administration.12 Volunteers with loss-of-function OCT1 polymorphisms had significantly higher plasma concentrations of O-desmethyltramadol and significantly prolonged miosis.13 Therefore, the role of UGT, P-gp, and OCT1 in opioids-associated DDIs should also be given importance.
Metabolism and transporter profiles of common opioid analgesics are listed in Table 1.
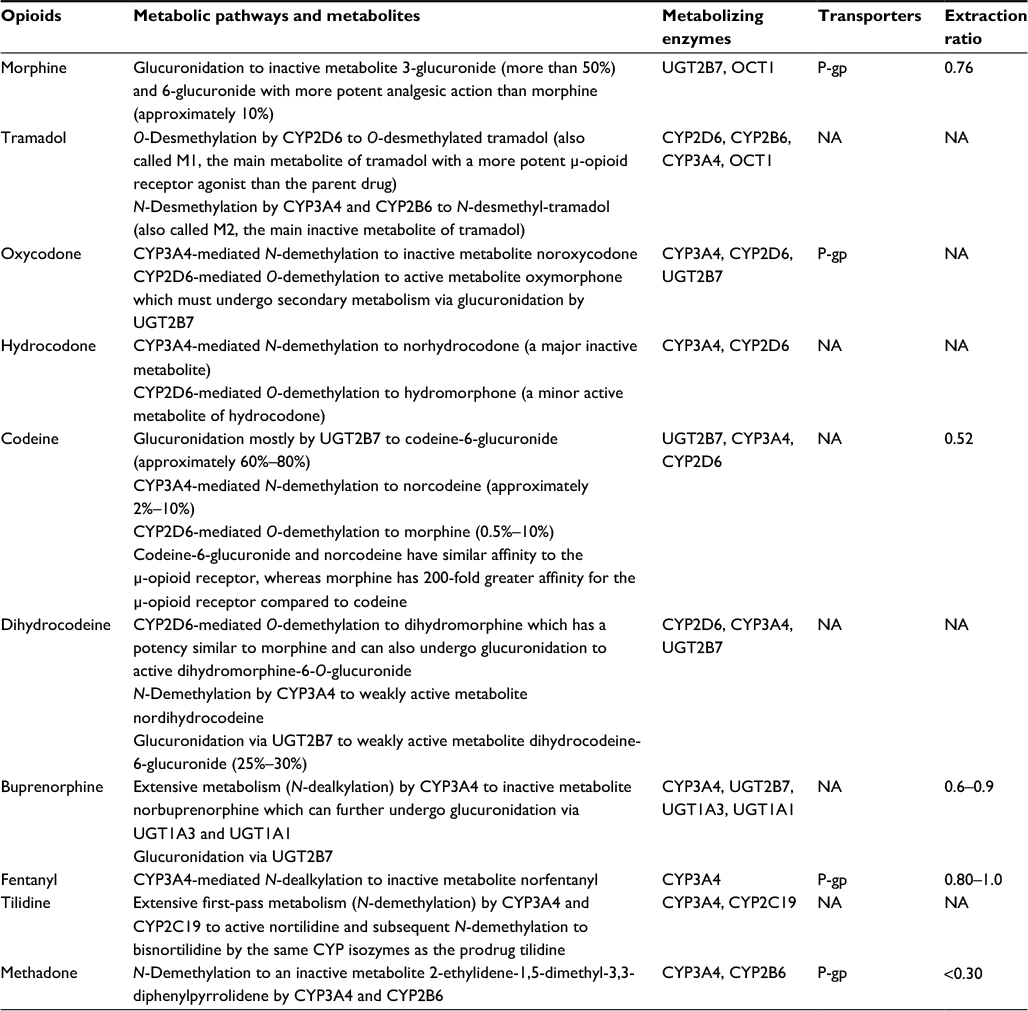 | Table 1 Metabolism and transporter profiles of common opioid analgesics Abbreviation: NA, not applicable. |
General information for opioid pharmacokinetic DDIs in RCTs
None of the opioid–drug/herb pairs was listed as contraindications of opioids involved in this review. RCTs investigating the same combination, almost unanimously, drew consistent conclusion except two RCTs on amantadine–intravenous (i.v.) morphine combination where a different amantadine dose was used, and two RCTs on morphine–ticagrelor combination where healthy volunteers and true patients were enrolled, respectively. Among 52 RCTs included under this review’s inclusion/exclusion criteria, 42 (80.8%) were conducted in healthy volunteers, whereas 10 (19.2%) enrolled true patients, involving nine drug pairs (i.e., morphine–ticagrelor, methadone–zidovudine, methadone–buprenorphine, quinidine–codeine, voriconazole–methadone, fluconazole–methadone, metoclopramide–morphine, amantadine–morphine, and levomepromazine–codeine).
For each DDI, the object drug is defined as the medication whose pharmacokinetics and/or pharmacodynamics may be modified by the drug interaction process whereas the precipitant drug is defined as the medication responsible for affecting the pharmacological action or the pharmacokinetic properties of the object drug.14 In this review, opioid pharmacokinetic DDIs were categorized into two circumstances. Circumstances in which opioid is comedicated as a precipitant drug include morphine–P2Y12 inhibitors (clopidogrel, prasugrel, ticagrelor), morphine–gabapentin, and methadone–zidovudine. Circumstances in which opioid is comedicated as an object drug include rifampin–opioids (morphine, tramadol, oxycodone, methadone), quinidine–opioids (morphine, fentanyl, oxycodone, codeine, dihydrocodeine, methadone), antimycotics–opioids (buprenorphine, fentanyl, morphine, oxycodone, methadone, tilidine, tramadol), protease inhibitors–opioids (ritonavir, ritonavir/lopinavir–oxycodone, ritonavir–fentanyl, ritonavir–tilidine), grapefruit juice–opioids (oxycodone, fentanyl, methadone), antidepressants–opioids (paroxetine–tramadol, paroxetine–hydrocodone, paroxetine–oxycodone, escitalopram–tramadol), metoclopramide–morphine, amantadine–morphine, sumatriptan–butorphanol nasal sprays, ticlopidine–tramadol, St John’s wort–oxycodone, macrolides/ketolides–oxycodone, and levomepromazine–codeine.
Opioid-related pharmacokinetic DDI mechanisms are illustrated in Figure 2.
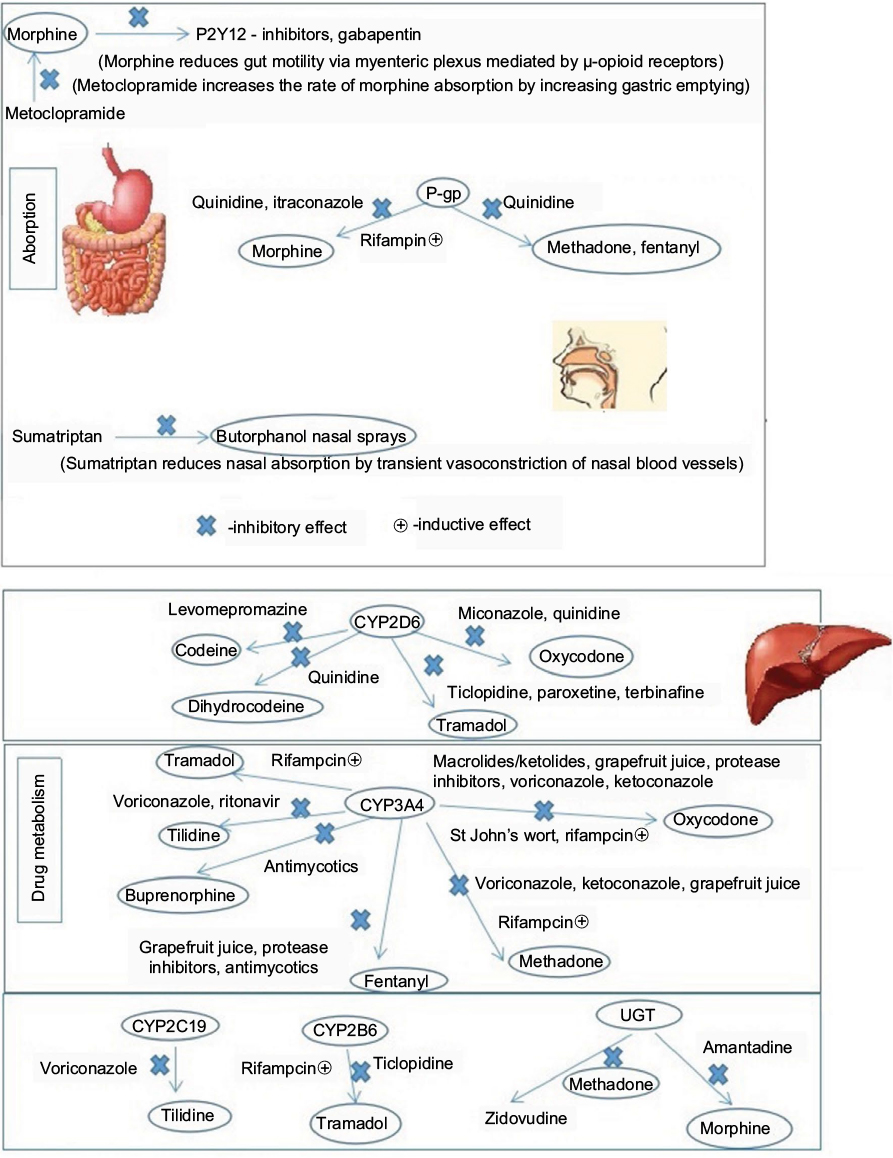 | Figure 2 Opioid-related pharmacokinetic drug–drug interaction mechanisms. |
Circumstances in which opioid is comedicated as a precipitant drug
Morphine–P2Y12 inhibitors
Clopidogrel
An RCT in healthy subjects showed that 5 mg of i.v. morphine could delay clopidogrel absorption, decrease the area under the concentration–time curve (AUC) of clopidogrel active metabolite by 34%, and retard and diminish clopidogrel effects on platelet function following a loading dose of 600 mg clopidogrel.15
Prasugrel
An RCT in healthy volunteers showed that 5 mg of i.v. morphine could reduce the peak concentration (Cmax) of prasugrel active metabolite by 31% (P<0.05) despite that it did not significantly alter the AUC and drug absorption of 60-mg prasugrel and its effects on platelets.16
Ticagrelor
An RCT in healthy subjects also showed pharmacokinetic but not pharmacodynamic DDI between i.v. morphine and ticagrelor. Concomitant 5-mg i.v. morphine could slow the drug absorption of ticagrelor by 1 h followed by a 180-mg loading dose, and decrease plasma levels of ticagrelor and its active metabolite ARC124910XX by 25%–31% and the AUC by 22%–23% (P<0.05). However, morphine was not found to affect the pharmacodynamic effects of ticagrelor on platelet aggregation in whole blood, platelet plug formation, and vasodilator-stimulated phosphoprotein phosphorylation in healthy volunteers within 6 h after drug administration.17
Another RCT in patients with myocardial infarction showed that 5 mg of i.v. morphine could statistically significantly lower the AUC values of ticagrelor and its active metabolite by 36% and 37%, respectively, followed by a 180-mg loading dose, with a concomitant delay in ticagrelor Tmax (4 vs 2 h; P=0.004). The placebo group exhibited a stronger antiplatelet effect, whereas patients receiving morphine were observed with a greater prevalence of high platelet reactivity, indicating that morphine could delay and attenuate ticagrelor action.18
Taken together, RCTs investigating DDIs between morphine and P2Y12 inhibitors in true patients may reflect a realistic emergency scenario and overcome the limitation of RCTs performed in healthy volunteers under standardized conditions. The most likely mechanism for an appreciable delay in the onset of effect seems to be that morphine impairs gut absorption of orally administered P2Y12 receptor inhibitors by reducing gut motility via myenteric plexus mediated by μ-opioid receptors.19 It is necessary to develop strategies overcoming or at least diminishing the negative impact of morphine on the antiplatelet effect of oral P2Y12 receptor inhibitors in patients with acute myocardial infarction. i.v. cangrelor can provide a rapid onset and maximal platelet inhibition and thus might be an alternative of other oral P2Y12 antagonists for use in combination with morphine. Ticagrelor administered as a crushed tablet was bioequivalent to whole-tablet administration, but it took a shorter time to reach the peak plasma concentration (Tmax) compared to whole-tablet administration (1 vs 2 h) and an increased plasma concentration of ticagrelor and ARC124910XX at early time points was noted, which may offset the adverse effect of morphine on efficacy of oral P2Y12 receptor inhibitors.20 Such management should be evaluated in further head-to-head RCTs in true patients.
Morphine–gabapentin
An RCT in healthy male volunteers revealed a DDI between gabapentin 600 mg and controlled-release morphine 60 mg. The combination of morphine and gabapentin resulted in a significant increase in pain tolerance compared to morphine plus placebo. Pharmacokinetics of morphine and its glucuronides were not altered in the presence of gabapentin, whereas the AUC of gabapentin significantly increased by 44.4% (P<0.05) when morphine was coadministered.21 The pharmacokinetic changes in gabapentin may be due to increased absorption caused by reduced intestinal motility induced by morphine. Unlike the results of morphine–gabapentin DDI, another RCT confirmed that there was no significant pharmacokinetic DDI between morphine and gabapentin enacarbil, a prodrug of gabapentin designed for improving absorption by high-capacity transporters expressed throughout the intestine.22
Methadone–zidovudine
Zidovudine is primarily metabolized to an inactive glucuronide form via UGT enzymes. An RCT confirmed that methadone-maintained patients receiving standard zidovudine doses exhibited greater zidovudine exposure. Acute methadone treatment significantly increased oral and i.v. zidovudine AUC by 41% and 19%, respectively, while decreasing zidovudine clearance by approximately 20% for both routes. Chronic methadone treatment significantly decreased zidovudine clearance by 26% for both routes. These pharmacokinetic changes may be primarily due to the inhibition of zidovudine glucuronidation by methadone and the decreased renal clearance of zidovudine.23 It would be necessary to increase toxicity surveillance and reduce zidovudine dose when the two agents are given concomitantly. Another RCT showed that the opioid-dependent pharmacotherapeutic agent buprenorphine had no statistically significant effect on zidovudine disposition,24 and would not produce zidovudine toxicity. Therefore, buprenorphine may be used as an alternative to methadone to be coadministered with zidovudine.
Circumstances in which opioid is comedicated as an object drug
Rifampin–opioids
Morphine
An RCT in healthy male volunteers investigated the influence of rifampin 600 mg/day for 13 days on analgesic effects and pharmacokinetics of oral morphine 10 mg. Coadministration of rifampin considerably significantly reduced the mean AUC and Cmax of morphine by 27.7% and 40.7%, respectively, and consequently abolished the analgesic effect of morphine. Interestingly, rifampin treatment also reduced the AUC values and urinary recoveries of morphine-3-glucuronide and morphine-6-glucuronide,25 indicating that the rifampin–morphine DDI could not be attributed to induction of UGTs although rifampin is a potent UGT inducer and morphine is a UGT2B7 substrate. A hypothesis was proposed in the case report by Fudin et al; that is, rifampin-mediated intestinal P-gp induction diminished morphine absorption and ultimately decreased plasma free morphine and metabolites. Therapeutic regimen for a complex patient could be successfully titrated from oral morphine to a buprenorphine transdermal patch by balancing managed pain, history of drug abuse, completion of rifampin therapy, and a reasonable pain regimen upon discharge.26
Tramadol
An RCT in healthy subjects evaluated the effect of enzyme induction with rifampicin 600 mg once daily for 5 days on the pharmacokinetics and pharmacodynamics of oral (100 mg) and i.v. (50 mg) tramadol. Rifampicin significantly decreased the exposure to tramadol and M1 by 43%–59% after both oral and i.v. administration, increased the clearance of i.v. tramadol by 67%, and reduced bioavailability of oral tramadol from 66% to 49%.27 However, rifampicin pretreatment could not interfere with the pharmacological effects of tramadol in this sample population. Rifampicin’s induction of CYP3A4 and CYP2B6, rather than CYP2D6, is the most plausible explanation of the observed reduction in tramadol and M1 AUC and the shortened apparent half-life of M1.
Oxycodone
An RCT in healthy volunteers showed that oral rifampin 600 mg once daily for 7 days significantly decreased the AUC values of 0.1 mg/kg i.v. and oral oxycodone 15 mg by 53% and 86%, respectively, reduced oral bioavailability of oxycodone from 69% to 21%, and attenuated pharmacologic effects of oral oxycodone.28 Induction of CYP3A by rifampin is the underlying mechanism for pharmacokinetic changes. To maintain adequate analgesia, dose adjustment of oxycodone may be necessary when used in combination with rifampicin.
Methadone
An RCT in healthy volunteers showed that rifampin decreased bioavailability and plasma concentrations of methadone, and increased oral (1.7-fold) and i.v. (3.2-fold) clearance, CYP3A4-mediated 2-ethylidine-1,5-dimethyl-3,3-diphenylpyrrolidine (EDDP) formation clearances, and hepatic extraction (all P<0.05).29
Taken together, given that four RCTs have revealed clinically important pharmacokinetic interaction between rifampicin and morphine, tramadol, oxycodone, and methadone, it would seem prudent to avoid using this drug concomitantly with these four opioids in clinical settings. Regarding rifampicin–opioid combination, it is challenging to find an opioid analgesic for patients receiving rifampicin, since the major enzymes involved (CYPs other than CYP2D6, and UGTs) in the metabolism of all classes of opioids are significantly induced by rifampicin. Rifabutin and rifampicin are two rifamycins that are structurally similar and share mechanisms of action. Rifabutin is a less potent inducer than rifampicin. Evidence show that rifabutin could not produce any significant changes in the pharmacokinetics of methadone in HIV-infected former injecting drug users, and opiate withdrawal in buprenorphine-maintained patients.30,31 Further studies may be conducted to confirm whether rifabutin is an alternative to rifampicin coadministered with opioids.
Quinidine–opioids
Morphine
An RCT in normal healthy volunteers showed that oral quinidine altered subjective self-assessments of the effects of oral but not i.v. morphine. Quinidine 600 mg significantly increased the oral morphine Cmax by 88% and AUC by 60% rather than significantly affecting elimination rate.9 The underlying mechanism for such pharmacokinetic changes may be that morphine is a P-gp substrate and quinidine affects the absorption and bioavailability of orally administered morphine by inhibition of intestinal P-gp efflux.
Fentanyl
An RCT in normal healthy volunteers showed that quinidine 600 mg significantly increased oral fentanyl effects as measured by the change in dark-adapted pupil diameter (miosis) and subjective self-assessment, but it did not alter the magnitude or time to maximum miosis, time-specific pupil diameter, or subjective self-assessments after i.v. administration of fentanyl (2.5 μg/kg). Quinidine increased oral fentanyl plasma Cmax by 162% and AUC by 171% (both P<0.05) but had no effect on apparent elimination, suggesting that intestinal P-gp could affect the absorption and bioavailability of oral fentanyl.32 Quinidine had less effect on i.v. fentanyl pharmacodynamics, suggesting that P-gp has less effect on brain access of fentanyl.
Oxycodone
An RCT showed that premedication with 200 mg quinidine in healthy CYP2D6 extensive metabolizers (EMs) could prevent the production of active oxymorphone and significantly reduce plasma oxymorphone levels, without substantially affecting the pharmacodynamic effects of 20 mg controlled-release oxycodone (e.g., psychomotor or subjective drug effects, adverse effects).33 An RCT by Samer et al showed that blocking CYP2D6 (with quinidine) reduced Cmax of oxymorphone and noroxymorphone by 40% and 80%, and increased noroxycodone AUC by 70%. The magnitude of quinidine–oxycodone DDI depended on CYP2D6 genotype. The increase in noroxycodone exposure in the presence of quinidine was more pronounced in ultrarapid metabolizers (UMs) than EMs, and in EMs than in poor metabolizers (PMs).34
Codeine
An RCT in healthy subjects compared the pharmacokinetics and pharmacodynamics of codeine 120 mg with and without quinidine 100 mg between Caucasian and Chinese CYP2D6 EMs. Quinidine significantly reduced codeine O-demethylation in both ethnic groups; however, the extent of quinidine–codeine interaction was ethnically dependent. The absolute decrease was greater in Caucasians (115.8±25.9 vs 46.8±10.6 mL/min, respectively, P<0.03). Quinidine diminished morphine production by CYP2D6 inhibition and markedly reduced codeine’s analgesic, respiratory, psychomotor, and miotic effects (P<0.01) in the Caucasians rather than in the Chinese.35 Another RCT in drug-experienced but never-dependent subjects showed that short-term quinidine pretreatment (50 mg four times daily for 4 days) inhibited codeine O-demethylation more than did single-dose (50 mg) quinidine pretreatment (P<0.01).36
Dihydrocodeine
An RCT in healthy subjects showed that blockade of the CYP2D6-dependent metabolism of dihydrocodeine to dihydromorphine with quinidine 50 mg premedication significantly reduced the production of active metabolite dihydromorphine, but it did not attenuate the analgesic effects of a single dose of slow-release dihydrocodeine 60 mg. It indicated that the systemic morphine metabolites of dihydrocodeine are not of major importance for the analgesic effect of dihydrocodeine and that CYP2D6 PMs could gain similar analgesia as EMs.37 However, it is necessary to confirm these results with repeated dosing in patients with pain.
Methadone
An RCT in healthy subjects showed that quinidine 600 mg could increase the plasma concentrations of oral methadone 10 mg in the absorptive phase and the miosis caused by methadone, suggesting that intestinal P-gp could affect oral methadone absorption and hence its clinical effects. Quinidine had no effect on pharmacodynamics of i.v. methadone 10 mg, suggesting that P-gp seems unlikely to be a determinant of the access of methadone to the brain.38
Taken together, the mechanism for quinidine–opioids DDIs may involve P-gp inhibition (morphine, fentanyl, and methadone) or CYP2D6 inhibition (oxycodone, codeine). Combination use of quinidine and opioids is uncommon in clinical practice. Quinidine is more likely used as a known probe inhibitor of CYP2D6 and P-gp to investigate the possibility of DDI between opioid and inhibitor of CYP2D6 or P-gp.
Antimycotics–opioids
Buprenorphine
An RCT in healthy male volunteers revealed the DDI risk precipitated by voriconazole and posaconazole during therapy with sublingual buprenorphine. A 5-day oral pretreatment with voriconazole (400 mg twice daily on the first day and 200 mg twice daily thereafter) increased the mean AUC(0–infinity) of buprenorphine 1.80-fold (P<0.001), Cmax 1.37-fold (P<0.013), and half-life (t½) 1.37-fold (P<0.001). Posaconazole appeared to have a weaker DDI with buprenorphine compared to voriconazole because its treatment (400 mg twice daily for 5 days) increased the AUC0–∞ of buprenorphine 1.25-fold (P<0.001).39 The underlying DDI mechanism may be probably via inhibition of CYP3A4-mediated N-dealkylation of buprenorphine.
Another RCT in healthy subjects showed negligible effects of ketoconazole 200 mg administered twice daily on the pharmacokinetics of buprenorphine following the administration of a buprenorphine transdermal system 10 μg/h for 7 days, without apparent safety concerns with buprenorphine transdermal system–ketoconazole combination.40
Care should be exercised in the combined use of buprenorphine with triazole antimycotics, particularly with voriconazole. Metabolism of buprenorphine during therapy with buprenorphine transdermal system seems unlikely to be affected by coadministration of CYP3A4 inhibitors. It is interesting to further investigate the underlying mechanism for difference in extent of pharmacokinetic DDIs related with different formulation characteristics of buprenorphine and different triazole antimycotics.
Fentanyl
An RCT studied the interactions of i.v. fentanyl 5 μg/kg with voriconazole and fluconazole in healthy volunteers. The mean plasma clearance of i.v. fentanyl was decreased by 23% (P<0.05) and 16% (P<0.05) after administration of voriconazole (400 mg twice on the first day and 200 mg twice on the second day) and fluconazole (400 mg once on the first day and 200 mg once on the second day), respectively.41 Another RCT investigated the influence of CYP3A inhibition by ketoconazole 200 mg administered orally twice daily over 2 days on the single-dose pharmacokinetics of i.v. fentanyl 5 μg/kg in healthy volunteers. Results showed that ketoconazole could significantly increase fentanyl AUC(0–infinity) by 33% and reduce systemic clearance by 22%.42 However, another RCT showed neutral DDI between itraconazole (200 mg once daily for 4 days) and i.v. fentanyl (single dose of 3 μg/kg) in healthy volunteers.43
Caution should be exercised, especially in patients who are given ketoconazole, voriconazole, or fluconazole during long-lasting fentanyl treatment to avoid respiratory depression induced by insidiously elevated fentanyl concentration.
Morphine
An RCT revealed that itraconazole 200 mg once daily for 4 days could moderately increase plasma concentrations of oral 0.3 mg/kg morphine by 28% in healthy male volunteers, probably by enhancing its absorption by inhibiting intestinal wall P-gp. However, itraconazole did not significantly affect the pharmacokinetic variables of M3G or M6G or the pharmacodynamic effects of morphine.44
Oxycodone
An RCT assessed the effect of miconazole oral gel 85 mg administered three times daily (t.i.d.) on the pharmacokinetics of oral immediate-release oxycodone 10 mg in healthy volunteers. Pretreatment with miconazole oral gel caused a strong inhibition of the CYP2D6-dependent metabolism and moderate inhibition of the CYP3A4-dependent metabolism of oxycodone, resulting in increased exposure to oral oxycodone and moderate clinical relevance of the interaction.45
Another RCT investigated the effect of voriconazole on the pharmacokinetics and pharmacodynamics of single-dose oxycodone 10 mg in healthy subjects. Pretreatment of voriconazole (two doses of 400 mg twice daily, followed by five doses of 200 mg twice daily) drastically increased the mean AUC(0–infinity) of oxycodone by 3.6-fold, Cmax by 1.7-fold, and elimination half-life by 2.0-fold (P<0.001). The AUC(0–infinity) ratio of noroxycodone to oxycodone was decreased by 92% (P<0.001), and that of oxymorphone increased by 108% (P<0.01), indicating that inhibition of CYP3A-mediated oxycodone N-demethylation by voriconazole resulted in shunting to CYP2D6 pathway of the opioid. Pharmacodynamic effects of oxycodone were modestly increased by voriconazole.46 Lower doses of oxycodone may be needed during voriconazole comedication to avoid opioid-related adverse effects especially after repeated dosing.
Another RCT in healthy CYP2D6 EMs showed that pretreatment with ketoconazole increased the AUC of oral oxycodone (0.2 mg/kg) 2.0- to 3.0-fold compared with placebo or paroxetine. Decrease in pupil diameter induced by oxycodone together with placebo could be accentuated in the presence of ketoconazole. Ketoconazole, rather than paroxetine, increased the analgesic effect and adverse events (e.g., nausea, drowsiness, and pruritus) of oxycodone. Pharmacodynamic changes associated with CYP3A4 inhibition may be clinically important in patients treated with oxycodone.34
Methadone
An RCT in male patients on methadone therapy evaluated the pharmacokinetic interaction between voriconazole and methadone at steady state and the safety and tolerability profile during the coadministration. Voriconazole (200 mg twice daily administered following 400-mg twice-daily loading doses on the first day) increased the steady-state exposure of both methadone enantiomers, but the magnitude of increase in (S)-methadone exposure was twice as much as that of pharmacologically active enantiomer (R)-methadone. Methadone patients receiving voriconazole showed no signs or symptoms of significant opioid withdrawal or overdose.47
Another RCT determined the effect of 200 mg/day oral fluconazole on methadone disposition in volunteers receiving methadone maintenance therapy. Fluconazole pretreatment for 14 days significantly increased serum methadone AUC by 35%, and serum methadone peak and trough concentrations by 27% and 48%, respectively, and reduced oral clearance of methadone by 24%; however, patients cotreated with fluconazole did not exhibit signs or symptoms of significant narcotic overdose.48
Methadone has a wide therapeutic window, and therefore, the addition of voriconazole or fluconazole to methadone therapy is not likely to require adjustment of methadone dose. However, caution should be still exercised because cessation of voriconazole or fluconazole therapy may lead to withdrawal symptoms caused by a decrease in methadone concentrations after tolerance had developed to higher ones.
Tilidine
An RCT investigated the influence of the potent CYP2C19 and CYP3A4 inhibitor voriconazole (400 mg) on the pharmacokinetics and analgesic effects of tilidine 100 mg in healthy volunteers. Voriconazole caused a 20-fold increase in serum tilidine AUC and delayed the onset of analgesic activity. Also, it significantly inhibited the sequential metabolism of tilidine and increased exposure of the active nortilidine. Combination use of voriconazole and tilidine almost doubled the incidence of adverse events.49
Oral tramadol
An RCT investigated the possible interaction of tramadol with the antifungal agents terbinafine (CYP2D6 inhibitor) and itraconazole (CYP3A4 inhibitor) in healthy subjects including eight CYP2D6 EMs and four CYP2D6 UMs. Terbinafine 250 mg once daily for 4 days increased the AUC(0–infinity), Cmax, and elimination half-life of oral tramadol 50 mg by 115%, 53%, and 48%, respectively (P<0.001), decreased the AUC(0–infinity), Cmax, and elimination half-life of M1 by 64%, 79%, and 50%, respectively (P<0.001). Terbinafine reduced subjective drug effect of tramadol (P<0.001). In contrast, itraconazole had minor effects on tramadol pharmacokinetics.50
Protease inhibitors–opioids
Oxycodone
An RCT investigated the effect of ritonavir 300 mg and lopinavir/ritonavir 400/100 mg administered for 4 days on the pharmacokinetics and pharmacodynamics of oral oxycodone 10 mg in healthy volunteers. Ritonavir and lopinavir/ritonavir strongly inhibited the CYP3A-mediated N-demethylation of oxycodone, and significantly increased the AUC of oxycodone by 3.0-fold and 2.6-fold, the elimination half-life, and the self-reported drug effect (all P<0.001).51 Reductions in oxycodone dose may be needed to avoid opioid-related adverse effects when oxycodone is used clinically in patients during ritonavir and lopinavir/ritonavir treatment.
Fentanyl
An RCT investigated possible interactions between multidose ritonavir and i.v. fentanyl 5 μg/kg in healthy volunteers. By inhibiting the CYP3A4-mediated metabolism of fentanyl, ritonavir reduced fentanyl clearance by 67% (P<0.01), increased the AUC(0–18 h) by 81% (P<0.01), and prolonged elimination half-life from 9.4±4.6 to 20.1±8.4 h (P<0.01).52 Caution should be exercised if fentanyl is given to patients receiving ritonavir therapy.
Tilidine
An RCT in healthy volunteers revealed that ritonavir treatment (300 mg twice daily) for 3 days altered the sequential metabolism of tilidine, substantially reducing the partial metabolic clearances of tilidine to nortilidine and nortilidine to bisnortilidine and resulting in a 2.0-fold increase in the exposure of the active nortilidine.53
Taken together, three RCTs investigated DDIs between protease inhibitors and opioids (i.e., oxycodone, fentanyl, and tilidine), involving the mechanism of CYP3A4 inhibition.
Grapefruit juice–opioids
Grapefruit juice is a selective intestinal CYP3A and P-gp inhibitor. There were three RCTs investigating DDIs between grapefruit juice and opioids, involving the mechanism of CYP3A inhibition.
Oral oxycodone
An RCT in healthy volunteers examined the effect of grapefruit juice 200 mL t.i.d. taken for 5 days on the pharmacokinetics and pharmacodynamics of oral oxycodone 10 mg. Grapefruit juice statistically significantly increased the AUC(0–infinity) by 1.7-fold, the Cmax by 1.5-fold, and the half-life of oxycodone by 1.2-fold as compared to water. The formation of CYP3A4-dependent metabolites noroxycodone and noroxymorphone decreased, while CYP2D6-mediated oxymorphone formation increased in the presence of grapefruit juice. These pharmacokinetic changes were accompanied by modest alterations in the pharmacodynamic effects of oxycodone (e.g., the impairing effect of oxycodone on self-rated performance was enhanced, whereas the analgesic effect of oxycodone in the cold pressor test was not affected).54
Fentanyl
An RCT in healthy volunteers revealed that peak fentanyl concentrations, time to peak, AUC, and maximum pupil diameter change from baseline following administration of oral transmucosal fentanyl citrate were minimally affected by grapefruit juice.55 When treating breakthrough pain, with careful attention to maximal mucosal absorption and minimal swallowing, CYP3A-related drug interactions are unlikely to affect the onset or magnitude of oral transmucosal fentanyl analgesia.
Methadone
An RCT in healthy volunteers showed that grapefruit juice decreased the EDDP/methadone AUC ratio by 48% (P<0.05) after oral methadone but not i.v. methadone administration, with minimal effect on plasma concentrations and clearance of methadone following oral and i.v. administration.29
Antidepressants–opioids
Paroxetine–tramadol
An RCT in healthy CYP2D6 EMs showed that potent CYP2D6 inhibitor paroxetine was a dose-dependent dilator of the pupil and a dose-dependent inhibitor of (+)-tramadol’s O-demethylation.56 Another RCT in healthy CYP2D6 EMs investigated the influence of paroxetine pretreatment (20 mg daily for 3 days) on the biotransformation and the hypoalgesic effect of tramadol 150 mg. Paroxetine pretreatment increased the AUC of (+)- and (−)-tramadol by 37% and 32%, respectively, and decreased the AUC of (+)- and (−)-M1 by 67% and 40%, respectively. The hypoalgesic effect of tramadol was reduced by paroxetine in human experimental pain models, particularly in opioid-sensitive tests.57
Paroxetine–hydrocodone
An RCT in healthy subjects showed that coadministration of paroxetine 20 mg once daily for 12 days did not alter systemic exposure to hydrocodone and occurrence of adverse events following single-dose once-daily administration of extended-release formulation of hydrocodone bitartrate 20 mg, suggesting that hydrocodone can be coadministered with CYP2D6 inhibitors at therapeutic doses without dosage adjustment.58
Paroxetine–oxycodone
Two RCTs determined whether inhibition of CYP2D6 alone by paroxetine (20 mg once daily) or inhibition of both CYP2D6 and CYP3A4 by a combination of paroxetine (20 mg once daily) and itraconazole (200 mg once daily) administered for 5 days could alter the pharmacokinetics of and pharmacological response to i.v. oxycodone 0.1 mg/kg or oral immediate-release oxycodone 10 mg in healthy subjects. The effect of paroxetine on the plasma concentrations of i.v. oxycodone was negligible, whereas the combination of paroxetine and itraconazole prolonged the mean elimination half-life of i.v. oxycodone from 3.8 to 6.6 h (P<0.001) and increased the exposure to i.v. oxycodone 2.0-fold (P< 0.001), but did not result in pharmacodynamic changes.59
Paroxetine alone reduced the AUC(0–48 h) of the CYP2D6-dependent metabolite oxymorphone by 44% (P<0.05), but had no significant effects on the plasma concentrations of oxycodone or its pharmacological effects following oral administration of oxycodone in healthy volunteers. Pretreatment of paroxetine and itraconazole increased the mean AUC(0–infinity) of oxycodone by 2.9-fold (P<0.001), its Cmax by 1.8-fold (P<0.001), and visual analog scores for subjective drug effects, drowsiness, and deterioration of performance (P<0.05).60 It indicates that inhibition of both CYP3A4 and 2D6, rather than only CYP2D6, could precipitate substantial pharmacokinetic interactions with i.v. and oral oxycodone.
Escitalopram–tramadol
An RCT investigated the pharmacokinetic and pharmacodynamic effect of escitalopram 20 mg/day on tramadol 150 mg in healthy subjects. Escitalopram could significantly decrease the median AUC(0–infinity) of (+)-M1 by 29%, but it did not impair the analgesic effect of tramadol assessed by the cold pressor test.61 The underlying mechanism may be that escitalopram is a weak inhibitor of CYP2D6 whereas paroxetine is a strong CYP2D6 inhibitor.
Amantadine–morphine
Amantadine is a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist and can decrease pain and analgesic requirements. An RCT in patients undergoing radical prostatectomy showed that perioperative oral amantadine (200 mg on the evening before surgery, 200 mg at 1 h before surgery, and 100 mg at 8, 20, and 32 h after surgery) significantly reduced four outcome measures (i.e., plasma clearance of morphine at 22–24 h after surgery, cumulative morphine consumption for i.v. patient-controlled analgesia up to 48 h after surgery, intense mechanical sensitivity around the surgical wound, and incidence of bladder spasm pain). Lower postoperative morphine requirements may be due to a pharmacokinetic interaction between amantadine and morphine resulting in inhibition of the 3-glucuronidation of morphine, although additional pharmacodynamic effects involving the NMDA receptor may also be involved.62 Another RCT confirmed the beneficial effect of perioperative administration of amantadine (50 or 100 mg 1 h before surgery and 8, 20, and 32 h after operation) on postoperative pain, but it did not support the pharmacokinetic DDI mechanism because action of amantadine was not associated with significant changes in plasma levels of morphine, morphine-3-glucuronide and morphine-6-glucuronide. The difference between the findings derived from the two RCTs may be due to the amantadine dose difference.63
Sumatriptan–butorphanol nasal sprays
Sumatriptan and butorphanol nasal sprays are commonly used agents for the management of migraine headaches. An RCT in healthy subjects showed that plasma concentrations of butorphanol were reduced when it was administered 1 min, but not 30 min, after sumatriptan, whereas the pharmacokinetics of sumatriptan were not substantially altered by butorphanol. The analgesic effect of butorphanol may be diminished due to reduced nasal absorption resulting from probable transient vasoconstriction of nasal blood vessels by sumatriptan if butorphanol nasal spray is administered <30 min after sumatriptan nasal spray.64
Metoclopramide–morphine
An RCT in patients undergoing gynecological, orthopedic, and general surgical procedures revealed that concurrent administration of metoclopramide 10 mg and oral controlled-release morphine 20 mg resulted in significantly shorter Tmax of morphine, faster onset, and higher level of sedation in patients compared with ingestion of oral controlled-release morphine alone.65 The enhanced rate of morphine absorption may be due to increase in gastric emptying by metoclopramide.
Ticlopidine–tramadol
An RCT assessed possible drug interactions of tramadol 50 mg after 4 days of pretreatment with ticlopidine (250 mg twice daily) alone or along with itraconazole (200 mg once daily) in healthy subjects. Results showed that ticlopidine could significantly increase the mean AUC(0–infinity) of tramadol by 2.0-fold and Cmax by 1.4-fold, reduce its oral and renal clearance, and inhibit the formation of active M1 from tramadol. The ticlopidine–tramadol pharmacokinetic DDI is most likely due to inhibition of CYP2B6 and/or CYP2D6 by ticlopidine. Itraconazole has no marked effect on the ticlopidine–tramadol interaction.66 Concomitant use of ticlopidine and tramadol may enhance the risk of serotonergic effects, especially when higher doses of tramadol are used.
St John’s wort–oral oxycodone
Combination use of opioids and herbal antidepressant St John’s wort is possible because chronic pain is usually associated with depression. An RCT in healthy participants assessed the effects of a high hyperforin-containing product of St John’s wort 300 mg t.i.d. for 15 days on the CYP3A-mediated metabolism and behavioral and analgesic effects of oral oxycodone hydrochloride 15 mg. Results showed that St John’s wort could significantly reduce oxycodone AUC by 50%, shorten elimination half-life, and decrease the self-reported drug effect of oxycodone despite no alternation in cold pain threshold.67 Due to risk of subtherapeutic concentrations of oxycodone, attention should be drawn to the concomitant use of St John’s wort and oxycodone. Dose titration may be needed when St John’s wort treatment is added or discontinued. Further studies are needed to extend the results to chronic pain patients receiving oxycodone or verify the findings when low hyperforin-containing St John’s wort extracts are comedicated.
Macrolides/ketolides–oxycodone
There were two RCTs identified to address macrolides/ketolides–opioid DDIs. Liukas et al investigated the effect of CYP3A4 inhibitor clarithromycin 500 mg administered twice daily for 5 days on the pharmacokinetics and pharmacodynamics of oral oxycodone 10 mg in young and elderly subjects. Clarithromycin significantly decreased the apparent clearance of oxycodone by approximately 50%, prolonged its elimination half-life, and increased the mean AUC0–∞ of oxycodone by approximately 2.0-fold; however, it did not significantly alter the pharmacological response to oxycodone.68
An RCT in healthy subjects showed that oral telithromycin 800 mg administered for 4 days could significantly increase the oxycodone AUC(0–infinity) and reduce the noroxycodone AUC(0–infinity) by 46% following administration of immediate-release oxycodone 10 mg while modestly enhancing pharmacodynamic effects of oxycodone. Telithromycin clearly reduced the N-demethylation of oxycodone to noroxycodone by inhibiting CYP3A4.69
To avoid adverse effects when oxycodone is used concomitantly with clarithromycin or telithromycin, it might be appropriate to cut down oxycodone dose by 25%–50% followed by readjustment according to the clinical response.
Levomepromazine–codeine
An RCT in patients hospitalized for acute back pain revealed that the addition of low-dose neuroleptic levomepromazine could significantly inhibit the CYP2D6-mediated O-demethylation of codeine to morphine in homozygous CYP2D6 EMs receiving codeine/paracetamol (P<0.05).70 In patients prescribed this drug combination, the amount of morphine generated by the O-demethylation of codeine may be insufficient for effective pain relief.
Further research opportunities
First, the clinical validity or relevance of the DDIs between opioid analgesics and some comedications should be further addressed in patients receiving repeated doses of regimens.
Second, it is necessary to investigate the role of transporters (e.g., P-gp) and DDIs in mediating brain access and biodistribution of opioid analgesics because the underlying mechanism is still unclear for explaining results of some DDIs with significant pharmacokinetic changes rather than pharmacodynamic changes with clinical relevance. Also, it is interesting to explore the DDIs mediated by drug transporters other than P-gp. Tzvetkov et al observed that OCT1 plays an important role in the hepatocellular uptake of morphine and OCT1-mediated morphine uptake could be strongly inhibited by DDIs with irinotecan, verapamil, and ondansetron in HEK293 cells overexpressing human OCT1.12 However, OCT1-relevant clinical DDI study of opioid analgesic has not been available.
Third, the factors determining the degree of DDI should be addressed in more studies, and they are as follows: (1) Does the precipitant drug exhibit dose-dependent modulatory effects on pharmacokinetics and pharmacodynamics of the object drug? Paroxetine–tramadol DDI is a good example listed in this review.56 (2) Does different administration route or administration strategy affect the consequences of DDIs? DDI between grapefruit juice and oral rather than i.v. methadone29 and DDI between quinidine and oral rather than i.v. fentanyl32 are two good examples listed in this review. (3) Are DDI magnitudes ethnicity- or genotype-dependent? Quinidine–codeine DDI and quinidine–oxycodone DDI exhibiting the effect of CYP2D6 genotype on DDI magnitude are two good examples listed in this review.34,35 (4) Does the extraction ratio of opioid analgesics affect the extent of opioid-related DDIs? Extraction ratio is a measure of the organ’s relative efficiency in eliminating the drug from the systemic circulation over a single pass through the organ. Medications with a high extraction ratio are identified as having an extraction ratio of >0.7, while an intermediate has between 0.3 and 0.7 and a low has <0.3. Morphine, fentanyl, and buprenorphine are categorized as high-extraction ratio agents, while methadone is of a low-extraction ratio and codeine is an intermediate-hepatic-extraction ratio agent.71 Ibrahim et al observed that CYP3A inhibition could decrease alfentanil clearance more than fentanyl clearance and confirmed that high-extraction fentanyl would be theoretically less vulnerable to CYP3A4-mediated DDIs.72 However, there is little literature in this field. (5) Is there a difference in DDI strength observed in different types of subjects? RCTs investigating in true patients may reflect a realistic clinical scenario and overcome the limitation of RCTs performed in healthy volunteers under standardized conditions. The impact of morphine on the antiplatelet effect of ticagrelor observed in patients with acute myocardial infarction rather than in healthy volunteers was a good example. However, the results of opioid-related pharmacokinetic DDIs derived from RCTs even in healthy participants would enhance awareness of medication therapy management among clinicians. (6) The DDI strength would increase when an opioid is coadministered with inhibitors of multiple metabolizing enzymes primarily responsible for its biotransformation. For example, combination of paroxetine (CYP2D6 inhibitor) and itraconazole (CYP3A4 inhibitor), rather than paroxetine itself, produces significant DDI with oxycodone which undergoes CYP3A4-mediated N-demethylation to inactive metabolite noroxycodone and CYP2D6-mediated O-demethylation to active metabolite oxymorphone.59,60
Fourth, it is necessary to conduct more studies seeking alternatives to medications which exhibit significant DDI potential. Evidence derived from RCTs included in this review were as follows: (1) morphine has a significant DDI with gabapentin rather than gabapentin enacarbil;21,22 (2) itraconazole rather than terbinafine has minimal effects on tramadol pharmacokinetics;50 (3) escitalopram rather than paroxetine less likely affects the pharmacokinetic and pharmacodynamic effect of tramadol;57,61 (4) buprenorphine rather than methadone has minimal effect on zidovudine disposition;23,24 (5) buprenorphine transdermal system rather than sublingual buprenorphine seems unlikely to be affected by coadministration of CYP3A4 inhibitors;39,40 and (6) tramadol rather than extended-release formulation of hydrocodone bitartrate and oxycodone interact with paroxetine.57–60
Conclusion
In this review, we specifically addressed the opioid analgesics-related pharmacokinetic DDIs from the perspective of RCTs, presenting interesting research findings from recent years, such as the circumstances under which DDIs occur when opioid is the precipitant drug, the factors determining the magnitude of DDI, and the corresponding clinical risk management. None of the opioid–drug/herb pairs was listed as contraindications of opioids involved in this review. Effective and safe combination therapy of opioids can be achieved by promoting the awareness of potential changes in therapeutic efficacy and toxicities, prescribing alternatives or changing administration strategy, tailoring dose, reviewing the appropriateness of orders, and paying attention to medication monitoring.
Acknowledgments
This work was supported by the Zhejiang Provincial Bureau of Education (grant N20140209). The authors would also like to thank Professor Yong-bin Zhu (vice dean of Division of Medical Administration), Mr Guang-wu Li (inpatient pharmacy supervisor), and Ye-zhen Lu (clinical coordinator) for their kind help in medication management and use of opioid analgesics.
Disclosure
The authors report no conflicts of interest in this work.
References
Pergolizzi JV Jr, Ma L, Foster DR, et al. The prevalence of opioid-related major potential drug-drug interactions and their impact on health care costs in chronic pain patients. J Manag Care Spec Pharm. 2014;20(5):467–476. | ||
Summers KH, Puenpatom RA, Rajan N, Ben-Joseph R, Ohsfeldt R. Economic impact of potential drug-drug interactions in opioid analgesics. J Med Econ. 2011;14(4):390–396. | ||
Akobeng AK. Understanding randomised controlled trials. Arch Dis Child. 2005;90(8):840–844. | ||
Kotlinska-Lemieszek A, Klepstad P, Haugen DF. Clinically significant drug-drug interactions involving opioid analgesics used for pain treatment in patients with cancer: a systematic review. Drug Des Devel Ther. 2015;9:5255–5267. | ||
Overholser BR, Foster DR. Opioid pharmacokinetic drug-drug interactions. Am J Manag Care. 2011;17 (Suppl 11):S276–S287. | ||
Gharavi R, Hedrich W, Wang H, Hassan HE. Transporter-mediated disposition of opioids: implications for clinical drug interactions. Pharm Res. 2015;32(8):2477–2502. | ||
Mercadante S. Opioid metabolism and clinical aspects. Eur J Pharmacol. 2015;769:71–78. | ||
Mercer SL, Coop A. Opioid analgesics and P-glycoprotein efflux transporters: a potential systems-level contribution to analgesic tolerance. Curr Top Med Chem. 2011;11(9):1157–1164. | ||
Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of P-glycoprotein in the intestinal absorption and clinical effects of morphine. Clin Pharmacol Ther. 2003;74(6):543–554. | ||
Park HJ, Shinn HK, Ryu SH, Lee HS, Park CS, Kang JH. Genetic polymorphisms in the ABCB1 gene and the effects of fentanyl in Koreans. Clin Pharmacol Ther. 2007;81(4):539–546. | ||
Zwisler ST, Enggaard TP, Noehr-Jensen L, et al. The antinociceptive effect and adverse drug reactions of oxycodone in human experimental pain in relation to genetic variations in the OPRM1 and ABCB1 genes. Fundam Clin Pharmacol. 2010;24(4):517–524. | ||
Tzvetkov MV, dos Santos Pereira JN, Meineke I, Saadatmand AR, Stingl JC, Brockmöller J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem Pharmacol. 2013;86(5):666–678. | ||
Tzvetkov MV, Saadatmand AR, Lötsch J, Tegeder I, Stingl JC, Brockmöller J. Genetically polymorphic OCT1: another piece in the puzzle of the variable pharmacokinetics and pharmacodynamics of the opioidergic drug tramadol. Clin Pharmacol Ther. 2011;90(1):143–150. | ||
Li W, Zeng S, Yu LS, Zhou Q. Pharmacokinetic drug interaction profile of omeprazole with adverse consequences and clinical risk management. Ther Clin Risk Manag. 2013;9:259–271. | ||
Hobl EL, Stimpfl T, Ebner J, et al. Morphine decreases clopidogrel concentrations and effects: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2014;63(7):630–635. | ||
Hobl EL, Reiter B, Schoergenhofer C, et al. Morphine interaction with prasugrel: a double-blind, cross-over trial in healthy volunteers. Clin Res Cardiol. 2016;105(4):349–355. | ||
Hobl EL, Reiter B, Schoergenhofer C, et al. Morphine decreases ticagrelor concentrations but not its antiplatelet effects: a randomized trial in healthy volunteers. Eur J Clin Invest. 2016;46(1):7–14. | ||
Kubica J, Adamski P, Ostrowska M, et al. Morphine delays and attenuates ticagrelor exposure and action in patients with myocardial infarction: the randomized, double-blind, placebo-controlled IMPRESSION trial. Eur Heart J. 2016;37(3):245–252. | ||
Kubica J, Kubica A, Jilma B, et al. Impact of morphine on antiplatelet effects of oral P2Y12 receptor inhibitors. Int J Cardiol. 2016;215:201–208. | ||
Teng R, Carlson G, Hsia J. An open-label, randomized bioavailability study with alternative methods of administration of crushed ticagrelor tablets in healthy volunteers. Int J Clin Pharmacol Ther. 2015;53(2):182–189. | ||
Eckhardt K, Ammon S, Hofmann U, Riebe A, Gugeler N, Mikus G. Gabapentin enhances the analgesic effect of morphine in healthy volunteers. Anesth Analg. 2000;91(1):185–191. | ||
Chen C, Upward J, Arumugham T, Stier B, Davy M. Gabapentin enacarbil and morphine administered in combination versus alone: a double-blind, randomized, pharmacokinetic, and tolerability comparison. Clin Ther. 2015;37(2):349–357. | ||
McCance-Katz EF, Rainey PM, Jatlow P, Friedland G. Methadone effects on zidovudine disposition (AIDS Clinical Trials Group 262). J Acquir Immune Defic Syndr Hum Retrovirol. 1998;18(5):435–443. | ||
McCance-Katz EF, Rainey PM, Friedland G, Kosten TR, Jatlow P. Effect of opioid dependence pharmacotherapies on zidovudine disposition. Am J Addict. 2001;10(4):296–307. | ||
Fromm MF, Eckhardt K, Li S, et al. Loss of analgesic effect of morphine due to coadministration of rifampin. Pain. 1997;72(1–2):261–267. | ||
Fudin J, Fontenelle DV, Payne A. Rifampin reduces oral morphine absorption: a case of transdermal buprenorphine selection based on morphine pharmacokinetics. J Pain Palliat Care Pharmacother. 2012;26(4):362–367. | ||
Saarikoski T, Saari TI, Hagelberg NM, et al. Rifampicin markedly decreases the exposure to oral and intravenous tramadol. Eur J Clin Pharmacol. 2013;69(6):1293–1301. | ||
Nieminen TH, Hagelberg NM, Saari TI, et al. Rifampin greatly reduces the plasma concentrations of intravenous and oral oxycodone. Anesthesiology. 2009;110(6):1371–1378. | ||
Kharasch ED, Hoffer C, Whittington D, Sheffels P. Role of hepatic and intestinal cytochrome P450 3A and 2B6 in the metabolism, disposition, and miotic effects of methadone. Clin Pharmacol Ther. 2004;76(3):250–269. | ||
Brown LS, Sawyer RC, Li R, Cobb MN, Colborn DC, Narang PK. Lack of a pharmacologic interaction between rifabutin and methadone in HIV-infected former injecting drug users. Drug Alcohol Depend. 1996;43(1–2):71–77. | ||
McCance-Katz EF, Moody DE, Prathikanti S, Friedland G, Rainey PM. Rifampin, but not rifabutin, may produce opiate withdrawal in buprenorphine-maintained patients. Drug Alcohol Depend. 2011;118(2–3):326–334. | ||
Kharasch ED, Hoffer C, Altuntas TG, Whittington D. Quinidine as a probe for the role of p-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44(3):224–233. | ||
Heiskanen T, Olkkola KT, Kalso E. Effects of blocking CYP2D6 on the pharmacokinetics and pharmacodynamics of oxycodone. Clin Pharmacol Ther. 1998;64(6):603–611. | ||
Samer CF, Daali Y, Wagner M, et al. The effects of CYP2D6 and CYP3A activities on the pharmacokinetics of immediate release oxycodone. Br J Pharmacol. 2010;160(4):907–918. | ||
Caraco Y, Sheller J, Wood AJ. Impact of ethnic origin and quinidine coadministration on codeine’s disposition and pharmacodynamic effects. J Pharmacol Exp Ther. 1999;290(1):413–422. | ||
Kathiramalainathan K, Kaplan HL, Romach MK, et al. Inhibition of cytochrome P450 2D6 modifies codeine abuse liability. J Clin Psychopharmacol. 2000;20(4):435–444. | ||
Wilder-Smith CH, Hufschmid E, Thormann W. The visceral and somatic antinociceptive effects of dihydrocodeine and its metabolite, dihydromorphine. A cross-over study with extensive and quinidine-induced poor metabolizers. Br J Clin Pharmacol. 1998;45(6):575–581. | ||
Kharasch ED, Hoffer C, Whittington D. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. Br J Clin Pharmacol. 2004;57(5):600–610. | ||
Fihlman M, Hemmilä T, Hagelberg NM, et al. Voriconazole more likely than posaconazole increases plasma exposure to sublingual buprenorphine causing a risk of a clinically important interaction. Eur J Clin Pharmacol. 2016;72(11):1363–1371. | ||
Kapil RP, Cipriano A, Michels GH, et al. Effect of ketoconazole on the pharmacokinetic profile of buprenorphine following administration of a once-weekly buprenorphine transdermal system. Clin Drug Investig. 2012;32(9):583–592. | ||
Saari TI, Laine K, Neuvonen M, Neuvonen PJ, Olkkola KT. Effect of voriconazole and fluconazole on the pharmacokinetics of intravenous fentanyl. Eur J Clin Pharmacol. 2008;64(1):25–30. | ||
Ziesenitz VC, König SK, Mahlke NS, Skopp G, Haefeli WE, Mikus G. Pharmacokinetic interaction of intravenous fentanyl with ketoconazole. J Clin Pharmacol. 2015;55(6):708–717. | ||
Palkama VJ, Neuvonen PJ, Olkkola KT. The CYP 3A4 inhibitor itraconazole has no effect on the pharmacokinetics of i.v. fentanyl. Br J Anaesth. 1998;81(4):598–600. | ||
Heiskanen T, Backman JT, Neuvonen M, Kontinen VK, Neuvonen PJ, Kalso E. Itraconazole, a potent inhibitor of P-glycoprotein, moderately increases plasma concentrations of oral morphine. Acta Anaesthesiol Scand. 2008;52(10):1319–1326. | ||
Grönlund J, Saari TI, Hagelberg N, Neuvonen PJ, Olkkola KT, Laine K. Miconazole oral gel increases exposure to oral oxycodone by inhibition of CYP2D6 and CYP3A4. Antimicrob Agents Chemother. 2011;55(3):1063–1067. | ||
Hagelberg NM, Nieminen TH, Saari TI, et al. Voriconazole drastically increases exposure to oral oxycodone. Eur J Clin Pharmacol. 2009;65(3):263–271. | ||
Liu P, Foster G, Labadie R, Somoza E, Sharma A. Pharmacokinetic interaction between voriconazole and methadone at steady state in patients on methadone therapy. Antimicrob Agents Chemother. 2007;51(1):110–118. | ||
Cobb MN, Desai J, Brown LS Jr, Zannikos PN, Rainey PM. The effect of fluconazole on the clinical pharmacokinetics of methadone. Clin Pharmacol Ther. 1998;63(6):655–662. | ||
Grün B, Krautter S, Riedel KD, Mikus G. Inhibition of the active principle of the weak opioid tilidine by the triazole antifungal voriconazole. Br J Clin Pharmacol. 2009;68(5):712–720. | ||
Saarikoski T, Saari TI, Hagelberg NM, et al. Effects of terbinafine and itraconazole on the pharmacokinetics of orally administered tramadol. Eur J Clin Pharmacol. 2015;71(3):321–327. | ||
Nieminen TH, Hagelberg NM, Saari TI, et al. Oxycodone concentrations are greatly increased by the concomitant use of ritonavir or lopinavir/ritonavir. Eur J Clin Pharmacol. 2010;66(10):977–985. | ||
Olkkola KT, Palkama VJ, Neuvonen PJ. Ritonavir’s role in reducing fentanyl clearance and prolonging its half-life. Anesthesiology. 1999;91(3):681–685. | ||
Grün B, Merkel U, Riedel KD, Weiss J, Mikus G. Contribution of CYP2C19 and CYP3A4 to the formation of the active nortilidine from the prodrug tilidine. Br J Clin Pharmacol. 2012;74(5):854–863. | ||
Nieminen TH, Hagelberg NM, Saari TI, et al. Grapefruit juice enhances the exposure to oral oxycodone. Basic Clin Pharmacol Toxicol. 2010;107(4):782–788. | ||
Kharasch ED, Whittington D, Hoffer C. Influence of hepatic and intestinal cytochrome P4503A activity on the acute disposition and effects of oral transmucosal fentanyl citrate. Anesthesiology. 2004;101(3):729–737. | ||
Nielsen AG, Pedersen RS, Noehr-Jensen L, Damkier P, Brosen K. Two separate dose-dependent effects of paroxetine: mydriasis and inhibition of tramadol’s O-demethylation via CYP2D6. Eur J Clin Pharmacol. 2010;66(7):655–660. | ||
Laugesen S, Enggaard TP, Pedersen RS, Sindrup SH, Brøsen K. Paroxetine, a cytochrome P450 2D6 inhibitor, diminishes the stereoselective O-demethylation and reduces the hypoalgesic effect of tramadol. Clin Pharmacol Ther. 2005;77(4):312–323. | ||
Kapil RP, Friedman K, Cipriano A, et al. Effects of paroxetine, a CYP2D6 inhibitor, on the pharmacokinetic properties of hydrocodone after coadministration with a single-entity, once-daily, extended-release hydrocodone tablet. Clin Ther. 2015;37(10):2286–2296. | ||
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Laine K, Olkkola KT. Effect of inhibition of cytochrome P450 enzymes 2D6 and 3A4 on the pharmacokinetics of intravenous oxycodone: a randomized, three-phase, crossover, placebo-controlled study. Clin Drug Investig. 2011;31(3):143–153. | ||
Grönlund J, Saari TI, Hagelberg NM, Neuvonen PJ, Olkkola KT, Laine K. Exposure to oral oxycodone is increased by concomitant inhibition of CYP2D6 and 3A4 pathways, but not by inhibition of CYP2D6 alone. Br J Clin Pharmacol. 2010;70(1):78–87. | ||
Noehr-Jensen L, Zwisler ST, Larsen F, Sindrup SH, Damkier P, Brosen K. Escitalopram is a weak inhibitor of the CYP2D6-catalyzed O-demethylation of (+)-tramadol but does not reduce the hypoalgesic effect in experimental pain. Clin Pharmacol Ther. 2009;86(6):626–633. | ||
Snijdelaar DG, Koren G, Katz J. Effects of perioperative oral amantadine on postoperative pain and morphine consumption in patients after radical prostatectomy: results of a preliminary study. Anesthesiology. 2004;100(1):134–141. | ||
Bujak-Giżycka B, Kącka K, Suski M, et al. Beneficial effect of amantadine on postoperative pain reduction and consumption of morphine in patients subjected to elective spine surgery. Pain Med. 2012;13(3):459–465. | ||
Vachharajani NN, Shyu WC, Nichola PS, Boulton DW. A pharmacokinetic interaction study between butorphanol and sumatriptan nasal sprays in healthy subjects: importance of the timing of butorphanol administration. Cephalalgia. 2002;22(4):282–287. | ||
Manara AR, Shelly MP, Quinn K, Park GR. The effect of metoclopramide on the absorption of oral controlled release morphine. Br J Clin Pharmacol. 1988;25(4):518–521. | ||
Hagelberg NM, Saarikoski T, Saari TI, et al. Ticlopidine inhibits both O-demethylation and renal clearance of tramadol, increasing the exposure to it, but itraconazole has no marked effect on the ticlopidine-tramadol interaction. Eur J Clin Pharmacol. 2013;69(4):867–875. | ||
Nieminen TH, Hagelberg NM, Saari TI, et al. St John’s wort greatly reduces the concentrations of oral oxycodone. Eur J Pain. 2010;14(8):854–859. | ||
Liukas A, Hagelberg NM, Kuusniemi K, Neuvonen PJ, Olkkola KT. Inhibition of cytochrome P450 3A by clarithromycin uniformly affects the pharmacokinetics and pharmacodynamics of oxycodone in young and elderly volunteers. J Clin Psychopharmacol. 2011;31(3):302–308. | ||
Grönlund J, Saari T, Hagelberg N, et al. Effect of telithromycin on the pharmacokinetics and pharmacodynamics of oral oxycodone. J Clin Pharmacol. 2010;50(1):101–108. | ||
Vevelstad M, Pettersen S, Tallaksen C, Brørs O. O-demethylation of codeine to morphine inhibited by low-dose levomepromazine. Eur J Clin Pharmacol. 2009;65(8):795–801. | ||
Gelot S, Nakhla E. Opioid dosing in renal and hepatic impairment. US Pharm. 2014;39(8):34–38. | ||
Ibrahim AE, Feldman J, Karim A, Kharasch ED. Simultaneous assessment of drug interactions with low- and high-extraction opioids: application to parecoxib effects on the pharmacokinetics and pharmacodynamics of fentanyl and alfentanil. Anesthesiology. 2003;98(4):853–861. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.