")
Back to Journals » Research Reports in Clinical Cardiology » Volume 11
Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia: Mechanisms and Management
Authors AlTurki A, Alotaibi B , Joza J, Proietti R
Received 26 November 2019
Accepted for publication 5 March 2020
Published 18 March 2020 Volume 2020:11 Pages 19—29
DOI https://doi.org/10.2147/RRCC.S198185
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Kones
Ahmed AlTurki,1 Bader Alotaibi,1 Jacqueline Joza,1 Riccardo Proietti2
1Division of Cardiology, McGill University Health Center, Montreal, QC, Canada; 2Department of Cardiac, Thoracic, Vascular Sciences, and Public Health, University of Padua, Padua, Italy
Correspondence: Ahmed AlTurki
Division of Cardiology, McGill University Health Center, Montreal, QC H3G 1A4, Canada
Tel +1 514-934-1934
Fax +1 514-934-8569
Email [email protected]
Abstract: Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) is a primary cardiac myocytes disorder that predominantly affects the right ventricle. It is mainly inherited as autosomal dominant with variable expressivity; it also has been recognized as one of the major genetic causes of sudden cardiac death in the young and in athletes. The desmosomal protein is the most commonly affected structure and around 60% of diagnosed cases have an identified genetic mutation. ARVC is characterized histologically by the replacement of cardiomyocytes with fibro-fatty tissue that is progressive over time. The most commonly involved sites in the heart are right ventricle inflow tract, right ventricular outflow tract and posterolateral wall of the left ventricle. New diagnostic criteria have increased the sensitivity and specificity for ARVC/D. These include imaging evidence of RV regional wall motion abnormalities and RV dilatation using echocardiography, cardiac magnetic resonance imaging and angiography. Other diagnostic criteria include fibrous replacement of the RV-free wall on biopsy, repolarization and conduction abnormalities on the electrocardiogram as well as ventricular tachyarrhythmias and significant family history. Management involves assessing for implantable-cardioverter implantation, pharmacological therapy for prevention of ventricular arrhythmias and treatment of any ventricular dysfunction. Patients with ARVC/D who are engaged in strenuous/endurance/competitive physical activity have 2– 5-fold increased risk of sudden cardiac death and restriction from competitive/endurance sports is important. Family screening is important to identify asymptomatic patients.
Keywords: arrhythmogenic right ventricular dysplasia, sudden cardiac death, ventricular tachycardia, arrhythmogenic cardiomyopathy
Introduction
Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) also known as arrhythmogenic right ventricular dysplasia (ARV) was first well-described in 1982 in a case series of 24 patients by Marcus et al.1 ARVC/D is an inherited, autosomal dominant disease with variable expressivity and penetrance,2 that is characterized histologically by the replacement of cardiomyocytes with fibro-fatty tissue and is progressive over time. The new infiltrates act as a substrate for ventricular arrhythmias and eventually lead to right ventricular failure and sudden cardiac death (SCD).3 While the disease predominantly affects the right ventricle, recent studies and improved imaging has allowed the discernment of left ventricular involvement in a substantial proportion of patients.4
Due to variable expression, it is difficult to diagnose, and thus, to estimate the incidence and prevalence of ARVC/D. The disease spectrum varies between silent clinical features such as electrocardiographic changes, to florid right ventricular failure, malignant arrhythmias and SCD.5,6 Around 40% of cases are diagnosed through testing of first-degree relatives7 The estimated prevalence of ARVC/D is 1:1000 to 1:5000.8,9 This disease might be responsible for about 20% of cases of SCDs in the young across Europe and up to 27% of sports-related SCD.6,10 A recently published nationwide study on SCD in patients aged 13–25 years from USA national registry between 1980 and 2011 suggested that ARVC is responsible for about 5% of SCD.11 The disease is more prevalent in men with a ratio of 3:1, and a family history is present in 30–50% of cases.12–15
Efforts have been made to improve our understanding of this disease, resulting in new strategies that focus on early diagnosis based on evaluating the functional and structural cardiac alterations characteristic of this entity, as well as genetic testing for patients considered at high risk. This culminated in the development of the updated International Task Force Criteria for the clinical diagnosis of ARVC/D in 2010.16 The main objective of treatment is to reduce the risk of sudden cardiac death and improve the quality of life for patients suffering from this disease. This is achieved mainly by appropriate pharmacological and device therapy, as well as restriction of strenuous physical activity. The latter has a direct relationship with disease progression and severity of arrhythmias.17 ARVC is a common cause of sudden cardiac death (SCD) in athletes.18 This review focuses on mechanisms underlying ARVC and how they relate to the clinical presentation and diagnostic criteria. In addition, all aspects of the clinical management of ARVC will be addressed.
Mechanisms of ARVD/C
Genetics
It is now well established that the primary-diseased structure is a desmosomal protein in most cases.19–21 It was later discovered that other non-desmosomal proteins play a role in the pathogenesis of this disease. This is evident by the number of isolated disease-causing genes (Table 1). Of all patients diagnosed with ARVC, causative mutations can be identified in 60% of patients, with the majority involving genes that encode mechanical junction proteins in the intercalated disk.22
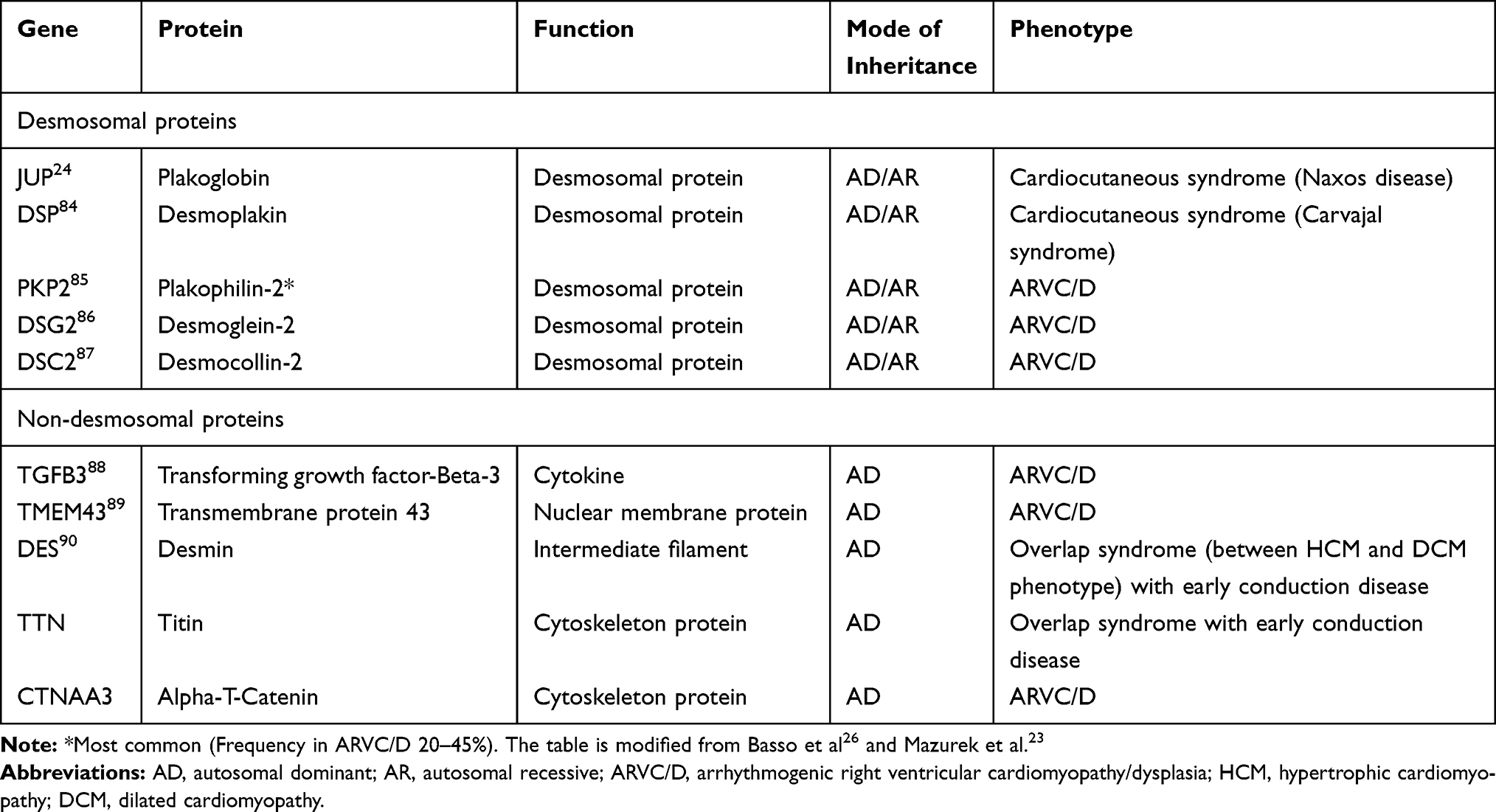 |
Table 1 Some of the Principal Disease-Associated Genes in Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia |
Pathogenesis
Desmosomal proteins are specialized proteins that span the cell membrane, facilitate communication and provide structural support between myocytes. Desmosomal proteins perform an important role in 1) gene transcription and regulation of differentiation 2) electrical current conduction through regulation of gap junction and 3) calcium homeostasis.21,23 Multiple theories have been proposed to explain the mechanism by which desmosomal abnormalities are related to the pathogenesis of ARVC/D and creation of fibrofatty infiltrates instead of the normal myocardium (Figure 1A).
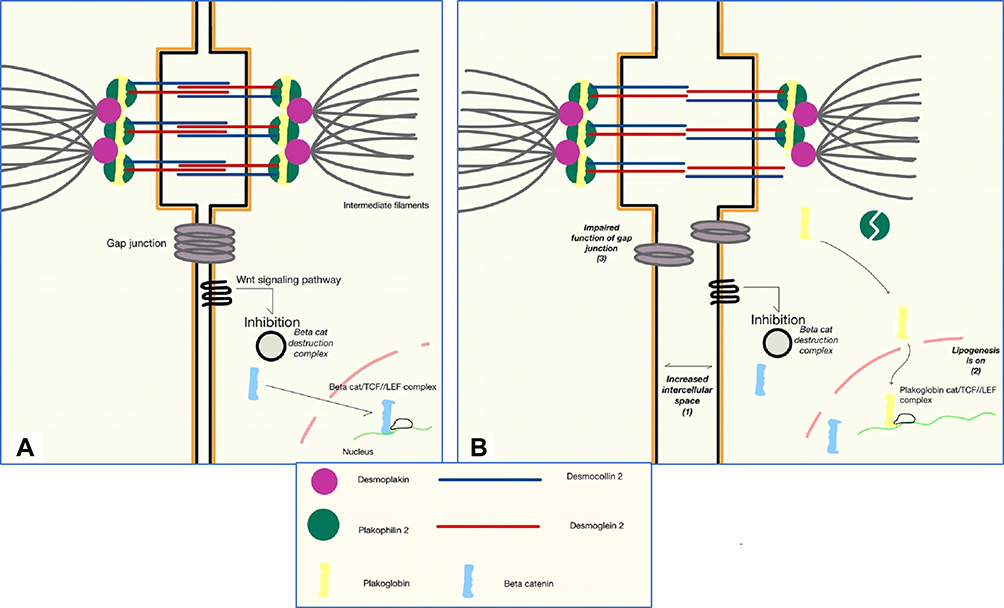 |
Figure 1 Schematic representation of molecular basis and pathogenesis of Arrhythogenic right ventricular cardiomyopathy/dysplasia. (A) Tight gap junctions and close intercellular connection between cardiomyocyte is essential in normal function and signal transduction between cardiac cells. (B) When there is a defective plakophilin 2, plakoglobin is freed to localize the nucleus and replaces beta-catenin in the TCF/LEF to form a complex that inhibits canonical Wnt signaling pathway thus increasing beta-catenin destruction and switching the cell to lipogenesis. This event will result in increased intercellular space, impaired cell-to-cell adhesion and malfunctioning of gap junctions. The steps of pathogenesis are numbered 1–3. Abbreviations: TCF, t-cell factor; LEF, lymphoid enhancer factor. |
An example of this pathogenesis is better understood in Naxos disease, a cardiocutaneous syndrome characterized by a triad of non-epidermolytic palmoplantar keratoderma, woolly hair and ARVC/D.24 The single causative protein in common between the triad is a mutant plakoglobin protein which is a part of cell-to-cell adhesion complex. Abnormalities in adhesion would promote myocytes detachment, apoptosis25 and subsequently, replacement by fibrofatty tissue.26 It is important to note that exercise increases the rate of myocytes detachment; athletic mutation-carriers become symptomatic earlier than non-athletics, and have a worse phenotype.27
Desmosomes are important proteins in transcription regulation. In normal myocardial cells, Canonical Wnt signaling is active and it prevents the degradation of cytoplasmic β-catenin protein. β-catenin is a protein that travels to the nucleus to facilitate and aid in the recognition and transcription of target genes by binding to TCF/LEF complexes.28 Plakoglobin (AKA γ-catenin) is one of the desmosomal proteins similar in properties to β-catenin, but with antagonizing effects.21,23,26 In patients with defective Desmoplakin (another desmosomal protein), free Plakoglobin is enabled to travel and localizes the nucleus where it replaces β-catenin, and switches transcription from myogenesis to adipogenesis29 (Figure 1B).
Arrhythmia is a common feature of dilated cardiomyopathy, and patients are at increased risk of developing sudden cardiac death with worsening structural abnormalities.30 However, arrhythmias in ARVC/D are one of the early manifestations of the disease, which can develop even before any obvious cardiac pathology.31,32 This is likely secondary to gap-junction remodeling associated with the ARVC/D. For example, there is a clear evidence of downregulation of Connexin 43, one of the major gap-junction proteins,33,34 this phenomena is not specific to ARVC/D, but also found in other cardiomyopathies.35 Another observation is the decreased INa secondary to decreased formation of sodium pores evident by paucity of Nav1.5 (a pore-forming protein) at the intercalated disks in patients with ARVC/D.36,37
Pathology
ARVC/D is a misleading name, as the disease may also affect the left ventricle (LV) instigating a shift to the use of the term arrhythmogenic ventricular cardiomyopathy.5 Right ventricular (RV) involvement can be generalized or localized as well. Grossly there will be dilation and thinning of the affected myocardium. The most commonly involved sites in the heart are right ventricle inflow tract, RV outflow tract and posterolateral wall of the LV. These three sites have been termed “the new triangle of dysplasia”.38 The RV apex, which was part of the “original triangle of dysplasia”,32 is also frequently affected but commonly only seen in severe and end-stage disease. Its worth mentioning that in some cases LV involvement may precede RV dysfunction.39 ARVC/D is a progressive disease, and typically starts in the RV and, with age, continues to invade the LV.5,40 LV involvement is commonly seen in patients with PKP2 mutation, which is the most frequently seen genotype in ARVC/D.2 On a microscopic level, the main characteristic of the disease is the replacement of normal myocardium by fibrofatty or fatty infiltrates, which are inherently a substrate for macro-re-entrant circuits.41,42 Frequently noted, and specially in autopsies of patients who had sudden cardiac death, the presence of inflammatory infiltrates in areas of dying myocardium.5,41
Clinical Features and Diagnostic Criteria
One of the challenging features of ARVC/D is that it may remain silent for decades. In addition, it has variable presentations ranging from totally asymptomatic patients, palpitations and sudden cardiac death. Many cases of ARVC/D come to medical attention following screening because of ARVC/D diagnosis in a relative.43 The mean age at diagnosis is 30 years with a range of 10 years to 50 years of age. Presentation appears to be earlier in athletes, and more malignant in males. There are specific features of some mutations and these are listed in Table 2.
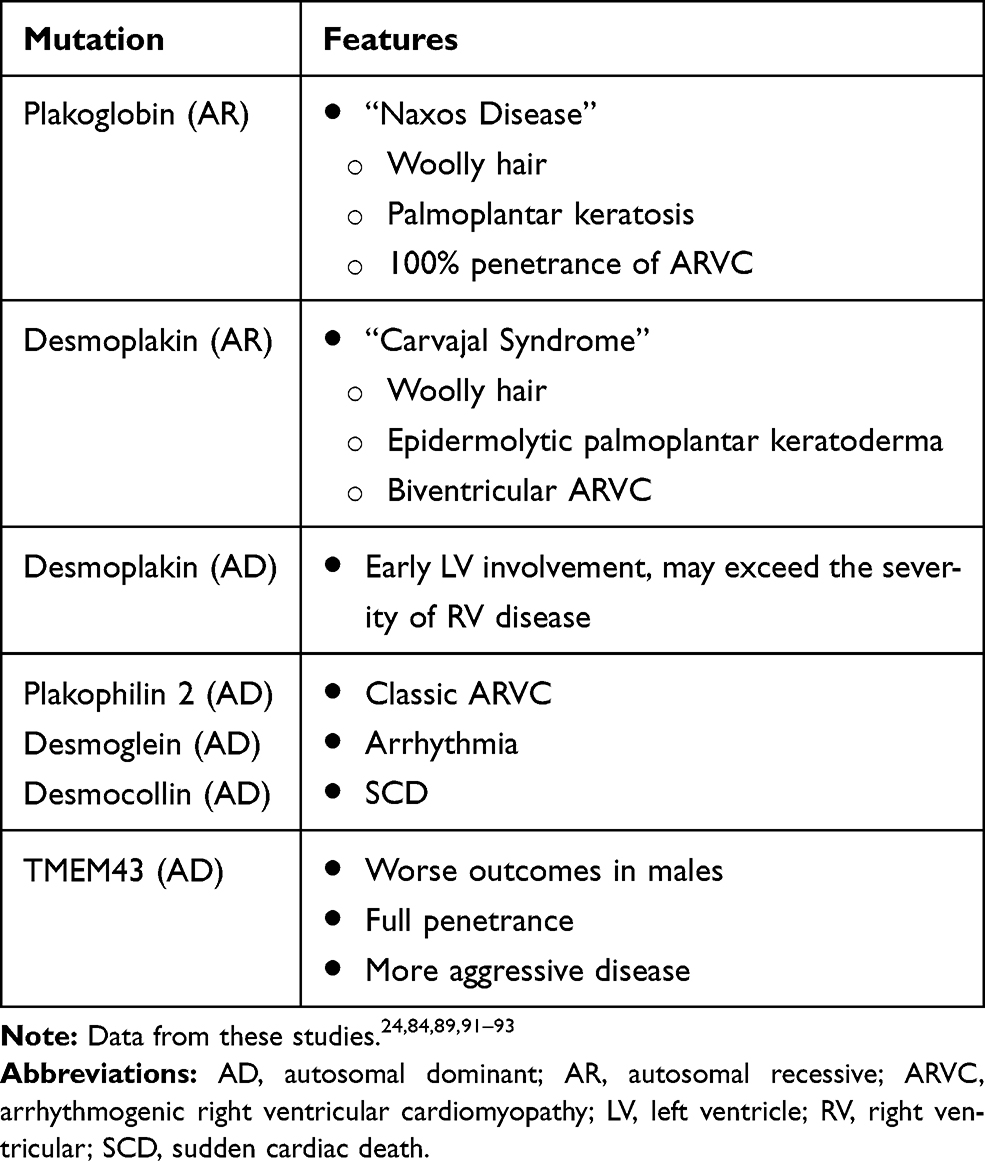 |
Table 2 Mutations and Associated Features |
Palpitations, presyncope and syncope are the most common complaints occurring in >90% of patients. Oher symptoms include atypical chest pain, dyspnea and right ventricular failure.44 Palpitations and presyncope/syncope are most commonly associated with ventricular arrhythmia, which can be premature ventricular beats, or even ventricular tachycardia, most commonly originating from the RV (LBBB morphology).7 In addition, ARVC/D patients are at increased risk of SCD and this can be the initial presentation.7,13 Given the non-specific signs and symptoms of ARVC/D, the original Task Force criteria in 1994 was updated in 2010.16 The new criteria increased the sensitivity and specificity of ARVC/D diagnosis. These include imaging evidence of RV regional wall motion abnormalities and RV dilatation using echocardiography, cardiac magnetic resonance imaging and angiography. Other diagnostic criteria include fibrous replacement of the RV-free wall on biopsy, repolarization and conduction abnormalities on the electrocardiogram as well as ventricular tachyarrhythmias and significant family history.16
Management
In patients with ARVC/D, the main goal of clinical management is to prevent sudden cardiac death and slow the progression of disease. Other aims include anticipation and management of complications as they arise in order to improve quality of life as well as the early detection of asymptomatic ARVC/D patients once a proband is identified to trigger cascade screening in first-degree relatives. In patients with symptomatic ventricular arrhythmias or those experiencing ICD therapies, interventions to reduce the burden of ventricular arrhythmias are needed. The avoidance of strenuous physical activity is essential to prevent progression of the disease. One should consider a multidisciplinary approach to manage ARVC/D patients, as this disease affects patient’s activity, habits, and mental status.
Once diagnosis is confirmed, a stepwise evaluation of the patient’s risks and clinical situation should be commenced. This would serve in determining prognosis, anticipate adverse outcomes and appropriateness of ICD therapy. Patients who have had a prior SCD or a hemodynamically significant sustained ventricular tachycardia have a clear indication for an ICD; in this population, the annual risk of requiring appropriate ICD therapy is around 7% to 10%.45 If a patient has not had a prior SCD or VT, then a risk stratification approach is needed to identify those who would benefit from an ICD; in high-risk patients, appropriate ICD therapy can occur in around 5% of patients per year, a substantial risk. Interestingly, in a recent study by Zorzi and colleagues, found that phenotypic expression of ARVC/D as defined by the presence of syncope, non-sustained ventricular arrhythmias or ventricular dysfunction as compared to gene-positive non-phenotypic patients portends a substantially greater risk of sustained VT and SCD.46 The incidence of VT or SCD was 2.3%/year among patients with definite ARVC with phenotypic expression and 0.2%/year in phenotype negative or borderline patients (P = 0.002).46
Risk Stratification
Most of the information on ARVC/D is from retrospective studies, which makes it difficult to create a precise risk stratification model. A recently developed risk model published by an international collaboration in six countries (18 centers) from Europe and North America reported that variables associated with higher risk for VT/VF are: male sex, young age, extent of T-wave inversion across precordial and inferior leads on ECG, imaging features reflecting more extensive RV disease and severity of ventricular arrhythmia.47
Multiple studies examined independent risk factors associated with poor outcomes (defined as malignant arrhythmia, appropriate ICD interventions, death or heart transplant) in ARVC/D by the means of multivariable analysis. Most commonly reported significant (or at borderline statistical significance) factors are aborted SCD, either VT or VF related;48–50 Unexplained syncope;48,50,51 non-sustained ventricular tachycardia;51,52 premature ventricular contraction frequency >1000/24 hrs;53 atrial fibrillation;54 inducible VT in electrophysiology studies;52,53,55,56 proband status;57 young age at first presentation;48,49,53 male sex;54,57–59 multiple genetic mutations;58 amount of myocardial scarring;60–62 extent of T-wave inversion on ECG;49,57,63 LV dysfunction44,48,55,64,65 and RV dysfunction or dilatation.55,66
Electrophysiologic study (EPS) is a powerful diagnostic tool in situations where there is a doubt about the diagnosis, specially between ARVC/D and idiopathic right ventricular outflow tract tachycardia.67 EPS can also serve as a prognostic tool, as mentioned above. Even with the presence of conflicting data regarding usefulness of VT-inducibility as a risk factor to predict long-term outcomes in ARVC/D, EPS can be helpful in optimization of the detection/discrimination algorithms and anti-tachycardia pacing protocols in ICD recipients.68 It is recommended that EPS should be considered in the diagnosis and/or evaluation of patients with suspected ARVC/D, it is also reasonable for risk stratification in asymptomatic patients.68
ICDs to Prevent SCD
Initial attempts at prevention of SCD through surgical fulguration were unsuccessful. ICDs made it possible to prevent SCD in patients with ARVC/D; however, it is important to target patients at risk of developing lethal arrhythmias (VT/VF) to avoid unneeded short or long-term complications of ICD therapy.69 Deciding on ICD placement is usually the best next logical step after patient’s presentation and risk stratification. It can be placed for primary or secondary prevention. ICD placement should be a shared decision made between patients and treating physicians. No randomized trials have yet been performed to compare ICDs with medical therapy in patients with ARVC/D. Patients with ARVC/D have a high burden of VT requiring ICD therapy or leading to SCD in those without an ICD. This is compounded by a prolonged period of exposure given the young age at diagnosis. On the other hand, the young age of ICD implantation also exposes patients to ICD complications, inappropriate ICD shocks and the requirement for multiple generator replacements. Appropriateness of ICD therapy in high-risk patients has been demonstrated in multiple studies. In a cohort of 131 patients with ARVC/D who did not have an ICD, 29% experienced significant ventricular tachycardia including SCD with an annualized rate of 5.6%.70 In a meta-analysis reported by Schinkel71 it was found that in high-risk ARVC/D patients, the rate of appropriate ICD intervention was 9.5%/year.
Recently, the Heart Rhythm Society published their expert consensus72 on ICD placement in arrhythmogenic cardiomyopathy; ARVC/D-specific recommendations were as follows: class I recommendation for patients with ARVC/D who had experienced SCD or hemodynamically not tolerated VT; class IIa recommendation for patients with ARVC/D that had stable sustained VT, or patients with multiple risk factors (Figure 2) (three major, two major and two minor, or one major and four minor); class IIb recommendation for patients with multiple risk factors (two major, one major and two minor, or four minor).72
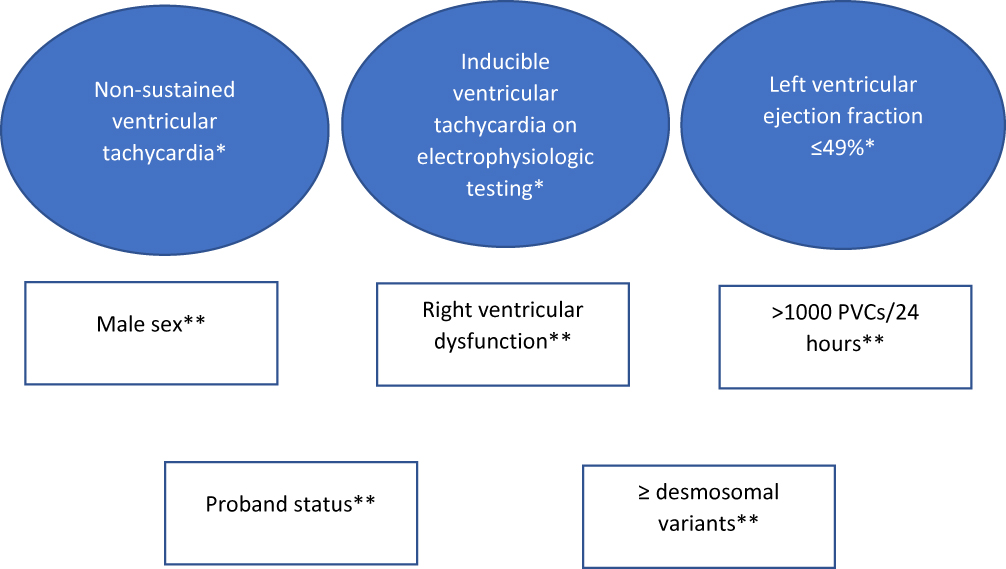 |
Figure 2 Risk factors for sudden cardiac death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia who have not experienced sudden cardiac death or sustained ventricular tachycardia. Criteria from 2019 Heart Rhythm Society expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy.72 *Major criteria; **Minor criteria. Abbreviation: PVC, premature ventricular contraction. |
Another set of recommendations for ICD therapy in ARVC/D patients were published in 2015 by Corrado et al68 with similar recommendations. Class I indications for ICD placements were in patients who have: 1)  1 episode of unstable sustained VT/VF (secondary prevention) or 2) Severe systolic dysfunction of the RV, LV, or both regardless of the presence of arrhythmia (primary prevention). Class IIa indications for ICD placement were in patients who have 1)
1 episode of unstable sustained VT/VF (secondary prevention) or 2) Severe systolic dysfunction of the RV, LV, or both regardless of the presence of arrhythmia (primary prevention). Class IIa indications for ICD placement were in patients who have 1)  1 episode of stable sustained VT or 2) Major risk factors (NSVT, moderate ventricular dysfunction, unexplained syncope). Class IIb indication is reserved for asymptomatic patients with minor risk factors. In patients with low risk for SCD (asymptomatic, no risk factors and healthy gene carriers), ICD implantation is not indicated and thus given a class III recommendation as risks of ICD placement outweighs the benefits. Device selection should be based on individualized clinical scenario; for example, cardiac resynchronization therapy (CRT) is reasonable in cases where there is EF of <35% and LBBB morphology on 12-lead ECG. Figure 3 depicts our current approach to the management of ARVC/D.
1 episode of stable sustained VT or 2) Major risk factors (NSVT, moderate ventricular dysfunction, unexplained syncope). Class IIb indication is reserved for asymptomatic patients with minor risk factors. In patients with low risk for SCD (asymptomatic, no risk factors and healthy gene carriers), ICD implantation is not indicated and thus given a class III recommendation as risks of ICD placement outweighs the benefits. Device selection should be based on individualized clinical scenario; for example, cardiac resynchronization therapy (CRT) is reasonable in cases where there is EF of <35% and LBBB morphology on 12-lead ECG. Figure 3 depicts our current approach to the management of ARVC/D.
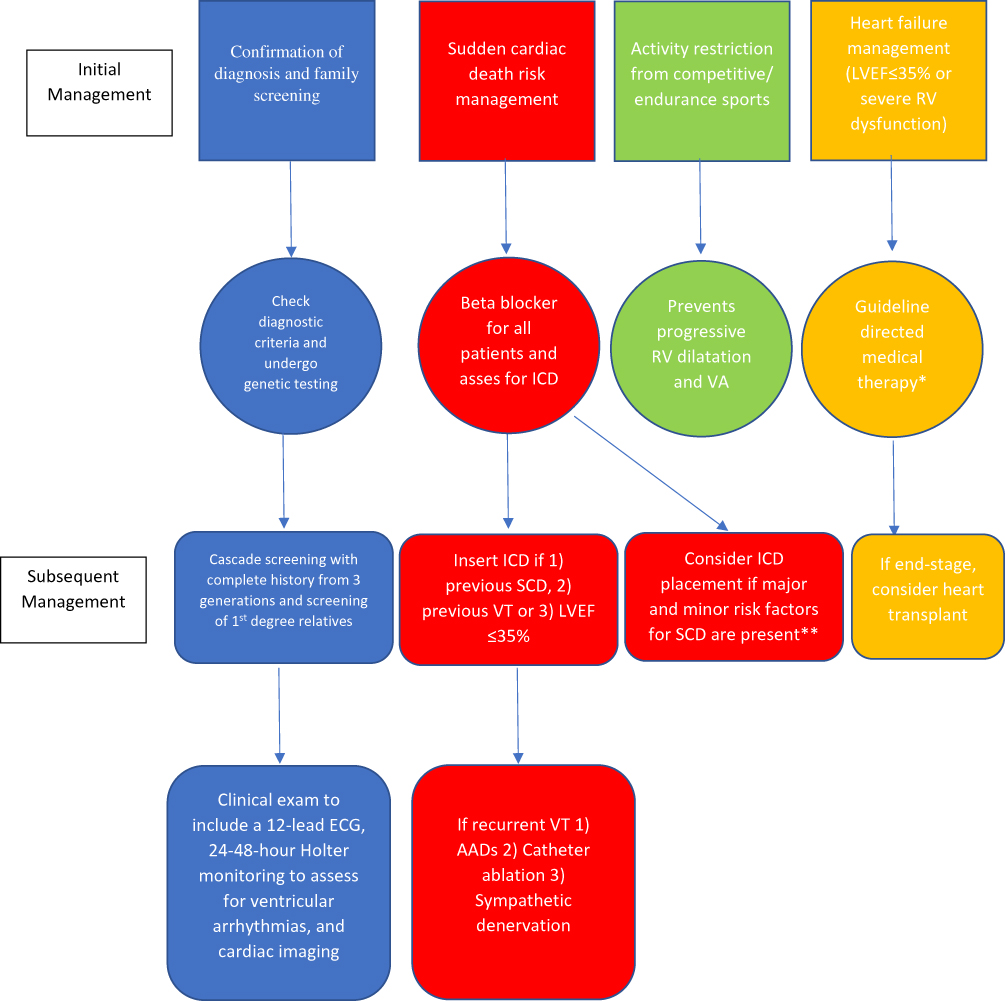 |
Figure 3 Suggested algorithm for the management of arrhythmogenic right ventricular cardiomyopathy/dysplasia. *Treatment with angiotensin converting enzyme inhibitor/angiotensin receptor blocker, beta blocker and mineralocorticoid receptor antagonists. **See Figure 2. Abbreviations: AAD, anti-arrhythmic drugs; ECG, electrocardiogram; ICD, implantable-cardioverter defibrillators; LVEF, left ventricular ejection fraction; RV, right ventricle; SCD, sudden cardiac death; VA, ventricular arrhythmias; VT, ventricular tachycardia. |
Activity Restriction and Lifestyle Modification
One of the modifiable risk factors is engagement in competitive/endurance sports. The link between high-intensity exercise and early onset of ARVC/D symptoms has been demonstrated in prospective, retrospective and animal studies. Patients with ARVC/D who are engaged in strenuous/endurance/competitive physical activity have 2-5-fold increased risk of sudden cardiac death.73,74 In addition, in the population of ARVC/D, athletes showed reduced LV and RV function compared with non-athletes in a mutation-positive family.17 Exercise restriction is associated with a 70–80% reduction in the risk of VT and ICD therapy including patients who never had ventricular arrhythmias beforehand.75 Given the clear association, it is recommended that all patients diagnosed with ARVC/D not participate in competitive or endurance exercises (Class I recommendation).72 It is also recommended for those patients to abstain from non-competitive high-intensity sports (such as hockey, football, basketball, and activities that require sprinting). Patients who are engaged in low-intensity recreational activities did not show a higher risk for early onset phenotype or SCD compared with patients who are inactive.74
Pharmacological Therapy
There is limited data for the use of anti-arrhythmic drugs (AAD) in patients with ARVC/D; however, most recommendations are based on clinical experience, registries and retrospective studies. AAD should not be prescribed prophylactically for asymptomatic patients. They are only indicated in patients who experience symptoms such as frequent PVCs and/or non-sustained VT, or patients who have an existing ICD and experience frequent appropriate device discharges.68
The choice of AAD is variable across literature, most commonly used medications are amiodarone or sotalol.76 Wichter et al assessed AAD efficacy in low and intermediate-risk patients with ARVC/D and found that sotalol was more effective in preventing ventricular arrhythmia than amiodarone.77 Amiodarone was more effective (although only in smaller number of patients) in another study published by Marcus et al.78 These conflicting results could be explained by the differences in the target dose used in both studies, specially sotalol dose.
Amiodarone and sotalol are both class III AAD, and both have a degree of beta blockade. In addition, both block potassium channels which slows repolarization of cardiac cells and increases the effective refractory period in ventricular cardiomyocytes thus increasing the action potential period. These effects are key features in the mechanism by which class III AADs can suppress arrhythmias. Interestingly, the distribution of the fibrosis with lower fibrotic density compared to ischemic scar and the cellular electrical uncoupling linked to molecular disjunction indicate that arrhythmic mechanisms such as abnormal automaticity and trigger activity are ore likely causative compared to re-entry in the genesis of ventricular arrhythmias.32
The current guidelines recommended the use of amiodarone when indicated and acknowledged the conflicting evidence for AAD.76 However, they recommend using a beta-blocker as a first-line agent in ARVC/D patients with either atrial or ventricular arrhythmia, and AADs should be an adjunct therapy, it is also recommended to avoid prophylactic use of beta blockers or AADs in asymptomatic patients.68 For patients with concomitant heart failure, pharmacological therapy should be based on guide-directed medical therapy.72
Catheter Ablation
VT ablation (both endocardial and epicardial) is not a curative solution for VT in patients with ARVC/D due to the progressive nature of the disease and patchy distribution. Multiple studies demonstrated high recurrence rate of VT in both short and long term follow up.79–82 It is recommended however, in cases refractory to maximal pharmacological therapy or if there is a contraindication to medical therapy.
Heart Transplant
If indicated heart transplant is a final therapeutic option in ARVC/D patients who are eligible for the procedure. Most common indications are severe heart failure or interactable ventricular arrhythmias.83
Family Screening
Current guidelines recommend that all ARVC/D patients should be seen by a genetic counselor or by an experienced clinician to obtain a comprehensive family history spanning three generations. In addition, first-degree relatives should submit to clinical evaluation every 1–3 beginning at the early age of 12 years. The cardiovascular evaluation should include a 12-lead ECG, 24–48 hr Holter monitoring to assess for ventricular arrhythmias, and cardiac imaging such as echocardiography or cardiac magnetic resonance imaging to assess for RV regional wall motion abnormalities or dilatation. As an adjunct to cardiovascular evaluation, exercise stress testing to provoke arrhythmias may be considered.
Conclusions
ARVC is as one of the major genetic causes of sudden cardiac death in the young and in athletes. The desmosomal protein is the most commonly affected structure and around 60% of diagnosed cases have an identified genetic mutation. New diagnostic criteria have improved the sensitivity of testing for ARVC. Management revolves around ICD implantation for primary or secondary prevention of ventricular arrhythmias. Future research should focus on curative approaches as well as interventions to improve quality of life.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Marcus FI, Fontaine GH, Guiraudon G, et al. Right ventricular dysplasia: a report of 24 adult cases. Circulation. 1982;65(2):384–398. doi:10.1161/01.CIR.65.2.384
2. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur Heart J. 2015;36(14):847–855. doi:10.1093/eurheartj/ehu509
3. Calkins H. Arrhythmogenic Right ventricular dysplasia/cardiomyopathy– three decades of progress –three decades of progress. Circ J. 2015;79(5):901–913. doi:10.1253/circj.CJ-15-0288
4. Berte B, Denis A, Amraoui S, et al. Characterization of the Left-sided substrate in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2015;8(6):1403–1412. doi:10.1161/CIRCEP.115.003213
5. Corrado D, Basso C, Thiene G, et al. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: a multicenter study. J Am Coll Cardiol. 1997;30(6):1512–1520. doi:10.1016/S0735-1097(97)00332-X
6. Corrado D, Fontaine G, Marcus FI, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: need for an international registry. Study Group on Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy of the Working Groups on Myocardial and Pericardial Disease and Arrhythmias of the European Society of Cardiology and of the Scientific Council on Cardiomyopathies of the World Heart Federation. Circulation. 2000;101(11):E101–6. doi:10.1161/01.cir.101.11.e101
7. Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2000;36(7):2226–2233. doi:10.1016/S0735-1097(00)00997-9
8. Sen-Chowdhry S, Morgan RD, Chambers JC, et al. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61(1):233–253. doi:10.1146/annurev.med.052208.130419
9. Kies P, Bootsma M, Bax J, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy: screening, diagnosis, and treatment. Heart Rhythm. 2006;3(2):225–234. doi:10.1016/j.hrthm.2005.10.018
10. Sadjadieh G, Jabbari R, Risgaard B, et al. Nationwide (Denmark) study of symptoms preceding sudden death due to arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2014;113(7):1250–1254. doi:10.1016/j.amjcard.2013.12.038
11. Maron BJ, Haas TS, Ahluwalia A, et al. Demographics and epidemiology of sudden deaths in young competitive athletes: from the United States National Registry. Am J Med. 2016;129(11):1170–1177. doi:10.1016/j.amjmed.2016.02.031
12. Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation. 2006;113(13):1634–1637. doi:10.1161/CIRCULATIONAHA.105.616490
13. Dalal D, Nasir K, Bomma C, et al. Arrhythmogenic right ventricular dysplasia: a United States experience. Circulation. 2005;112(25):3823–3832. doi:10.1161/CIRCULATIONAHA.105.542266
14. van Tintelen JP, Entius MM, Bhuiyan ZA, et al. Plakophilin-2 mutations are the major determinant of familial arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2006;113(13):1650–1658. doi:10.1161/CIRCULATIONAHA.105.609719
15. Te Riele AS, James CA, Groeneweg JA, et al. Approach to family screening in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Eur Heart J. 2016;37(9):755–763. doi:10.1093/eurheartj/ehv387
16. Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur Heart J. 2010;31(7):806–814. doi:10.1093/eurheartj/ehq025
17. Saberniak J, Hasselberg NE, Borgquist R, et al. Vigorous physical activity impairs myocardial function in patients with arrhythmogenic right ventricular cardiomyopathy and in mutation positive family members. Eur J Heart Fail. 2014;16(12):1337–1344. doi:10.1002/ejhf.2014.16.issue-12
18. Hsu JJ, Nsair A, Aboulhosn JA, et al. Monomorphic ventricular arrhythmias in athletes. Arrhythm Electrophysiol Rev. 2019;8(2):83–89. doi:10.15420/aer.2019.19.3
19. Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of right ventricular cardiomyopathy. J Cardiovasc Electrophysiol. 2005;16(8):927–935. doi:10.1111/jce.2005.16.issue-8
20. Moric-Janiszewska E, Markiewicz-Loskot G. Review on the genetics of arrhythmogenic right ventricular dysplasia. Europace. 2007;9(5):259–266. doi:10.1093/europace/eum034
21. Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5(5):258–267. doi:10.1038/ncpcardio1182
22. De Bortoli M, Postma AV, Poloni G, et al. Whole-exome sequencing identifies pathogenic variants in TJP1 gene associated with arrhythmogenic cardiomyopathy. Circ Genom Precis Med. 2018;11(10):e002123. doi:10.1161/CIRCGEN.118.002123
23. Mazurek S, Kim GH. Genetic and epigenetic regulation of arrhythmogenic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):2064–2069. doi:10.1016/j.bbadis.2017.04.020
24. McKoy G, Protonotarios N, Crosby A, et al. Identification of a deletion in plakoglobin in arrhythmogenic right ventricular cardiomyopathy with palmoplantar keratoderma and woolly hair (Naxos disease). Lancet. 2000;355(9221):2119–2124. doi:10.1016/S0140-6736(00)02379-5
25. Valente M, Calabrese F, Thiene G, et al. In vivo evidence of apoptosis in arrhythmogenic right ventricular cardiomyopathy. Am J Pathol. 1998;152(2):479–484.
26. Basso C, Pilichou K, Bauce B, et al. Diagnostic criteria, genetics, and molecular basis of arrhythmogenic cardiomyopathy. Heart Fail Clin. 2018;14(2):201–213. doi:10.1016/j.hfc.2018.01.002
27. James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62(14):1290–1297. doi:10.1016/j.jacc.2013.06.033
28. MacDonald BT, Tamai K, He X. Wnt/β-Catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. doi:10.1016/j.devcel.2009.06.016
29. Garcia-Gras E, Lombardi R, Giocondo MJ, et al. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Invest. 2006;116(7):2012–2021. doi:10.1172/JCI27751
30. Sen-Chowdhry S, McKenna WJ. Sudden death from genetic and acquired cardiomyopathies. Circulation. 2012;125(12):1563–1576. doi:10.1161/CIRCULATIONAHA.111.025528
31. Basso C, Bauce B, Corrado D, et al. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2011;9(4):223–233. doi:10.1038/nrcardio.2011.173
32. Corrado D, Link MS, Calkins H. Arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2017;376(1):61–72. doi:10.1056/NEJMra1509267
33. Asimaki A, Tandri H, Huang H, et al. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360(11):1075–1084. doi:10.1056/NEJMoa0808138
34. Kaplan SR, Gard JJ, Protonotarios N, et al. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease). Heart Rhythm. 2004;1(1):3–11. doi:10.1016/j.hrthm.2004.01.001
35. Peters NS, Green CR, Poole-Wilson PA, et al. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88(3):864–875. doi:10.1161/01.CIR.88.3.864
36. Rizzo S, Lodder EM, Verkerk AO, et al. Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein-2 mutant mice prior to cardiomyopathic changes. Cardiovasc Res. 2012;95(4):409–418. doi:10.1093/cvr/cvs219
37. Noorman M, Hakim S, Kessler E, et al. Remodeling of the cardiac sodium channel, connexin43, and plakoglobin at the intercalated disk in patients with arrhythmogenic cardiomyopathy. Heart Rhythm. 2013;10(3):412–419. doi:10.1016/j.hrthm.2012.11.018
38. Te Riele AS, James CA, Philips B, et al. Mutation-positive arrhythmogenic right ventricular dysplasia/cardiomyopathy: the triangle of dysplasia displaced. J Cardiovasc Electrophysiol. 2013;24(12):1311–1320. doi:10.1111/jce.2013.24.issue-12
39. Sen-Chowdhry S, Syrris P, Ward D, et al. Clinical and genetic characterization of families with arrhythmogenic right ventricular dysplasia/cardiomyopathy provides novel insights into patterns of disease expression. Circulation. 2007;115(13):1710–1720. doi:10.1161/CIRCULATIONAHA.106.660241
40. Pinamonti B, Sinagra G, Salvi A, et al. Left ventricular involvement in right ventricular dysplasia. Am Heart J. 1992;123(3):711–724. doi:10.1016/0002-8703(92)90511-S
41. Basso C, Thiene G, Corrado D, et al. Arrhythmogenic right ventricular cardiomyopathy. Dysplasia, dystrophy, or myocarditis? Circulation. 1996;94(5):983–991. doi:10.1161/01.CIR.94.5.983
42. Burke AP, Farb A, Tashko G, et al. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: are they different diseases? Circulation. 1998;97(16):1571–1580. doi:10.1161/01.CIR.97.16.1571
43. Groeneweg JA, Bhonsale A, James CA, et al. Clinical presentation, long-term follow-up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet. 2015;8(3):437–446. doi:10.1161/CIRCGENETICS.114.001003
44. Hulot JS, Jouven X, Empana J-P, et al. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004;110(14):1879–1884. doi:10.1161/01.CIR.0000143375.93288.82
45. AlTurki A, Proietti R, Russo V, et al. Anti-arrhythmic drug therapy in implantable cardioverter-defibrillator recipients. Pharmacol Res. 2019;143:133–142. doi:10.1016/j.phrs.2019.03.020
46. Zorzi A, Rigato I, Pilichou K, et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. Europace. 2016;18(7):1086–1094. doi:10.1093/europace/euv205
47. Cadrin-Tourigny J, Bosman LP, Nozza A, et al. A new prediction model for ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2019;40(23):1850–1858. doi:10.1093/eurheartj/ehz103
48. Corrado D, Leoni L, Link MS, et al. Implantable cardioverter-defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108(25):3084–3091. doi:10.1161/01.CIR.0000103130.33451.D2
49. Link MS, Laidlaw D, Polonsky B, et al. Ventricular arrhythmias in the North American multidisciplinary study of ARVC: predictors, characteristics, and treatment. J Am Coll Cardiol. 2014;64(2):119–125. doi:10.1016/j.jacc.2014.04.035
50. Watkins DA, Hendricks N, Shaboodien G, et al. Clinical features, survival experience, and profile of plakophylin-2 gene mutations in participants of the arrhythmogenic right ventricular cardiomyopathy registry of South Africa. Heart Rhythm. 2009;6(11 Suppl):S10–7. doi:10.1016/j.hrthm.2009.08.018
51. Corrado D, Calkins H, Link MS, et al. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010;122(12):1144–1152. doi:10.1161/CIRCULATIONAHA.109.913871
52. Bhonsale A, James CA, Tichnell C, et al. Incidence and predictors of implantable cardioverter-defibrillator therapy in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy undergoing implantable cardioverter-defibrillator implantation for primary prevention. J Am Coll Cardiol. 2011;58(14):1485–1496. doi:10.1016/j.jacc.2011.06.043
53. Orgeron GM, James CA, Te Riele A, et al. Implantable cardioverter-defibrillator therapy in arrhythmogenic right ventricular dysplasia/cardiomyopathy: predictors of appropriate therapy, outcomes, and complications. J Am Heart Assoc. 2017;6(6). doi:10.1161/JAHA.117.006242.
54. Mazzanti A, Ng K, Faragli A, et al. Arrhythmogenic right ventricular cardiomyopathy: clinical course and predictors of arrhythmic risk. J Am Coll Cardiol. 2016;68(23):2540–2550. doi:10.1016/j.jacc.2016.09.951
55. Wichter T, Paul M, Wollmann C, et al. Implantable cardioverter/defibrillator therapy in arrhythmogenic right ventricular cardiomyopathy: single-center experience of long-term follow-up and complications in 60 patients. Circulation. 2004;109(12):1503–1508. doi:10.1161/01.CIR.0000121738.88273.43
56. Saguner AM, Medeiros-Domingo A, Schwyzer MA, et al. Usefulness of inducible ventricular tachycardia to predict long-term adverse outcomes in arrhythmogenic right ventricular cardiomyopathy. Am J Cardiol. 2013;111(2):250–257. doi:10.1016/j.amjcard.2012.09.025
57. Bhonsale A, James CA, Tichnell C, et al. Risk stratification in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. Circ Arrhythm Electrophysiol. 2013;6(3):569–578. doi:10.1161/CIRCEP.113.000233
58. Rigato I, Bauce B, Rampazzo A, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6(6):533–542. doi:10.1161/CIRCGENETICS.113.000288
59. Kimura Y, Noda T, Otsuka Y, et al. Potentially lethal ventricular arrhythmias and heart failure in arrhythmogenic right ventricular cardiomyopathy: what are the differences between men and women? JACC Clin Electrophysiol. 2016;2(5):546–555. doi:10.1016/j.jacep.2016.02.019
60. Zorzi A, Migliore F, Elmaghawry M, et al. Electrocardiographic predictors of electroanatomic scar size in arrhythmogenic right ventricular cardiomyopathy: implications for arrhythmic risk stratification. J Cardiovasc Electrophysiol. 2013;24(12):1321–1327. doi:10.1111/jce.2013.24.issue-12
61. Migliore F, Zorzi A, Silvano M, et al. Prognostic value of endocardial voltage mapping in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Arrhythm Electrophysiol. 2013;6(1):167–176. doi:10.1161/CIRCEP.111.974881
62. Santangeli P, Dello Russo A, Pieroni M, et al. Fragmented and delayed electrograms within fibrofatty scar predict arrhythmic events in arrhythmogenic right ventricular cardiomyopathy: results from a prospective risk stratification study. Heart Rhythm. 2012;9(8):1200–1206. doi:10.1016/j.hrthm.2012.03.057
63. Saguner AM, Ganahl S, Baldinger SH, et al. Usefulness of electrocardiographic parameters for risk prediction in arrhythmogenic right ventricular dysplasia. Am J Cardiol. 2014;113(10):1728–1734. doi:10.1016/j.amjcard.2014.02.031
64. Lemola K, Brunckhorst C, Helfenstein U, et al. Predictors of adverse outcome in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy: long term experience of a tertiary care centre. Heart. 2005;91(9):1167–1172. doi:10.1136/hrt.2004.038620
65. Peters S. Long-term follow-up and risk assessment of arrhythmogenic right ventricular dysplasia/cardiomyopathy: personal experience from different primary and tertiary centres. J Cardiovasc Med (Hagerstown). 2007;8(7):521–526. doi:10.2459/01.JCM.0000278450.35107.b3
66. Saguner AM, Vecchiati A, Baldinger SH, et al. Different prognostic value of functional right ventricular parameters in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Cardiovasc Imaging. 2014;7(2):230–239. doi:10.1161/CIRCIMAGING.113.000210
67. O’Donnell D, Cox D, Bourke J, et al. Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract tachycardia. Eur Heart J. 2003;24(9):801–810. doi:10.1016/S0195-668X(02)00654-1
68. Corrado D, Wichter T, Link MS, et al. Treatment of arrhythmogenic right ventricular cardiomyopathy/dysplasia: an international task force consensus statement. Circulation. 2015;132(5):441–453. doi:10.1161/CIRCULATIONAHA.115.017944
69. Olde Nordkamp LR, Postema PG, Knops RE, et al. Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13(2):443–454. doi:10.1016/j.hrthm.2015.09.010
70. Wang W, Cadrin-Tourigny J, Bhonsale A, et al. Arrhythmic outcome of arrhythmogenic right ventricular cardiomyopathy patients without implantable defibrillators. J Cardiovasc Electrophysiol. 2018;29(10):1396–1402. doi:10.1111/jce.2018.29.issue-10
71. Schinkel AF. Implantable cardioverter defibrillators in arrhythmogenic right ventricular dysplasia/cardiomyopathy: patient outcomes, incidence of appropriate and inappropriate interventions, and complications. Circ Arrhythm Electrophysiol. 2013;6(3):562–568. doi:10.1161/CIRCEP.113.000392
72. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e301–e372.
73. Corrado D, Basso C, Rizzoli G, et al. Does sports activity enhance the risk of sudden death in adolescents and young adults? J Am Coll Cardiol. 2003;42(11):1959–1963. doi:10.1016/j.jacc.2003.03.002
74. Ruwald AC, Marcus F, Estes NAM, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2015;36(27):1735–1743. doi:10.1093/eurheartj/ehv110
75. Wang W, Orgeron G, Tichnell C, et al. Impact of exercise restriction on arrhythmic risk among patients with arrhythmogenic right ventricular cardiomyopathy. J Am Heart Assoc. 2018;7(12). doi:10.1161/JAHA.118.008843.
76. Proietti R, Russo V, AlTurki A. Anti-arrhythmic therapy in patients with non-ischemic cardiomyopathy. Pharmacol Res. 2019;143:27–32. doi:10.1016/j.phrs.2019.03.004
77. Wichter T, Borggrefe M, Haverkamp W, et al. Efficacy of antiarrhythmic drugs in patients with arrhythmogenic right ventricular disease. Results in patients with inducible and noninducible ventricular tachycardia. Circulation. 1992;86(1):29–37. doi:10.1161/01.CIR.86.1.29
78. Marcus GM, Glidden DV, Polonsky B, et al. Efficacy of antiarrhythmic drugs in arrhythmogenic right ventricular cardiomyopathy: a report from the North American ARVC registry. J Am Coll Cardiol. 2009;54(7):609–615. doi:10.1016/j.jacc.2009.04.052
79. Verma A, Kilicaslan F, Schweikert RA, et al. Short- and long-term success of substrate-based mapping and ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia. Circulation. 2005;111(24):3209–3216. doi:10.1161/CIRCULATIONAHA.104510503
80. Marchlinski FE, Zado E, Dixit S, et al. Electroanatomic substrate and outcome of catheter ablative therapy for ventricular tachycardia in setting of right ventricular cardiomyopathy. Circulation. 2004;110(16):2293–2298. doi:10.1161/01.CIR.0000145154.02436.90
81. Dalal D, Jain R, Tandri H, et al. Long-term efficacy of catheter ablation of ventricular tachycardia in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2007;50(5):432–440. doi:10.1016/j.jacc.2007.03.049
82. Fontaine G, Tonet J, Gallais Y, et al. Ventricular tachycardia catheter ablation in arrhythmogenic right ventricular dysplasia: a 16-year experience. Curr Cardiol Rep. 2000;2(6):498–506. doi:10.1007/s11886-000-0034-1
83. Tedford RJ, James C, Judge DP, et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012;59(3):289–290. doi:10.1016/j.jacc.2011.09.051
84. Rampazzo A, Nava A, Malacrida S, et al. Mutation in human desmoplakin domain binding to plakoglobin causes a dominant form of arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2002;71(5):1200–1206. doi:10.1086/344208
85. Gerull B, Heuser A, Wichter T, et al. Mutations in the desmosomal protein plakophilin-2 are common in arrhythmogenic right ventricular cardiomyopathy. Nat Genet. 2004;36(11):1162–1164. doi:10.1038/ng1461
86. Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113(9):1171–1179. doi:10.1161/CIRCULATIONAHA.105.583674
87. Syrris P, Ward D, Evans A, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy associated with mutations in the desmosomal gene desmocollin-2. Am J Hum Genet. 2006;79(5):978–984. doi:10.1086/509122
88. Beffagna G, Occhi G, Nava A, et al. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovasc Res. 2005;65(2):366–373. doi:10.1016/j.cardiores.2004.10.005
89. Merner ND, Hodgkinson KA, Haywood AFM, et al. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–821. doi:10.1016/j.ajhg.2008.01.010
90. van Tintelen JP, Van Gelder IC, Asimaki A, et al. Severe cardiac phenotype with right ventricular predominance in a large cohort of patients with a single missense mutation in the DES gene. Heart Rhythm. 2009;6(11):1574–1583. doi:10.1016/j.hrthm.2009.07.041
91. Norgett EE, Hatsell SJ, Carvajal-Huerta L, et al. Recessive mutation in desmoplakin disrupts desmoplakin-intermediate filament interactions and causes dilated cardiomyopathy, woolly hair and keratoderma. Hum Mol Genet. 2000;9(18):2761–2766. doi:10.1093/hmg/9.18.2761
92. Bauce B, Basso C, Rampazzo A, et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur Heart J. 2005;26(16):1666–1675. doi:10.1093/eurheartj/ehi341
93. Castelletti S, Vischer AS, Syrris P, et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: genotype-phenotype correlation. Int J Cardiol. 2017;249:268–273. doi:10.1016/j.ijcard.2017.05.018
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.