")
Back to Journals » Biologics: Targets and Therapy » Volume 15
Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy
Authors Hawkins ER , D'Souza RR, Klampatsa A
Received 4 March 2021
Accepted for publication 6 April 2021
Published 14 April 2021 Volume 2021:15 Pages 95—105
DOI https://doi.org/10.2147/BTT.S291768
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shein-Chung Chow
Elizabeth R Hawkins, Reena R D’Souza, Astero Klampatsa
Division of Cancer Therapeutics, The Institute of Cancer Research, London, UK
Correspondence: Astero Klampatsa
Thoracic Oncology Immunotherapy Group, Division of Cancer Therapeutics, The Institute of Cancer Research, 15 Cotswold Road, Sutton, London, SM2 5NG, UK
Tel +44 208 722 4090
Email [email protected]
Abstract: Chimeric antigen receptor (CAR) T-cell therapy engineers T-cells to express a synthetic receptor which redirects effector function to the tumor, to improve efficacy and reduce toxicities associated with conventional treatments, such as radiotherapy and chemotherapy. This approach has proved effective in treating hematological malignancies; however, the same effects have not been observed in solid tumors. The immunosuppressive tumor microenvironment (TME) creates a significant barrier to solid tumor efficacy and reduces the anti-cancer activity of endogenous tumor-resident immune cells, enabling cancer progression. In recent years, researchers have attempted to enhance CAR T-cell function in the TME by engineering the cells to express various proteins alongside the CAR. Examples of this engineering include inducing CAR T-cells to secrete cytokines or express cytokine receptors to modulate the cytokine milieu of the TME. Alternatively, the CAR T-cell may secrete antibody-like proteins to target a range of tumor antigens. Collectively, these methods are termed armored CAR T-cell therapy, and in this review, we will discuss the range of armored CAR T-cell approaches which have been investigated to date.
Keywords: armored CAR T-cells, immunotherapy, tumor microenvironment, solid tumors, immunosuppression, cell therapy
Introduction
Immunotherapy
Conventional cancer therapies like radiotherapy and chemotherapy are known to be effective at killing cancer cells, however they cannot effectively distinguish between malignant tissue and fast proliferating healthy cells, resulting in a variety of severe adverse effects.1,2 Due to the lack of specificity of available treatments, the idea for cancer immunotherapy emerged. One immunotherapy approach is cellular therapy which takes advantage of useful immune functions including antigen specificity, ability to expand on activation, trafficking to the area of interest and forming memory toward the target antigen.
Adoptive T-cell therapy (ACT) isolates T-cells from patients, expands them ex vivo and then reinfuses them into the patient. This was first conducted with T-cells isolated from patients’ tumors, called tumor-infiltrating lymphocytes (TILs) which, when non-specifically expanded, have demonstrated some clinical efficacy in melanoma.3,4 However, this approach has important limitations: 1) the tumors need to be resectable and with enough T-cells present for harvesting, 2) the antigens that the re-infused T-cells target are unknown, and 3) ex-vivo expansion of TILs is a highly laborious and expensive process. To further increase efficacy and specificity, scientists instead inserted exogenous T-cell receptors (TCRs), which had specificity for known tumor antigens, into T-cells. This approach has shown clinical benefit in a small proportion of patients,5,6 but caused problems with major-histocompatibility complex (MHC) incompatibility due to the allogeneic TCR, resulting in adverse effects.7 Subsequently this led to the development of synthetic receptors, called chimeric antigen receptors (CARs), which are MHC-unrestricted to prevent associated toxicities.
CARs are designed to recognize specific cancer-associated antigens and comprise of an antibody-derived single-chain variable region (scFv) for antigen recognition and binding, a membrane-spanning domain and an intracellular signaling domain. Endogenous TCRs have an intracellular CD3ζ domain which initiates downstream killing pathways on receptor activation, in a process known as signal 1. In normal physiology, TCR engagement is not sufficient to activate T-cell signaling and activation of costimulatory molecules on the T-cell is also required, termed signal 2. This costimulation enables cross-linking between the TCR and costimulatory receptors, leading to T-cell activation.
Three generations of CAR T-cells have been extensively investigated, with the first-generation designed to mimic a TCR in function, using the intracellular CD3ζ domain. An initial study using a first-generation CAR T-cell found that CARs were capable of inducing antigen-specific cytotoxicity in vitro, but cytokine secretion was weak and transient in the absence of costimulatory ligands on target cells.8 Despite showing some promise in early investigations, follow-up studies found that first-generation CAR T-cells produced minimal or no clinical benefit, targeting different antigens in a variety of cancers, and also had limited persistence following patient infusion.9–11
Subsequently, the second-generation of CAR T-cell was developed, incorporating a costimulatory domain into the intracellular region of the CAR to combine signals 1 and 2, with the aim to enhance T-cell activation and persistence. These CARs were shown to mediate effective in vitro anti-tumor functions along with showing marked efficacy with enhanced expansion, persistence, and cytokine secretion in vivo.12,13 Further investigations in Phase I and II clinical trials have shown that second-generation CAR T-cells provide significant clinical benefit, with high proportions of patients experiencing complete responses.14–16
Different costimulatory domains were demonstrated to have different effects on T-cell function. For example, despite having similar clinical efficacy, a CAR with a 4–1BB costimulatory domain produced longer persisting T-cells than using CD28 costimulation.17 In contrast, another study found that the CD28 costimulatory domain significantly increased basal CAR T-cell proliferation compared to using 4–1BB.18 Thus, third-generation CARs were developed to combine the signaling capacity and T-cell functionality of two costimulatory domains within one CAR.
Investigations into third-generation CARs have provided evidence that having two costimulatory domains can enhance CAR T-cell anti-tumor function and increase T-cell persistence in vivo.19,20 In addition, third-generation CAR T-cells have been shown to enhance efficacy, proliferation, and cytokine production in the clinic.21 However, there is contradictory evidence as to whether second- or third-generation CAR T-cells produce more significant responses in patients.21,22
Tumor Microenvironment
Second and third generations of CAR T-cell therapies have shown promising clinical benefit in a range of hematological malignancies.14,15,21,23 This led to approval of two second-generation CD19-targeting CAR T-cells by the US Food and Drug Administration and the European Medicines Agency, to treat B-cell lymphomas and acute lymphoblastic leukemia. To date, this success has not been observed in solid tumors, despite attempts to target many different antigens across a range of tumor types.24–26 For hematological malignancies, CAR T-cell infusion delivers the therapy directly where it is required, allowing CAR T-cells to target single malignant cells. In solid tumors, the T-cells have to overcome multiple barriers simply to localize to the tumor site. Penetration of the solid tumor mass by the T-cells proves an additional difficult obstacle, but even if this is overcome the T-cells are then met by a highly immunosuppressive tumor microenvironment (TME) which impedes the anti-tumor effect that CAR T-cells can elicit.27
Within the mass of a solid tumor, there is a variety of cell types alongside the malignant cells which interact with each other using signals such as cytokines, chemokines, and growth factors (Figure 1). Collectively, they form a TME which is immunosuppressive and supports tumor survival, growth, and metastasis. Cells present in the solid tumor include immune cells, fibroblasts, and endothelial cells, each with different functions. Some immune cells within the TME are potent cytotoxic cells and high proportions of them have been associated with good prognosis in patients. These include CD8+ cytotoxic T-cells,28 CD4+ T helper 1 (Th1) cells,28 B-cells,29 and natural killer (NK) cells,30 which secrete cytokines to promote immune cell survival and function while aiding the anti-cancer immune response. Examples of these cytokines are interleukin-2 (IL-2) and interferon-γ (IFN-γ).
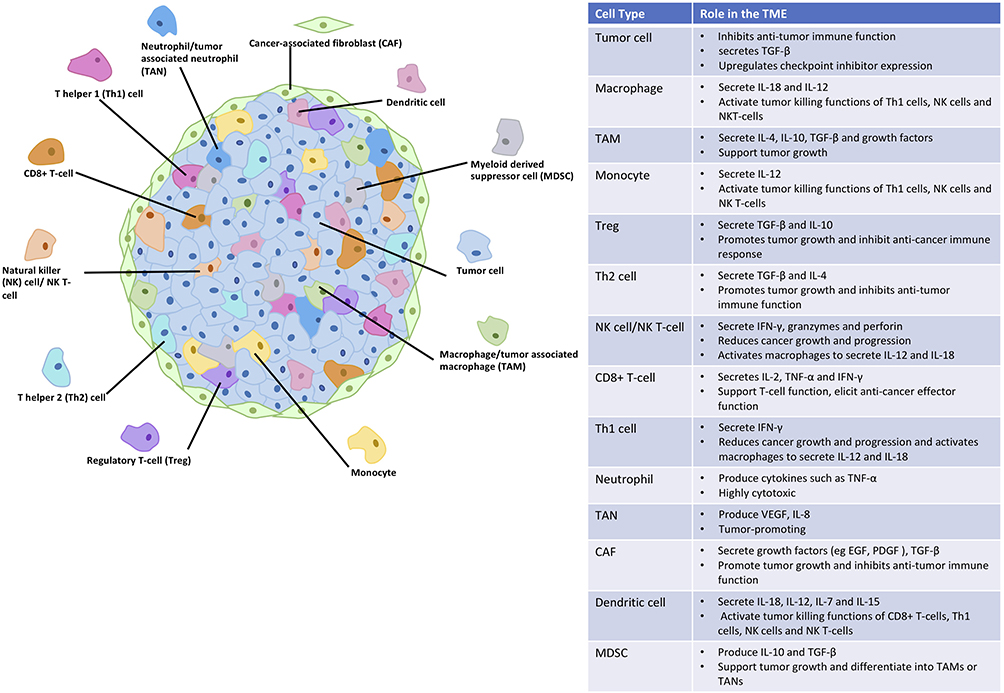 |
Figure 1 The cells found in solid tumors and their role in the regulation of the TME. |
A number of immune cells within the TME, however, secrete cytokines such as IL-10, transforming growth factor-β (TGF-β), and IL-4 which are suppressive and reduce the anti-cancer immune response of the cytotoxic cells present. These immunosuppressive effects are caused by regulatory T-cells (Tregs), CD4+ T helper 2 (Th2) cells, and tumor-associated macrophages (TAMs), all of which are associated with a poor prognosis when found at high levels in patient tumors.28,31,32 Additionally, cancer-associated fibroblasts (CAFs) secrete TGF-β along with mitogenic growth factors that activate cell growth and division in cancer cells,33 further promoting tumor progression.
The balance between the suppressor and activator immune cell activity within the TME is affected by many factors. Cancer cells proliferate rapidly resulting in hypoxia, as the surrounding vasculature is unable to support the tumor. Low oxygen levels impede immune function in a variety of ways, for example, evidence suggests that hypoxia increases Th2 cell activity while reducing the Th1 response in the tumor,34 and aids macrophage polarization to the immunosuppressive phenotype.35 Cancer cells can also upregulate expression of immune checkpoint ligands,36 which usually bind to receptors on T-cells to prevent immune overactivation. These checkpoints include programmed cell death protein 1 (PD-1) and its ligand PD-L1, along with cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and its B7 ligands. Increased expression in cancer cells can “switch off” the anti-cancer immune response and enable cancer growth.
Cells within the TME produce factors including adenosine and prostaglandin E2 (PGE2) which also prevent immune activation. Adenosine acts on effector T-cell A2 receptors to increase protein kinase A (PKA) which downregulates TCR signaling, reduces IL-2 secretion, and inhibits proliferation and cytokine production.37 Simultaneously, increased PKA enhances Treg activity and increases immune checkpoint expression.37 PGE2 also reduces immune function by downregulating Th1 cell function and enhancing Th2 cells and Tregs.38 Overall, this produces a hostile environment within the tumor, which prevents anti-cancer immune function.
Armored CAR T-Cells
To overcome the drawback of the TME on CAR T-cell therapy in solid tumors, a fourth generation of CAR T-cell has been developed. This generation, also known as armored CAR T-cells, are engineered to express proteins alongside a second- or third-generation CAR, shown in Figure 2, to reduce immunosuppression and further mediate anti-tumor efficacy. In this review, we will explore the types of armored CAR T-cells and how production of different proteins alters immune activity.
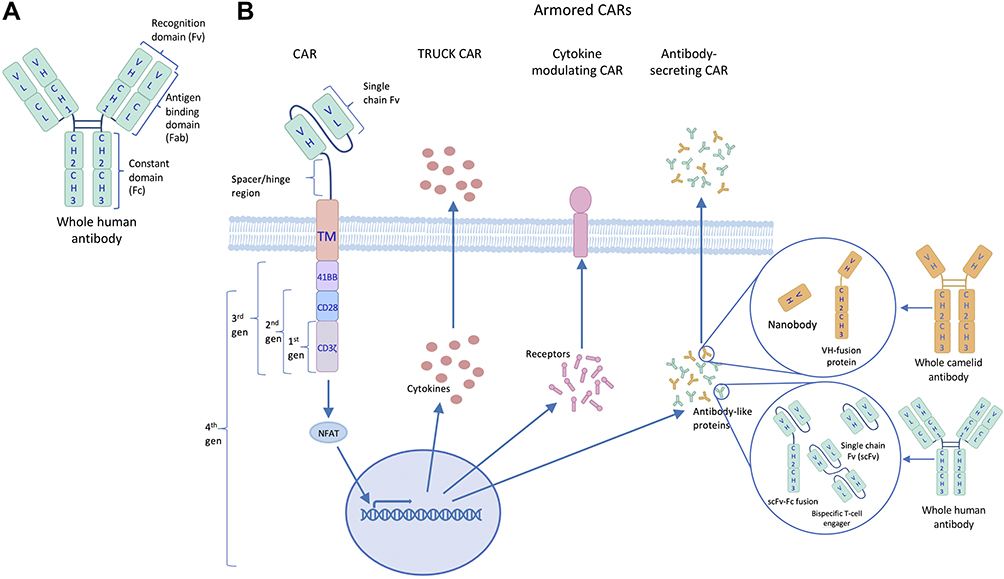 |
Figure 2 The evolutionary structure of chimeric antigen receptors (CARs). (A) The targeting moiety (scFv) of CARs derives from the recognition domain of human antibodies. (B) CAR designs have evolved beyond the 3 generations, which were based on added costimulatory domains, to include TRUCK, cytokine-modulating and antibody-secreting constructs, collectively known as 4th generation or armored CARs. |
Truck CAR T-Cells
The term armored CAR incorporates TRUCK (T-cells Redirected towards Universal Cytokine Killing) CAR T-cells, a specific type of armored CAR T-cells which secrete cytokines to interfere with the immunosuppressive cytokine profile within the solid tumor. By engineering CAR T-cells to modulate the cytokine milieu, it could be possible to turn the TME immune-activating and increase anti-cancer function of both the CAR T-cells and resident immune cells.
IL-12
IL-12 is a cytokine released from monocytes, macrophages, and dendritic cells (DCs) in response to infection. Secretion of this cytokine drives naïve T-cells to differentiate into Th1 cells involved in immune-stimulation, rather than Th2 cells which are immunosuppressive,39 while also stimulating NK cells. These differentiated Th1 cells and expanded NK cells secrete IFN-γ,40 which in turn induces further IL-12 release from macrophages,41 producing an immune-activating positive feedback loop. Thus, employing this signaling into CAR T-cells could enhance solid tumor efficacy by reactivating immune cells in the TME.
Studies of CD19-targeting CAR T-cells constitutively secreting IL-12 have shown that it enhances anti-tumor efficacy compared to the CD19 CAR alone, both in vitro and in vivo,42 and induces a bystander affect in host immune cells in vivo, providing immune memory to the cancer antigen.43 Additionally, this approach has been shown to enhance preclinical efficacy compared to T-cells expressing only a CAR in an ovarian cancer mouse model.44
Previous studies however, observed that, at effective doses systemic administration of IL-12 was highly toxic, including one study where these toxicities resulted in 2 patient deaths.45 Due to concerns about potential systemic toxicities from CAR T-cells constitutively secreting IL-12, T-cells with inducible IL-12 secretion upon CAR engagement were developed,46,47 to direct IL-12 secretion and function to the tumor site. Interestingly, one study found CAR induction of IL-12 not only reduced antigen-positive tumor growth in mice compared to CAR T-cells lacking the cytokine, but also prevented growth of antigen-negative tumor cells, however only in the presence of antigen-positive targets.46 On further investigation, this was found to be due to increased macrophage accumulation at the tumor site in the presence of IL-12, resulting in antigen-independent anti-tumor activity.46 This demonstrates the potential of TRUCK CAR T-cells to enhance tumor cytotoxicity, induce a bystander anti-tumor immune response, and enable cytotoxicity of the variety of cells within a solid tumor, irrespective of antigen expression.
IL-18
IL-18 is released primarily from macrophages and DCs as an inactive pro-form. The pro-form is constitutively expressed in these cells and, on cell activation, the protein is cleaved to produce functional IL-18. As with IL-12, IL-18 activates Th1 and NK cells to release pro-inflammatory cytokines, namely IFN-γ.48 Therefore, IL-18 secretion could enhance CAR T-cell function in a similar way to IL-12. Additionally, in the clinic, systemic IL-18 administration was shown to have minimal toxicity, with the main adverse effects being mild fever, headaches and nausea,49 suggesting that IL-18 is relatively safe to use.
By designing anti-CD19 CAR T-cells which constitutively secrete IL-18, it was shown that this enhanced CAR T-cell expansion and persistence and increased the survival of mice bearing CD19+ tumors, while also activating the endogenous immune system compared to CAR T-cells without cytokine secretion.50 Despite the previous safety of systemic IL-18, researchers have developed CAR T-cells with inducible IL-18 secretion which have been shown to reduce the size of advanced pancreatic tumors in mice and prolonged survival compared to treatment with the CAR alone.51 This study also found that IL-18 producing CAR T-cells were more effective at controlling late-stage disease than CAR T-cells producing IL-12.51
A study looking at TCR transfer therapy, rather than CAR T-cells, accompanied with the inducible secretion of either IL-12 or IL-18, found that IL-18 secretion induced no toxicities in vivo while IL-12 resulted in marked increases in systemic pro-inflammatory cytokines and associated severe toxicity.52 Taken together, these data suggest that IL-18 secretion by CAR T-cells could be more efficacious than IL-12 secretion and also safer for patient infusion.
IL-15
There is evidence that numerous cell types are responsible for the production of IL-15, including macrophages and DCs,53,54 and once released, IL-15 stimulates CD8+ T-cells and NK cells which increases their proliferation and cytotoxic capacity. Administration of IL-15 to mice has been shown to enhance anti-tumor activity of adoptively transferred CD8+ tumor-reactive T-cells,55 which suggests IL-15 could also enhance anti-tumor activity of CAR T-cell therapy.
CAR T-cells further engineered to secrete IL-15 have been shown to augment tumor cytotoxicity compared to using CAR alone T-cells in vitro and in vivo,56,57 and increase T-cell expansion on antigen recognition in vitro.56 Similarly, this can increase antigen-independent T-cell proliferation, while enabling T-cell persistence after tumor clearance.57 It was also observed that the secretion of IL-15 provided greater protection against tumor rechallenge in vivo than CAR T-cells not secreting IL-15.56 Interestingly, one of the studies used a form of IL-15 tethered to the membrane and found that this promoted the CAR T-cells to develop a memory phenotype.57 These data suggest that IL-15 could provide long-term CAR T-cell-mediated immunity toward the cancer antigen along with enhancing CAR T-cell function within the TME.
A clinical trial investigating systemic IL-15 administration for the treatment of metastatic cancers found that this resulted in toxicities including hypotension, thrombocytopenia, and liver toxicity.58 Thus, before IL-15 armored CAR T-cells are used in the clinic it is important to consider that inducible or tethered IL-15 may be better to avoid high systemic levels of the cytokine.
IL-7
When activated, CAR T-cells secrete IL-2 to maintain their survival and function. However, in solid tumors, IL-2 secretion also aids survival of Tregs, a T-cell subset which causes immunosuppression by secretion of TGF-β. Therefore, researchers have looked into replacing IL-2 secretion by CAR T-cells with secretion of another cytokine that can promote CAR T-cell survival and function without enhancing Treg activity. IL-7 has been shown to augment proliferation and function of CAR T-cells without affecting Tregs,59 meaning there is potential for this cytokine in enhancing CAR T-cell function in solid tumors.
IL-7 secretion induced by CAR T-cell engagement was found to amplify CAR T-cells in the presence of TGF-β, while CARs not secreting IL-7 were suppressed by the cytokine.60 Similar effects were observed when the same concept was trialed with IL-15 secretion,60 however IL-15 stimulates Tregs so this approach would potentially enhance TGF-β release as seen with IL-2. These data suggest that secretion of IL-7 by CAR T-cells not only enhances CAR T-cell function but also partly prevents suppression by immunosuppressive cytokines in the TME.
Modulating Cytokine Function
Altering the cytokine environment within a solid tumor may be critical for switching the TME from immunosuppressive to pro-inflammatory and thus enhance CAR T-cell function. As well as engineering CAR T-cells to secrete cytokines, it is possible to alter cytokine function by manipulating how the CAR T-cells respond to cytokines. This is another type of armored CAR T-cell which may increase solid tumor efficacy.
IL-7
As IL-7 is a T-cell stimulating cytokine, different armored CAR T-cell approaches have been taken to increase its proinflammatory functions within the tumor. The same group who studied IL-7 secretion by CAR T-cells also used a hybrid receptor combining IL-7 and IL-2 receptor domains, to convert an IL-7 ligand signal into IL-2 intracellular signaling pathways.60 This was found to counteract immunosuppression of CAR T-cell proliferation and cytotoxic capacity when IL-7 was supplied.60 This is a method of enabling stimulation of CAR T-cells via the IL-2 pathway, but without stimulating Tregs that express IL-2 receptors, allowing selective T-cell activation and preventing immune suppression.
TGF-β
TGF-β is produced as a pro-form in a variety of cell types, but in the TME this is mostly by cancer and stroma cells. When active, TGF-β prevents T-cell differentiation toward the Th1 and Th2 lineages,61 while promoting differentiation of Tregs.62 The Tregs then produce more TGF-β, further increasing immunosuppression within the tumor, which impedes CAR T-cell function. High levels of TGF-β within the TME have been shown to correspond to poor prognosis in patients,63–65 demonstrating that reducing TGF-β mediated immunosuppression in solid tumors may be key to enhancing CAR T-cell efficacy and clinical outcome.
One approach to overcome TGF-β immune suppression is by co-expressing the CAR with the dominant-negative TGF-β receptor II (dnTGF-βRII).66 This receptor acts as a decoy receptor for TGF-β, sequestering the cytokine without inducing downstream signaling, thus reducing the immunosuppressive functions within the TME. The co-expression of the dnTGF-βRII was found to increase CAR T-cell proliferation, reduce exhaustion, and enhance anti-tumor function and persistence in vivo.66 Led from these studies, the researchers are now recruiting for a phase I clinical trial, using this novel CAR T-cell approach in patients with castrate-resistant prostate cancer (clinical trial identifier NCT03089203).
It has also been attempted to overcome the function of TGF-β by co-expressing a CAR with constitutively active Akt (caAkt).67 In T-cells, caAkt enhances proliferation and cytokine secretion along with determining T-cell sensitivity to inhibitory factors such as TGF-β. It was observed that this engineering enhanced CAR T-cell activation, proliferation, and anti-tumor function while preventing T-cell suppression and Treg differentiation.67 These approaches show promise for increasing CAR T-cell function within solid tumors by regulating the local TGF-β activity, which could improve solid tumor responses in the clinic.
IL-4
IL-4 is produced within the TME by cells including Th2 cells and has been shown to support tumor growth and prevent anti-tumor immune functions.68 Importantly, the immunosuppressive function of IL-4 has been shown to be a key mediator of cancer-promoting TAMs.69 Therefore, altering the way CAR T-cells respond to IL-4 could be useful for enhancing their function.
To alter IL-4 immunosuppression within the tumor, Mohammed et al conducted similar research as discussed previously, with IL-7, making a hybrid IL-4/IL-7 receptor.70 In this case, the exodomain of the IL-4 receptor was fused to the IL-7 intracellular domain so that on stimulation of CAR T-cells by the immunosuppressive cytokine IL-4, the immune activation pathways of IL-7 are initiated, enhancing the CAR T-cell anti-tumor response. This research found that while expression of a CAR T-cell increased anti-tumor activity, these cells had poor expansion in vitro.70 Conversely, expression of the hybrid IL-4/IL-7 receptor enhanced proliferation of the T-cells, but also dampened their cytotoxic capacity.70 Combining both approaches into an armored CAR T-cell however, has shown to enhance both expansion in the presence of IL-4 and also increased antigen-specific tumor lysis in vitro, while producing greater anti-tumor activity in vivo.70 Altering IL-4 function could thus be useful in producing the desired anti-tumor CAR T-cell function.
CD40L
Cluster of differentiation 40 ligand (CD40L) is a transmembrane protein which is upregulated on T-cells upon activation. Binding of CD40L to its receptor (CD40) on DCs activates T-cells to secrete pro-inflammatory cytokines, such as IL-12 and IFN-γ, and induces T-cell proliferation.71 Additionally, when binding to CD40 on tumor cells, CD40L has been shown to facilitate tumor apoptosis.72 Due to the anti-tumor effects of the CD40L-CD40 interactions, the Brentjens group engineered a CD19 targeting CAR T-cell to also express CD40L, which was shown to enhance the in vitro CAR T-cell efficacy against CD19+ tumors and moderately, although significantly, increased survival of tumor bearing mice.73 This again demonstrates a novel approach to manipulate cytokine activity on CAR T-cells as a way to overcome suppression by the TME.
Secretion of Antibody-Like Proteins
The specificity of the antibody recognition domain makes it ideal for targeting antigens of interest on cancer cells, hence CARs were developed using this domain as the extracellular scFv part of the construct. This ability of antibodies to bind antigens also provides potential for dual targeting of antigens on a cancer cell using one therapy. Therefore, a CAR that secretes antibody-like proteins, may enhance CAR T-cell redirection to the tumor and recognition of cancer-associated antigens. Additionally, antibodies have the ability to mediate separate immune killing mechanisms, including antibody-dependent cell-mediated cytotoxicity (ADCC), meaning it is possible for this combination of T-cell engineering to enhance cancer cytotoxicity in multiple ways.
Checkpoint Inhibitors
Checkpoint inhibitors are antibodies specific to either immune checkpoint ligands on tumor cells or receptors on T-cells, working to prevent the receptor-ligand interaction which “switches off” the immune response to cancer. These checkpoint inhibitors have shown significant clinical benefits in melanoma74,75 and non-small-cell lung cancer,76 along with other cancer types.77 Furthermore, administration of both CAR T-cells and checkpoint inhibitors has been shown to increase clinical benefit compared to treatment with CAR T-cells alone in a small clinical study involving six patients.78 The synergistic effect of CAR T-cells with checkpoint inhibitor antibodies led to the development of armored CAR T-cells, which secrete checkpoint inhibitors to reduce immune suppression within the solid tumor.
Studies looking into checkpoint blockade secretion by CAR T-cells have mainly focused on secretion of anti-PD-1 scFvs. This approach takes the antigen recognition domain of the antibody, which is a small molecule for the CAR T-cell to produce and secrete, but can effectively bind to immune checkpoints to block interactions. One study found that anti-PD-1 scFv secretion by CAR T-cells enhanced proliferation of the CAR T-cells and also reduced PD-1 expression on CAR T-cells in vitro, which can further reduce inhibition.79 Additionally, anti-PD-1 scFv secretion was seen to enhance CAR T-cell anti-tumor function in a gastric cancer mouse model.79 Similar effects of anti-PD-1 scFv secretion by CAR T-cells were reported in another study, which showed this increased T-cell cytokine production and proliferation in vitro, while in vivo there was enhanced anti-tumor efficacy and CAR T-cell accumulation in the TME.80
Suarez et al used a CAR T-cell secreting an anti-PD-1 scFv-Fc fusion protein which was shown to reduce in vitro T-cell exhaustion.81 Depending on the antibody isoform used, the therapy was seen to induce ADCC of the targets through the Fc domain, demonstrating the potential to enhance the anti-tumor response to these CAR T-cells through multiple mechanisms.81 It was also observed to reduce in vivo tumor growth, recruit NK cells to the tumor, and resulted in CAR T-cells with reduced expression of exhaustion markers.81 Overall, this approach shows promise as it reduces suppression of CAR T-cells and subsequently enhances their function, while adding an Fc domain also enables activation of multiple immune killing mechanisms and induces a bystander effect to enhance cancer cytotoxicity.
Nanobodies
A nanobody is a small protein consisting of the single domain variable heavy chain (VH) derived from a camelid antibody. The small size of nanobodies means they are stable, but they still maintain high affinities for target antigens.82 Previously, nanobody therapies have been shown to be efficacious in cancer studies. One such investigation found that nanobodies controlled tumor growth with greater potency than using whole antibodies.83 This demonstrates potential for using nanobody secretion by CAR T-cells which are easier to introduce into the T-cell than other antibody structures due to the smaller size.
A recent study engineered CAR T-cells to secrete nanobodies targeting the antigen CD47 on cancer cells,84 which acts to “switch-off” phagocytosis. Despite the secretion of nanobodies not increasing tumor cytotoxicity in vitro compared to non-secreting CAR T-cells, it was shown to delay tumor growth and prolong survival in vivo.84 They also showed that due to the small size of nanobodies, CAR T-cells can be engineered to secrete both a nanobody and a VH-fusion protein (nanobody + Fc region), allowing targeting of multiple cancer antigens alongside the CAR.84 However, the functionality of these dual-secreting CAR T-cells was not assessed.84 Nevertheless this is an important finding as it could enable whole tumor targeting by CAR T-cells due to the heterogeneity of antigen expression throughout solid tumors.
BiTEs
Bispecific T-cell engagers (BiTEs) consist of two scFvs with different specificities joined together. One of these scFvs binds to CD3 on T-cells while the other can bind a target cancer antigen. By engaging both the T-cell and the cancer cell, the small BiTE brings the two close together, enabling them to form an immune synapse.85 This allows the TCR to bind antigens on the cancer cell, activating downstream killing pathways.
Blinatumomab is a BiTE targeting CD19, which is overexpressed in B-cell malignancies, along with CD3. In clinical trials, this BiTE has been shown to produce significant benefit in patients with non-Hodgkin’s lymphoma, where one study found very low doses produced remissions in all patients enrolled.86 Previously, T-cells have been engineered to secrete BiTEs and this was observed to enhance anti-tumor function of the engineered T-cells while recruiting bystander T-cells to the tumor.87 Therefore, engineering CAR T-cells to secrete BiTEs has the potential to enhance their anti-tumor efficacy by increasing contact between the CARs and tumor cells.
Recently, one study looked at the effect of BiTE secretion on CAR T-cell function and discovered that these BiTEs not only bound CAR T-cells, but also bystander non-engineered T-cells,88 demonstrating that BiTE-secreting CAR T-cells could activate a more global anti-cancer immune response. This dual engineering approach produced heterogenous anti-tumor activity in mouse models, and also produced a more favorable T-cell differentiation pathway, directing the CAR T-cells toward the effector memory rather than the central memory phenotype.88 This is interesting for the potential of long-term protection against cancer by CAR T-cells, along with activation of tumor-resident T-cells to increase the anti-cancer immune response.
Conclusion
At present, there is a significant research drive to increase the efficacy of CAR T-cell therapy in solid tumors, with many researchers focusing on the development of novel armored CAR T-cell approaches. This rapidly progressing field of immunotherapy is promising, with preclinical and clinical data suggesting this could bridge the gap for solid tumor treatment. Existing data have demonstrated beneficial effects against a range of problems associated with targeting the solid tumor including reducing immunosuppression within the TME, recruiting and activating endogenous immune cells against the tumor, development of anti-cancer immune memory, and targeting multiple antigens on the heterogenous tumor. Together, these exciting results encourage hope for clinical approval of one or more armored CAR T-cell in the future.
Author Contributions
EH, RRD and AK designed the manuscript, EH and AK wrote the manuscript; EH, RRD and AK edited, revised and reviewed the manuscript. All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Funding
EH is supported by a studentship award from a generous anonymous philanthropist. RRD and AK are supported by the Cris Cancer Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Nurgali K, Jagoe RT, Abalo R. Editorial: adverse effects of cancer chemotherapy: anything new to improve tolerance and reduce sequelae? Front Pharmacol. 2018;9:245. doi:10.3389/fphar.2018.00245
2. Berkey FJ. Managing the adverse effects of radiation therapy. Am Fam Physician. 2010;82(4):381–388, 394.
3. Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi:10.1056/NEJM198812223192527
4. Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86(15):1159–1166. doi:10.1093/jnci/86.15.1159
5. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi:10.1200/JCO.2010.32.2537
6. Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi:10.1038/mt.2010.272
7. Schendel DJ, Frankenberger B. Limitations for TCR gene therapy by MHC-restricted fratricide and TCR-mediated hematopoietic stem cell toxicity. Oncoimmunology. 2013;2(1):e22410. doi:10.4161/onci.22410
8. Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia. 1999;1(2):123–127. doi:10.1038/sj.neo.7900018
9. Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi:10.1038/sj.mt.6300104
10. Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi:10.1158/1078-0432.CCR-06-1183
11. Jensen MC, Popplewell L, Cooper LJ, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–1256. doi:10.1016/j.bbmt.2010.03.014
12. Davila ML, Kloss CC, Gunset G, Sadelain M. CD19 CAR-targeted T cells induce long-term remission and B Cell Aplasia in an immunocompetent mouse model of B cell acute lymphoblastic leukemia. PLoS One. 2013;8(4):e61338. doi:10.1371/journal.pone.0061338
13. Vera J, Savoldo B, Vigouroux S, et al. T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood. 2006;108(12):3890–3897. doi:10.1182/blood-2006-04-017061
14. Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi:10.1056/NEJMoa1407222
15. Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi:10.1056/NEJMoa1707447
16. Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-Cell Lymphoma. N Engl J Med. 2019;380(1):45–56. doi:10.1056/NEJMoa1804980
17. Ying Z, He T, Wang X, et al. Parallel Comparison of 4-1BB or CD28 Co-stimulated CD19-Targeted CAR-T Cells for B Cell Non-Hodgkin’s Lymphoma. Mol Ther Oncolytics. 2019;15:60–68. doi:10.1016/j.omto.2019.08.002
18. Quintarelli C, Orlando D, Boffa I, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. 2018;7(6):e1433518. doi:10.1080/2162402X.2018.1433518
19. Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106(9):3360–3365. doi:10.1073/pnas.0813101106
20. Zhao Z, Condomines M, van der Stegen SJC, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–428. doi:10.1016/j.ccell.2015.09.004
21. George P, Dasyam N, Giunti G, et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: a phase I clinical trial protocol (ENABLE). BMJ Open. 2020;10(2):e034629. doi:10.1136/bmjopen-2019-034629
22. Condomines M, Arnason J, Benjamin R, Gunset G, Plotkin J, Sadelain M. Tumor-Targeted Human T Cells Expressing CD28-Based Chimeric Antigen Receptors Circumvent CTLA-4 Inhibition. PLoS One. 2015;10(6):e0130518. doi:10.1371/journal.pone.0130518
23. Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi:10.1126/scitranslmed.aac5415
24. Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66(11):1425–1436. doi:10.1007/s00262-017-2034-7
25. Lamers CH, Sleijfer S, van Steenbergen S, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi:10.1038/mt.2013.17
26. Brown CE, Badie B, Barish ME, et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi:10.1158/1078-0432.CCR-15-0428
27. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ, Guedan S. CAR-T Cells hit the tumor microenvironment: strategies to overcome tumor escape. Front Immunol. 2020;11:1109. doi:10.3389/fimmu.2020.01109
28. Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi:10.1038/nrc3245
29. Coronella JA, Telleman P, Kingsbury GA, Truong TD, Hays S, Junghans RP. Evidence for an antigen-driven humoral immune response in medullary ductal breast cancer. Cancer Res. 2001;61(21):7889–7899.
30. Zhang S, Liu W, Hu B, et al. Prognostic significance of tumor-infiltrating natural killer cells in solid tumors: a systematic review and meta-analysis. Front Immunol. 2020;11:1242. doi:10.3389/fimmu.2020.01242
31. Hiraoka N, Onozato K, Kosuge T, Hirohashi S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res. 2006;12(18):5423–5434. doi:10.1158/1078-0432.CCR-06-0369
32. Xu J, Yu Y, He X, et al. Tumor-associated macrophages induce invasion and poor prognosis in human gastric cancer in a cyclooxygenase-2/MMP9-dependent manner. Am J Transl Res. 2019;11(9):6040–6054.
33. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel). 2015;7(4):2443–2458. doi:10.3390/cancers7040902
34. Lederer JA, Rodrick ML, Mannick JA. The effects of injury on the adaptive immune response. Shock. 1999;11(3):153–159. doi:10.1097/00024382-199903000-00001
35. Laoui D, Van Overmeire E, Di Conza G, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30. doi:10.1158/0008-5472.CAN-13-1196
36. Marwitz S, Scheufele S, Perner S, Reck M, Ammerpohl O, Goldmann T. Epigenetic modifications of the immune-checkpoint genes. Clin Epigenetics. 2017;9:51. doi:10.1186/s13148-017-0354-2
37. Leone RD, Emens LA. Targeting adenosine for cancer immunotherapy. J Immunother Cancer. 2018;6(1):57. doi:10.1186/s40425-018-0360-8
38. Kobayashi K, Omori K, Murata T. Role of prostaglandins in tumor microenvironment. Cancer Metastasis Rev. 2018;37(2–3):347–354. doi:10.1007/s10555-018-9740-2
39. Manetti R, Parronchi P, Giudizi MG, et al. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J Exp Med. 1993;177(4):1199–1204. doi:10.1084/jem.177.4.1199
40. Chan SH, Perussia B, Gupta JW, et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173(4):869–879. doi:10.1084/jem.173.4.869
41. Skeen MJ, Miller MA, Shinnick TM, Ziegler HK. Regulation of murine macrophage IL-12 production. Activation of macrophages in vivo, restimulation in vitro, and modulation by other cytokines. J Immunol. 1996;156(3):1196–1206.
42. Pegram HJ, Purdon TJ, van Leeuwen DG, et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia. 2015;29(2):415–422. doi:10.1038/leu.2014.215
43. Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate lymphoma in fully lymphoreplete mice through induction of host immunity. Mol Ther Oncolytics. 2018;8:41–51. doi:10.1016/j.omto.2017.12.003
44. Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors. Oncoimmunology. 2015;4(3):e994446. doi:10.4161/2162402X.2014.994446
45. Leonard JP, Sherman ML, Fisher GL, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90(7):2541–2548.
46. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697–5706. doi:10.1158/0008-5472.CAN-11-0103
47. Liu Y, Di S, Shi B, et al. Armored Inducible Expression of IL-12 enhances antitumor activity of glypican-3-targeted chimeric antigen receptor-engineered t cells in hepatocellular carcinoma. J Immunol. 2019;203(1):198–207. doi:10.4049/jimmunol.1800033
48. Takeda K, Tsutsui H, Yoshimoto T, et al. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity. 1998;8(3):383–390. doi:10.1016/S1074-7613(00)80543-9
49. Robertson MJ, Kirkwood JM, Logan TF, et al. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008;14(11):3462–3469. doi:10.1158/1078-0432.CCR-07-4740
50. Avanzi MP, Yeku O, Li X, et al. Engineered tumor-targeted T Cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23(7):2130–2141. doi:10.1016/j.celrep.2018.04.051
51. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet. Cell Rep. 2017;21(11):3205–3219. doi:10.1016/j.celrep.2017.11.063
52. Kunert A, Chmielewski M, Wijers R, Berrevoets C, Abken H, Debets R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology. 2017;7(1):e1378842. doi:10.1080/2162402X.2017.1378842
53. Stonier SW, Ma LJ, Castillo EF, Schluns KS. Dendritic cells drive memory CD8 T-cell homeostasis via IL-15 transpresentation. Blood. 2008;112(12):4546–4554. doi:10.1182/blood-2008-05-156307
54. Mortier E, Advincula R, Kim L, et al. Macrophage- and dendritic-cell-derived interleukin-15 receptor alpha supports homeostasis of distinct CD8+ T cell subsets. Immunity. 2009;31(5):811–822. doi:10.1016/j.immuni.2009.09.017
55. Klebanoff CA, Finkelstein SE, Surman DR, et al. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci U S A. 2004;101(7):1969–1974. doi:10.1073/pnas.0307298101
56. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-specific chimeric antigen receptor and Interleukin-15. Clin Cancer Res. 2019;25(9):2915–2924. doi:10.1158/1078-0432.CCR-18-1811
57. Hurton LV, Singh H, Najjar AM, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113(48):E7788–E7797. doi:10.1073/pnas.1610544113
58. Conlon KC, Lugli E, Welles HC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82. doi:10.1200/JCO.2014.57.3329
59. Perna SK, Pagliara D, Mahendravada A, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2014;20(1):131–139. doi:10.1158/1078-0432.CCR-13-1016
60. Golumba-Nagy V, Kuehle J, Hombach AA, Abken H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol Ther. 2018;26(9):2218–2230. doi:10.1016/j.ymthe.2018.07.005
61. Chen CH, Seguin-Devaux C, Burke NA, et al. Transforming growth factor beta blocks Tec kinase phosphorylation, Ca2+ influx, and NFATc translocation causing inhibition of T cell differentiation. J Exp Med. 2003;197(12):1689–1699. doi:10.1084/jem.20021170
62. Walker MR, Kasprowicz DJ, Gersuk VH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25- T cells. J Clin Invest. 2003;112(9):1437–1443. doi:10.1172/JCI19441
63. Zhou J, Jiang W, Huang W, Ye M, Zhu X. Prognostic values of transforming growth factor-beta subtypes in ovarian cancer. Biomed Res Int. 2020;2020:2170606.
64. Li J, Shen C, Wang X, et al. Prognostic value of TGF-β in lung cancer: systematic review and meta-analysis. BMC Cancer. 2019;19(1):691. doi:10.1186/s12885-019-5917-5
65. Reis ST, Pontes-Júnior J, Antunes AA, et al. Tgf-β1 expression as a biomarker of poor prognosis in prostate cancer. Clinics (Sao Paulo). 2011;66(7):1143–1147. doi:10.1590/s1807-59322011000700004
66. Kloss CC, Lee J, Zhang A, et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–1866. doi:10.1016/j.ymthe.2018.05.003
67. Sun J, Dotti G, Huye LE, et al. T cells expressing constitutively active Akt resist multiple tumor-associated inhibitory mechanisms. Mol Ther. 2010;18(11):2006–2017. doi:10.1038/mt.2010.185
68. Prokopchuk O, Liu Y, Henne-Bruns D, Kornmann M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: evidence for autocrine and paracrine actions. Br J Cancer. 2005;92(5):921–928. doi:10.1038/sj.bjc.6602416
69. Gocheva V, Wang HW, Gadea BB, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–255. doi:10.1101/gad.1874010
70. Mohammed S, Sukumaran S, Bajgain P, et al. Improving chimeric antigen receptor-modified t cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25(1):249–258. doi:10.1016/j.ymthe.2016.10.016
71. Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: t-T help via APC activation. J Exp Med. 1996;184(2):747–752. doi:10.1084/jem.184.2.747
72. Schattner EJ, Mascarenhas J, Bishop J, et al. CD4+ T-cell induction of Fas-mediated apoptosis in Burkitt’s lymphoma B cells. Blood. 1996;88(4):1375–1382. doi:10.1182/blood.V88.4.1375.bloodjournal8841375
73. Curran KJ, Seinstra BA, Nikhamin Y, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23(4):769–778. doi:10.1038/mt.2015.4
74. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi:10.1056/NEJMoa1003466
75. Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372(4):320–330. doi:10.1056/NEJMoa1412082
76. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
77. Lewis AL, Chaft J, Girotra M, Fischer GW. Immune checkpoint inhibitors: a narrative review of considerations for the anaesthesiologist. Br J Anaesth. 2020;124(3):251–260. doi:10.1016/j.bja.2019.11.034
78. Chong EA, Melenhorst JJ, Lacey SF, et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 2017;129(8):1039–1041. doi:10.1182/blood-2016-09-738245
79. Zhou JT, Liu JH, Song TT, Ma B, Amidula N, Bai C. EGLIF-CAR-T Cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res. 2020;12:8893–8902. doi:10.2147/CMAR.S260915
80. Ping Y, Li F, Nan S, et al. Augmenting the Effectiveness of CAR-T Cells by Enhanced Self-Delivery of PD-1-Neutralizing scFv. Front Cell Dev Biol. 2020;8:803. doi:10.3389/fcell.2020.00803
81. Suarez ER, Chang D, Sun J, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–34355. doi:10.18632/oncotarget.9114
82. Yang EY, Shah K. Nanobodies: next generation of cancer diagnostics and therapeutics. Front Oncol. 2020;10:1182. doi:10.3389/fonc.2020.01182
83. Roovers RC, Vosjan MJ, Laeremans T, et al. A biparatopic anti-EGFR nanobody efficiently inhibits solid tumour growth. Int J Cancer. 2011;129(8):2013–2024. doi:10.1002/ijc.26145
84. Xie YJ, Dougan M, Ingram JR, et al. Improved antitumor efficacy of chimeric antigen receptor t cells that secrete single-domain antibody fragments. Cancer Immunol Res. 2020;8(4):518–529. doi:10.1158/2326-6066.CIR-19-0734
85. Offner S, Hofmeister R, Romaniuk A, Kufer P, Baeuerle PA. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol Immunol. 2006;43(6):763–771. doi:10.1016/j.molimm.2005.03.007
86. Bargou R, Leo E, Zugmaier G, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321(5891):974–977. doi:10.1126/science.1158545
87. Iwahori K, Kakarla S, Velasquez MP, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol Ther. 2015;23(1):171–178. doi:10.1038/mt.2014.156
88. Choi BD, Yu X, Castano AP, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. 2019;37(9):1049–1058. doi:10.1038/s41587-019-0192-1
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.