")
Back to Journals » International Journal of Nanomedicine » Volume 13
Aptamer–drug conjugate: targeted delivery of doxorubicin in a HER3 aptamer-functionalized liposomal delivery system reduces cardiotoxicity
Authors Dou XQ, Wang H, Zhang J, Wang F, Xu GL, Xu CC, Xu HH, Xiang SS, Fu J, Song HF
Received 29 August 2017
Accepted for publication 7 December 2017
Published 5 February 2018 Volume 2018:13 Pages 763—776
DOI https://doi.org/10.2147/IJN.S149887
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Xiao-qian Dou,1 Hua Wang,2 Jing Zhang,3 Fang Wang,3 Gui-li Xu,1 Cheng-cheng Xu,1 Huan-hua Xu,1 Shen-si Xiang,1 Jie Fu,1 Hai-feng Song1
1Department of Pharmacology and Toxicology, Beijing Institute of Radiation Medicine, Beijing, China; 2Ophthalmology Department, Eye Hospital, China Academy of Chinese Medical Sciences, Beijing, China; 3Bioanalysis Department, United-Power Pharma Tech Co., Ltd., Beijing, China
Introduction: The toxic side effects of doxorubicin (DOX) have limited its use in chemotherapy. Neither liposomal DOX nor pegylated liposomal DOX are able to completely resolve this issue. This is a proof-of-concept study testing aptamer-drug conjugate (ApDC) targeted delivery systems for chemotherapeutic drugs.
Methods: Aptamer library targeting human epidermal growth factor receptor 3 (HER3) was screened and affinity was determined by enzyme-linked immunosorbent assay. Specificity was tested in MCF-7HER3-high, BT474HER3-high, and 293THER3-negative cells using flow cytometry and confocal microscopy. We further developed a HER3 aptamer-functionalized liposome encapsulating DOX and the efficiency of this ApDC was detected by cellular uptake analysis and cell viability assay. In MCF-7 tumor-bearing mice, tumor targeting evaluation, efficacy, toxicity and preliminary pharmocokinetic study was performed.
Results: The candidate #13 aptamer had highest affinity (Kd =98±9.7 nM) and specificity. ApDC effectively reduces the half maximal inhibitory concentration of DOX compared with lipsome-DOX and free DOX. In vivo imaging and preliminary distribution studies showed that actively targeted nanoparticles, such as Apt-Lip-DOX molecules, could facilitate the delivery of DOX into tumors in MCF-7–bearing mice. This targeted chemotherapy caused greater tumor suppression than other groups and alleviated side effects such as weight loss, low survival rate, and organ (heart and liver) injury demonstrated by H&E staining.
Conclusion: The results indicate that targeted chemotherapy using the aptamer–drug conjugate format could provide better tolerability and efficacy compared with non-targeted delivery in relatively low-dose toxic drugs.
Keywords: breast cancer, human epidermal growth factor receptor 3, aptamer–drug conjugate, doxorubicin, cardiac toxicity, targeted therapy
Introduction
Chemotherapy is still the preferred treatment strategy for cancer, along with surgery. Doxorubicin (DOX) is one of the most effective chemotherapeutic agents. Multiple cancers, such as breast cancer, gynecologic cancer, liver cancer, lung cancer, lymphoma, multiple myeloma, head and neck cancer, and soft tissue sarcoma, have shown good response to DOX.1 Unfortunately, treatment with this drug also causes cardiotoxicity, hair loss, and other side effects. Notably, congestive cardiomyopathy was reported in one-third to one-half of elderly patients with B-cell lymphoma treated with cyclophosphamide, DOX, vincristine, and prednisolone (CHOP) with or without rituximab.2
Nanoparticles are now widely used as nonviral delivery systems. The use of DOX encapsulated in nanoparticles is advantageous because of its high efficacy and reduced cardiac toxicity, due its sustained release effect. The pegylated liposome-encapsulated form of DOX entered the market after it obtained approval by the US Food and Drug Administration in 1995.3 In China, DOX hydrochloride liposome injections are also available (eg, Shanghai Fudan-zhangjiang Bio-Pharmaceutical Co.). The pegylated liposome-encapsulated DOX form has low cardiotoxicity, but high skin and oral toxicity (hand–foot syndrome [HFS]) are induced partly because of extravasation of the liposomes due to long circulation time.4,5 Therefore, in addition to pegylated liposome-encapsulated form of DOX, the use of non-pegylated liposomal (NPL)-DOX is also predominant in chemotherapy.6 NPL-DOX as a component of the R-CHOP regimen is a highly effective treatment in patients with diffuse large B cell lymphoma and pre-existing cardiac diseases.6 Though the median left ventricular ejection fraction did not change after chemotherapy, the cardiac events were, indeed, observed in 36% of patients in this cardiac high-risk group; most of these events occurred in the follow-up and, in some cases, a relationship to chemotherapy remains uncertain. A decreased left ventricular ejection fraction was observed in 28% of patients without a greater frequency in the group of patients with pre-existing reduced left ventricular ejection fractions. In summary, the potential cardiotoxicity of NPL-DOX need to be confirmed in a prospective randomized trial. Therefore, to date, issues regarding the enhanced efficacy and reduced toxicity of DOX have not been resolved.
Tumor-targeting strategies for the transportation of chemotherapeutic agents have been achieved via use of antibody–drug conjugates, as approved by the US Food and Drug Administration. Ado-trastuzumab emtansine (Kadcyla) is a human epidermal growth factor receptor 2 (HER2) extracellular domain (ECD)-targeting monoclonal antibody (trastuzumab) combined with mertansine (DM1), the indication of which is late, HER2-positive metastatic breast cancer.7 However, challenges still exist, such as preparation quality, the limitations of macromolecules such as their difficult penetration into the tumors, the immunogenicity of the naked antibody, and the FcγRIIa-mediated toxicologic effects.8 However, non-targeting toxicity is also caused by the shedding of chemotherapeutic agents. Complex pharmacokinetic profiles also contribute to unpredictable efficacy and toxicity. Therefore, other alternative strategies must be explored to fill this gap.
The area of aptamer–drug conjugates (ApDCs) is now emerging, based on the hypothesis that targeted delivery of toxic agents to tumors will provide better tolerability and efficacy. ApDCs can be realized either by covalent conjugation or by noncovalent conjugation through intercalation.9 Zhao et al10 prepared a CD117-targeting aptamer–methotrexate covalent conjugation system in acute myeloid leukemia. Targeted delivery of cytotoxic agents using aptamer-functionalized nanomaterials is also a promising strategy that has been shown to be flexible in its applications.11
Here, we developed a single-stranded (ss) DNA aptamer that could specifically bind to HER3, which has been recognized as a key signaling hub in several clinical contexts.12 High expression of HER3 is not only associated with resistance to HER2 inhibitors in HER2-amplified breast cancer, with anti-estrogen therapies in estrogen receptor-positive breast cancer, and with epidermal growth factor receptor (EGFR) inhibitors in head and neck cancer, but is also involved in a variety of other tumor types, such as melanoma.13 The HER3ECD-targeted aptamer-functionalized liposomal DOX in this study reduced cardiac toxicity and prolonged survival time in tumor-bearing mice (Figure S1). The proof-of-concept study here indicates the potential clinical value of Apt-Lip-DOX biotherapeutics for targeted HER3 (but not limited to HER3) therapy with minimal side effects in patients undergoing chemotherapy.
Materials and methods
Materials
Polyetherimide was obtained from Polysciences (Warrington, PA, USA). All the magnetic beads used in the experiment were purchased from BEAVERBIO (Suzhou, China). Streptavidin–horseradish peroxidase (Molecular Probes) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). 3,3′,5,5′-Tetramethylbenzidine was obtained from CWBIO (Beijing, China). The protein of GPC3 and EGFR were purchased from Sino Biological Inc. (Beijing, China). HER3ECD and HER2ECD (trastuzumab and pertuzumab binding epitopes antigens) were prepared in our laboratory. Bovine serum albumin (BSA) was sourced from VWR International (Radnor, PA, USA). Anti-human HER3 Ab and rhodamine-labeled goat anti-rabbit IgG were purchased from Biolegend (Santiago, CA, USA) and LifeSpan Biosciences (Seattle, WA, USA). 4′-6-Diamidino-2-phenylindole (DAPI) and lysosome marker LysoGreen were purchased separately from Beyotime Biotechnology (Shanghai, China) and KeyGEN BioTECH (Jiangsu, China). Quantum dots (QD) with a CdSe core and a ZnS shell were obtained from Nanjing Technology (Hangzhou, China). DOX was purchased from Hisun Pfizer (Hangzhou, China). Hydrogenated soy phosphatidyl choline (HSPC) was obtained from Tywei (Shanghai, China). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol 2000)] (DSPE-PEG [2000]) Carbox was sourced from Avanti Polar Lipids (Alabaster, AL, USA). N-hydroxysuccinimide (NHS) and 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) were both from Epicenter (Chicago, IL, USA). Cholesterol estradiol (Chol) was sourced from SIGMA (San Francisco, CA, USA). Matrigel matrix was purchased from BD Biosciences (San Jose, CA, USA). In addition, the aptamer library, primers, and subsequent modified sequences were synthesized by Sangon Biotech (Shanghai, China).
Cell lines and mice
The cell lines of human embryonic kidney (293T and 293E) and breast cancer (MCF-7, BT474) were purchased from American Type Culture Collection (Manassas, VA, USA). MCF-7 and 293T were cultured in the following medium: dulbecco’s modified eagle medium (DMEM) supplied with 10% fetal bovine serun (FBS), at 37°C with 5% CO2. BT474 cells were cultured in RPMI 1640 supplied with 10% FBS. Only 293E cells were suspension cells and cultured in Freestyle™ 293 expression medium. All the reagents used in cell culture were purchased from Thermo Fisher Scientific, including DMEM, RPMI 1640, trypsin, FBS, and Freestyle 293 expression medium. Female BALB/c nude mice were obtained from Beijing Vital River Laboratory Animal Technology Co. (Beijing, China). Female NOD-Prkdcem26Il2rgem26Nju (NCG) mice (5 weeks, 18–20 g, five animals/group) were obtained from the Model Animal Research Center of Nanjing University (Nanjing, China). All the mice were bred in a pathogen-free animal facility and provided with ad libitum access to a standard diet (PMI LabDiet®; LabDiet, St Louis, MO, USA) and water. The study was performed with the approval of the Ethical Committee of Beijing Institute of Radiation Medicine and according to the principles in the Declaration of Helsinki. The animal study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the Beijing Institute of Radiation Medicine. Animal welfare and experimental procedures were carried out in accordance with the National Research Council’s Guide for the use of laboratory animals.
Production of HER3ECD
HER3ECD was produced in 293E cells as described previously.14 Briefly, the ECDs of HER3 were cloned into HXT-chis, an expression vector that adds a His6-tag to the N-terminus of the encoded protein kept in our laboratory. Polyetherimide was used to transfect the HER3ECD expression vector into 293E cells. The cell supernatant was collected and HER3ECD was purified on liquid chromatography protein analyzer (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) through the nickel column.
Systematic evolution of ligands by exponential enrichment (SELEX)
The ssDNA library contained a central randomized sequence of 38 nucleotides: 5′-GGGAGCTCAGAATAAACGCTCAA-N38-TTCGACATGAGGCCCGGATC-3′. The constant regions included targets for polymerase chain reaction (PCR) primers. Protein-bound nickel-nitrilotriacetic acid (Ni-NTA) magnetic beads (BEAVERBIO) were prepared by first equilibrating 150 μL of a 10% slurry of Ni-NTA magnetic beads onto binding buffer (20 mM Na3PO4, 500 mM NaCl, 5 mM imidazole, pH =7.4). The equilibrated beads were resuspended in 1,250 μL of binding buffer containing 50 μL purified HER3ECD (1 mg/L) and rotated with a HulaMixer® sample mixer (Thermo Fisher Scientific) for 30 min at 4°C. The bead-bound HER3ECD was then washed three times with 1 mL of PBS Tween-20 (PBST: 50 mM K2HPO4, pH =7.5, 150 mM NaCl, 0.05% Tween-20), and then diluted to 0.25 mg/mL (2.5 pmol/mL of 100 kDa HER3ECD) with PBST and stored at 4°C.
In the initial round of selection, the “SelexApt” library was incubated with the bead-bound HER3ECD using a 10-fold molar excess of ssDNA in a volume that gave a 10 nM HER3ECD concentration. One nanomole of SelexApt was diluted with 100 μL of PBST in a PCR tube, heated to 95°C for 5 min and immediately cooled on ice. This material was added to 10 mL of PBST containing 1 mg/mL BSA. Bead-bound HER3ECD (100 pmol) was then added to this mixture and incubated with rotation for 30 min at 25°C. A magnet was then applied to the tubes, the supernatant was removed, and the beads were washed three times with 1 mL of PBST, mixing by inversion at each wash step. The proteins and bound aptamers were eluted from the Ni-NTA magnetic beads with 15 μL of elution buffer (20 mM Na3PO4, 500 mM NaCl, 500 mM imidazole, pH =7.4) and transferred to PCR tubes. Amplification conditions were: 15 min at 95°C, 28 cycles of 10 s at 95°C, 20 s at 58°C, 30 s at 72°C, and finally cooling for 30 s at 37°C. This protocol produced the correct size product as determined using PCR (Eastwin, Beijing, China). After the amplification step, 300 μL of the PCR product was mixed with 80 μL (10 mg/mL) streptavidin magnetic beads (BEAVERBIO) for 1 h at 25°C and then washed with 1 mL of PBST three times. Single-stranded aptamers (non-biotinylated strand) were separated from the immobilized complementary strands by 15 min incubation with 50 mL of fresh 100 mM NaOH. A magnet was applied to the tubes, and the ssDNA was removed and diluted with 1 mL of PBST. Next, the material was heated to 95°C for 5 min and then immediately placed on ice to begin the next round of SELEX.
For the next rounds of selection, the amount of protein was reduced to 50 pmol (rounds 2–7) and, subsequently, 10 pmol (rounds 8–10) in a binding volume of 10 mL, and the incubation time was reduced to 20 min. The PCR cycle number was reduced to 20 cycles as products of incorrect size were previously amplified. In order to increase the specificity of aptamers, we selected GPC3 as an anti-screen protein (rounds 5 and 7). Eventually, we identified a number of candidate sequences by sequencing. Secondary structures were predicted by DNAMAN software (DNAMAN 6.0; Lynnon Biosoft, San Ramon, CA, USA). Procedures of SELEX for aptamer selection are shown in Figure S2.
Aptamer-enzyme linked assay
To measure the binding of aptamers to HER3ECD immobilized on microtiter plates, six concentrations (ranging from 2.5 to 200 pmol) of purified HER3ECD and GPC3, HER2ECD (trastuzumab and pertuzumab binding epitopes antigens), EGFR and BSA were bound to wells in 100 μL of binding buffer overnight at 4°C. The wells were then washed three times with 300 μL of PBST. Biotinylated aptamers were diluted to 2.58 ng/mL in PBST, heated to 95°C, and then quickly cooled on ice. A 100 μL aliquot of the aptamer was incubated with proteins for 1 h at 25°C, shaking gently. The wells were washed five times with 300 μL of PBST for 3 min each on a plate vortex. Streptavidin–horseradish peroxidase (Molecular Probes) was diluted 1:8,000 with PBST and a 100 μL aliquot was incubated with the proteins and bound aptamers in the plate for 45 min at 25°C. The wells were washed again as described above, and then, 100 μL of 3,3′,5,5′-tetramethylbenzidine was added to each well and incubated for 25 min at 25°C in the dark. The reactions were stopped with the addition of 50 μL of 1 M H2SO4 and the protein-bound aptamer–streptavidin complex was quantified by determining the absorbance at 450 nm using a SpectraMax Plus (Thermo Fisher Scientific). The equilibrium dissociation constants (Kd) were calculated using the one site specific binding model; equation Y = BmaxX/(Kd + X); where X is the concentration of radioligand, Y is the specific binding, and Bmax is the maximum specific binding in the same units as Y.
Cellular binding of aptamers using flow cytometry
The MCF-7, BT474, and 293T were cultured and digested with 0.25% trypsin and washed with PBS three times. Approximately 1×106 cells were incubated with 2 nM fluorescein isothiocyanate (FITC)-aptamer for 1 h in the dark at 25°C. The cells were then washed three times with 1 mL of PBS and measured by flow cytometry (BD Biosciences, San Jose, CA, USA).
Confocal imaging
Co-location of HER3 Ab and aptamer was performed to verify the specificity. MCF-7 cells were seeded on round glass and grown overnight. The cell-filled dishes were washed with PBS and fixed with fresh 2% paraformaldehyde at 25°C for 30 min. Then the cell-filled dishes were washed three times with 1 mL of PBS. The MCF-7 line was incubated with anti-human HER3 Ab and FITC-Apt at the same time at 25°C for 1 h in the dark. Then the dishes were washed three times with PBS. Next, the MCF-7 cells were incubated with the rhodamine-labeled goat anti-rabbit IgG and incubated for 45 min in the dark as secondary antibody against HER3 Ab. After three washes with PBS, DAPI was added to the cells and incubated for 3 min. Finally, the cells were fixed and prepared for visualization by Nikon Ti A1 confocal laser scanning microscopy (Nikon Ti A1; Nikon Corporation, Tokyo, Japan).
Preparation of targeted delivery system
The schematic structure of several liposomes encapsulated with DOX in the market and in this study is shown in Figure S3. Liposomal formulations were prepared by the standard lipid hydration method. The molar ratio of HSPC to cholesterol and to DSPE-PEG (2000) was 10:2:1. Also, the DOX was dissolved in water for injection during hydration. The mass ratio of HSPC to DOX was 10:1. Then the aptamers with 5′-NH2 and 3′-thio-stability modification about 10 nucleotides (100 nmol) were conjugated by the amino carboxyl condensation reaction with a 10-fold excess of DSPE-PEG (2000) Carbox in the catalytic effect of NHS and EDC by gentle soaking for 30 min at 25°C. After the conjugation reaction, excess of NHS and EDC was removed clean by several times of ultrafiltration with double-distilled H2O, avoiding the binding efficiency of Apt-DSPE-PEG2000 and Lip-DOX. The next reaction of Apt-DSPE-PEG2000 was connected to Lip-DOX by the secondary hydration and post-insertion method (Figure S3C).15 The particle size and the zeta potential of Apt-Lip-DOX complexes were measured by a laser particle size analyzer (Malvern, Worcestershire, UK), and their morphology was observed by transmission electron microscopy (HITACHI, Tokyo, Japan).
In vitro DOX release
Apt-Lip-DOX, Lip-DOX, and free DOX (500 μg) were dissolved with 1 mL water in different dialysis bags, which were suspended in 50 mL of PBS buffer with 5% FBS (pH =7.4) at 37°C with constant stirring at 140 rpm. At a desired time point, 0.5 mL of the suspension was collected for high-performance liquid chromatography (HPLC; Waters 2695 High Performance Liquid Chromatograph, Waters 996 Detector; Waters, Milford, MA, USA) analysis to determine the DOX release curve among different groups. We selected a C18 column and the appropriate mobile phase, in which the molar ratio of 1.23% aqueous sodium acetate solution (pH =3.3) to methanol was 6:4.
Cellular uptake analysis
The efficiency of aptamer-liposome-carboxyfluorescein oligodeoxynucleotide (FAM-ODN) compound delivery into the cells was observed by confocal laser scanning microscopy. Approximately 1×105 MCF-7 and 293T cells were seeded into six-well plates and cultured for 24 h before transfection. Then, the same volume of aptamer-liposome-FAM-ODN (200 nM) and non-targeting liposome-FAM-ODN (200 nM) was added to the wells. The medium was discarded 3 h after transfection, and the cells were washed three times with PBS. Confocal images were collected by Nikon Ti A1 confocal laser scanning microscopy (Nikon Corporation, Tokyo, Japan).
Cell viability assay
In vitro cytotoxicities of Apt-Lip-DOX, Lip-DOX, and free DOX were analyzed in MCF-7/BT474 and 293T cells by real-time cell assay (ACEA Biosciences, Santiago, MN, USA). Blank liposomes were used as controls to rule out liposomal toxicity. Briefly, the cells (400 μL) were cultured in E-plates in duplicates at a density of 14,000 cells/well and were subsequently incubated for 24 h. DOX samples were added at varying concentrations (ranging from 0.3 to 5 μg/mL) and cells were observed for 96 h with a real-time monitor. The half maximal inhibitory concentration of the different groups was calculated using the cell proliferation curves.
In vivo targeting evaluation of ApDC
To investigate the in vitro targeting ability of aptamer in ApDC system, CdSe/ZnS QD, which is water soluble, was used as an alternative of DOX. For Apt-Lip-QD system, QDs with emission maxima at 655 nm (QD 655) were used with an ellipsoid core/shell with diameters of 6 nm (minor axis) by 12 nm (major axis) to ensure similar particle size and potential as Apt-Lip-DOX.
Apt-Lip-QD and Lip-QD were prepared and intravenously injected into the MCF-7 tumor-bearing BALB/C nude mice. One hour following the injection, mice were anesthetized and their bodies were scanned using an IVIS Spectrum CT (Caliper Life Sciences, Pittsburgh, PA, USA) with a laser of wavelength 430 nm and emission wavelength of 600 nm. Alternatively, the tumors were removed 1 h following the injection and the QD signals were collected using the IVIS Spectrum CT.
In vivo efficacy and safety experiments
Female NOD-Prkdcem26Il2rgem26Nju (NCG) mice (5 weeks, 18±2 g) were bred in a pathogen-free animal facility and were administered estradiol by intramuscular injection every 2 days. MCF-7 cells were harvested and resuspended in PBS and matrigel matrix to a final density of 5×107 cells/mL. When the tumors had grown to ~100 mm3, animals were distributed randomly into four treatment groups (n=5 per group). NaCl (control), free DOX, Lip-DOX, and Apt-Lip-DOX were injected intravenously via the tail vein at an equivalent dosage of 5 mg/kg DOX every 3 days for a total of eight times. Tumor volumes and body weights were measured, and the survival rate of the mice was monitored. Tissues were collected at the appropriate time point for H&E staining and analysis. Tumor volume was calculated according to the following formula: volume = (longest diameter × shortest diameter2)/2. Tissues were fixed in 2% paraformaldehyde to observe the toxicity of different drugs in various tissues.
Preliminary pharmocokinetic study in tumor-bearing mice
Preliminary pharmacokinetic (PK) study was applied to detect whether ApDC would enhance delivery efficacy and reduce toxicity. Validated HPLC was used to determine plasma pharmacokinetics and distribution of DOX with daunorubicin as an internal standard according to a previous report.16 Lip-DOX and Apt-Lip-DOX were injected at 5 mg/kg DOX and blood samples were collected up to 24 h (10 min, 1, 4, and 24 h) at different time points (n=5 at each time point). The main organs (tumors, heart, liver, spleen, and kidney) were collected from the 24 h time point mice. To all samples, an aqueous solution of daunorubicin (0.5 μg/mL in 1.5 mL H2O) was added as an internal standard for DOX quantification, followed by homogenization. The methods for DOX quantification using HPLC have been described above. DOX concentration in the organs was calculated as % of injected dose/g tissue (% ID/g).
Statistical analysis
Statistical significance was determined using a Student’s t-test with GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA). Comparison of multiple groups was performed using a one-way analysis of variance. Tumor sizes were analyzed and compared using the Mann–Whitney U test. A two-tailed value of P<0.05 was considered significant.
Results
Development of a HER3ECD-specific ssDNA aptamer
After 10 rounds of screening with decreasing HER3ECD concentrations and three rounds of negative screening with recombinant GPC3 protein, the resultant ssDNA pool was sequenced. Second-generation sequencing identified two dominant sequences (aptamer #7 and #13), and these two sequences accounted for 42% of the total reads with secondary structures shown in Figure 1A. The primary and secondary structures of the two aptamers showed no conserved regions, which might indicate that different domains of the HER3ECD were targeted.
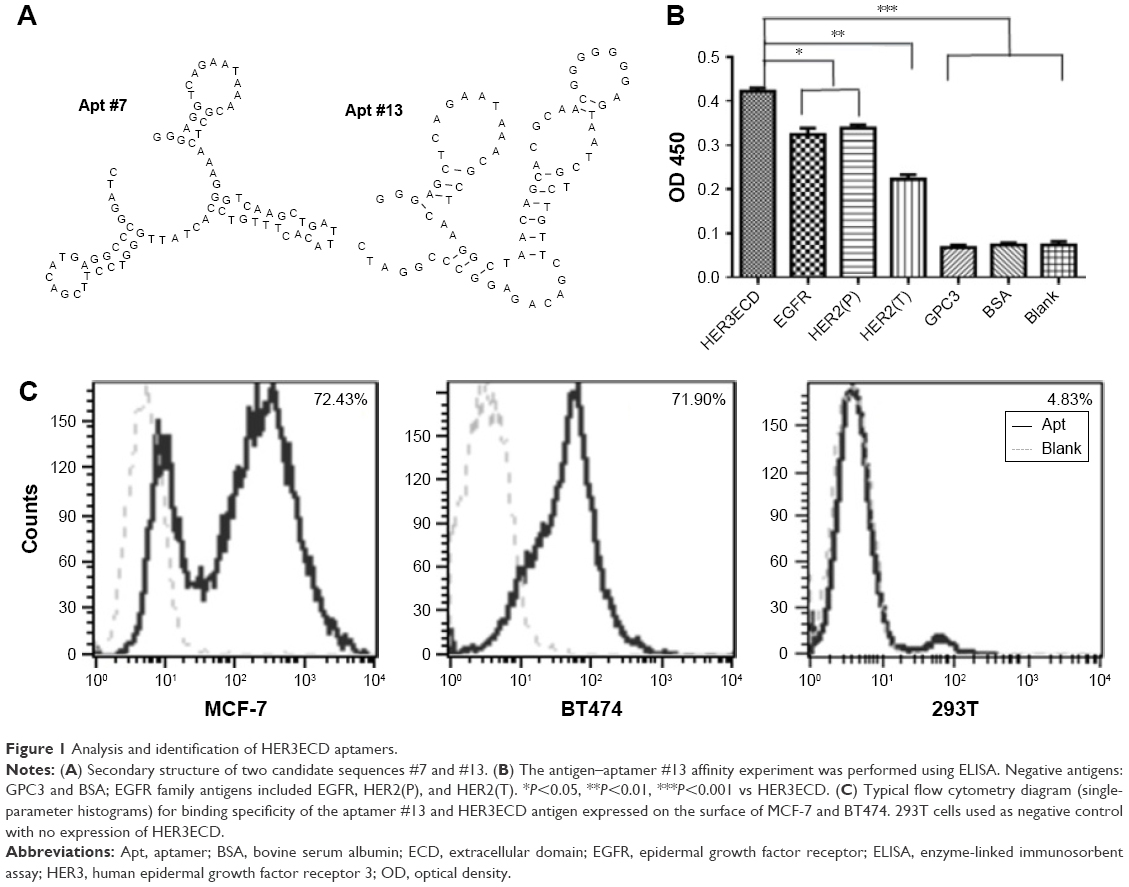 | Figure 1 Analysis and identification of HER3ECD aptamers. |
Specific HER3ECD binding capacity was determined using enzyme-linked immunosorbent assay with biotin-labeled aptamers #7 and #13. The binding capacities of aptamers #7 and #13 to HER3ECD protein were Kd =106±8.3 and 98±2.8 nM, respectively (data not shown). Therefore, we chose #13 as the aptamer of interest in the following experiments. Aptamer #13 showed specific binding to HER3ECD, rather than to GPC3 or BSA proteins (Figure 1B). Interestingly, #13 was also bound to HER2ECD and EGFR, though statistical differences existed between HER3ECD and HER2ECD or EGFR.
Flow cytometry analysis confirmed that Apt #13 was able to bind to HER3-expressing MCF-7 (72.43%) and BT474 cells (71.90%), but not to HER3-negative 293T cells (4.83%; Figure 1C). Confocal microscopy images demonstrated the co-location of FITC-Apt #13 and phycoerythrin-labeled HER3 antibody in MCF-7 cells (Figure 2A). The flow cytometry analysis combined with the co-localization experiment confirmed the key feature associated with the recognition of HER3 target by #13 aptamer.
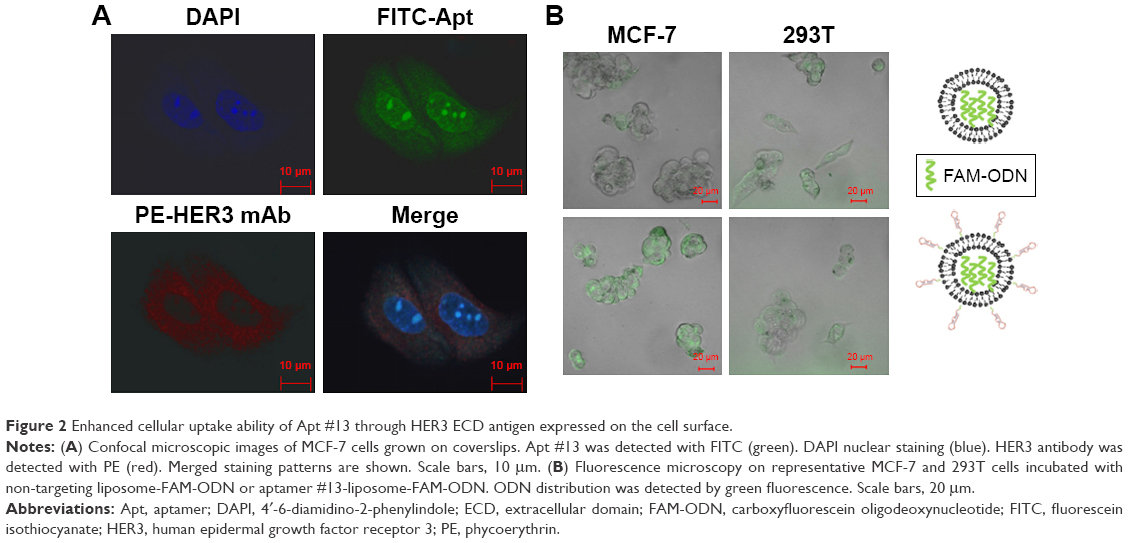 | Figure 2 Enhanced cellular uptake ability of Apt #13 through HER3 ECD antigen expressed on the cell surface. |
Formulation of ApDCs
For targeted chemotherapy, Apt-Lip-DOX conjugate system was constructed and validated, including physical characteristics, in vitro DOX release rate, enhanced cellular uptake rate, and cellular viability via aptamer. Physical characteristics, including the particle size, zeta potential, and morphology, were evaluated after preparing Apt-Lip-DOX conjugates. The average particle size of conjugates was 170±25 nm, and their zeta potential distribution was −45.9±6.31 mV. The conjugates were spherical with particle sizes as shown in Figure S4A and B.
Quantitative DOX concentration curves ranging from 2 to 64 μg/mL were constructed first using HPLC method. The inter- and intra-precision of low-, middle-, and high-quality controls was <2.5%. The formulations of ApDCs were optimized according to the maximal DOX encapsulation efficiency. Generally, the DOX encapsulation efficiency in our study was 55%±5%.
In the in vitro DOX release assay, release of DOX in the free DOX group was nearly 100% after 1 h and the release of DOX encapsulated in either liposomes or Apt-liposomes was only 20%–30% after 24 h, which demonstrates the advantages of the liposomal sustained release delivery system (Figure S4C).
Effective cellular uptake is the key feature for targeted chemotherapy. We assessed the aptamer #13-liposome-FAM-ODN for its ability to enhance cellular uptake efficacy in MCF-7HER3-high and 293THER3-negative cell lines. As shown in Figure 2B, FAM-ODN was efficiently distributed into the MCF-7 cells by the ApDC delivery system compared with the liposome-FAM-ODN delivery system. Weak fluorescence was observed in 293T cells in both the reagents treated, because of the liposome. It is obvious that the presence of Apt and the expression of HER3 on the cell surface contributed to the effective endocytosis capacity of ApDC.
Accordingly, the in vitro cell viability assays showed that the ApDC system could further inhibit the growth of MCF-7HER3+ and BT474HER3+ cells compared to the Lip-DOX and free DOX treatment (Figure 3; Figure S4D). Because of the presence of HER3 expression and enhanced cellular uptake ability, Apt-Lip-DOX was found to be advantageous over Lip-DOX in MCF-7 and BT474 cells; however, there was no significant difference observed in 293T cells. This study showed that small molecules of water-soluble drugs (DOX) can be encapsulated by liposomes to change the pathways in order to improve the ability of cells, and aptamers could further enhance the cellular uptake efficiency, which is consistent with the cellular imaging experiment (Figure 2B). Of course, the toxicity tests were performed to exclude cell viability effects due to liposome toxicity (Figure S4D).
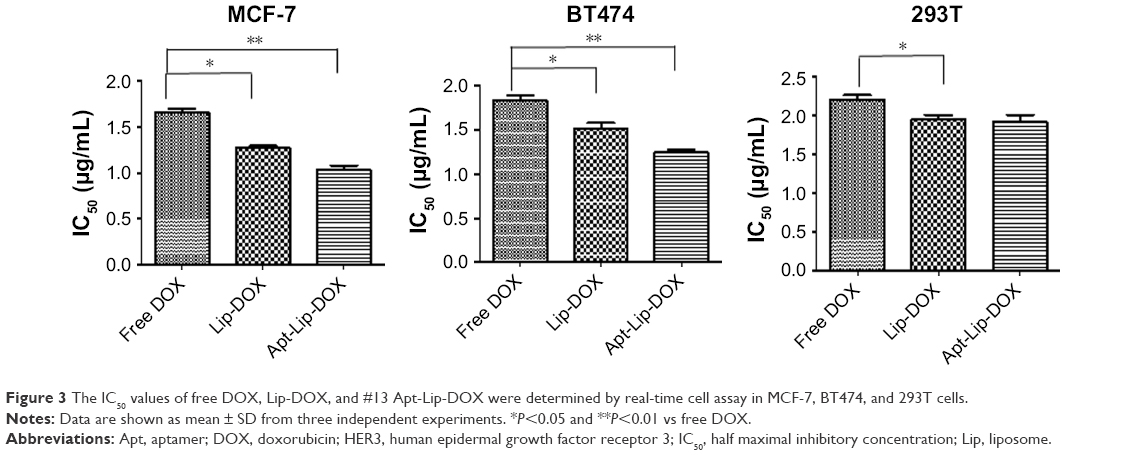 | Figure 3 The IC50 values of free DOX, Lip-DOX, and #13 Apt-Lip-DOX were determined by real-time cell assay in MCF-7, BT474, and 293T cells. |
Targeted chemotherapy of ApDC in MCF-7 tumor-bearing mice
First of all, targeting delivery and antitumor efficacy in vivo need to be confirmed. Whole body optical imaging was undertaken 4 h after injection. As shown in Figure 4A, noninvasive in vivo optical imaging demonstrated that as compared to Lip-DOX, Apt-Lip-DOX injected intravenously had higher uptake, prolonged retention, and markedly enhanced contrast in MCF-7 tumors.
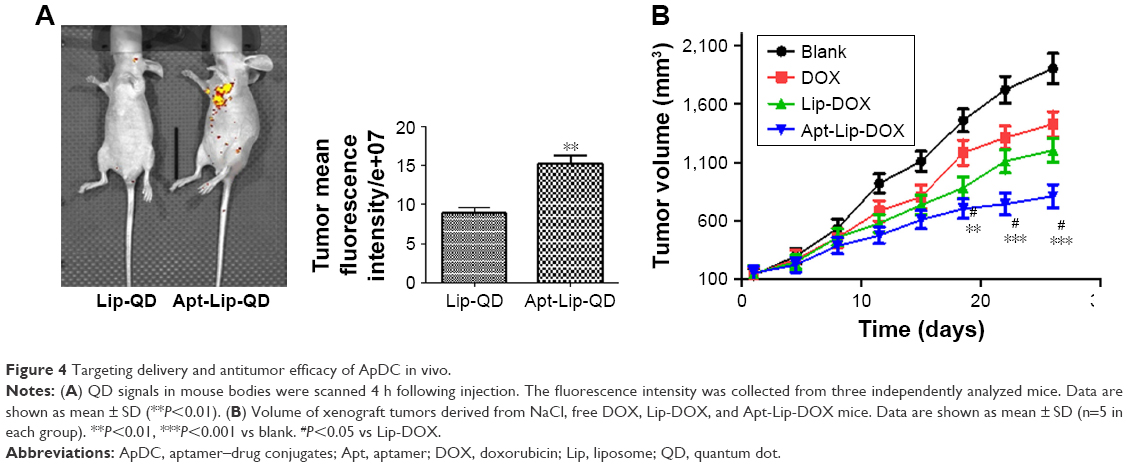 | Figure 4 Targeting delivery and antitumor efficacy of ApDC in vivo. |
To demonstrate the therapeutic effect of ApDC on tumor growth, MCF-7 tumor-bearing mice were intravenously injected with NaCl (blank), DOX only, Lip-DOX, and Apt-Lip-DOX successively (DOX dose: 5 mg/kg). In the control group receiving NaCl, tumors displayed rapid and continued outgrowth during the course of the experiment (about 30 days after the first administration). The mean tumor size of the ApDC treatment group was much smaller than those of the DOX and Lip-DOX treatment groups (Figure 4B). In conclusion, animals treated with ApDC exhibited a statistically significant suppression of tumor development compared with the other two DOX format-treated groups.
In addition to the therapeutic effects, we also analyzed whether the side effects could be reduced in the targeted chemotherapy strategy. Body weight, survival time and rate, and pathologic changes in the main organs using H&E staining of tissue sections were investigated. During 30 days post-chemotherapy, body weight of mice in the DOX groups reduced dramatically, especially in the last 10 days. Reduced body weight was also observed in mice treated with Lip-DOX; however, body weight did not show any statistical difference between the blank and Apt-Lip-DOX groups (Figure 5A). As for the survival index (Figure 5B), the survival time was gradually prolonged after treatment with DOX, Lip-DOX, and Apt-Lip-DOX. At 36 days post-chemotherapy, the survival rate was 0% in the DOX group, and at 60 days post-chemotherapy, the survival rate was 40% in the Lip-DOX group. Notably, the survival rate was 100% in the Apt-Lip-DOX and vehicle control groups.
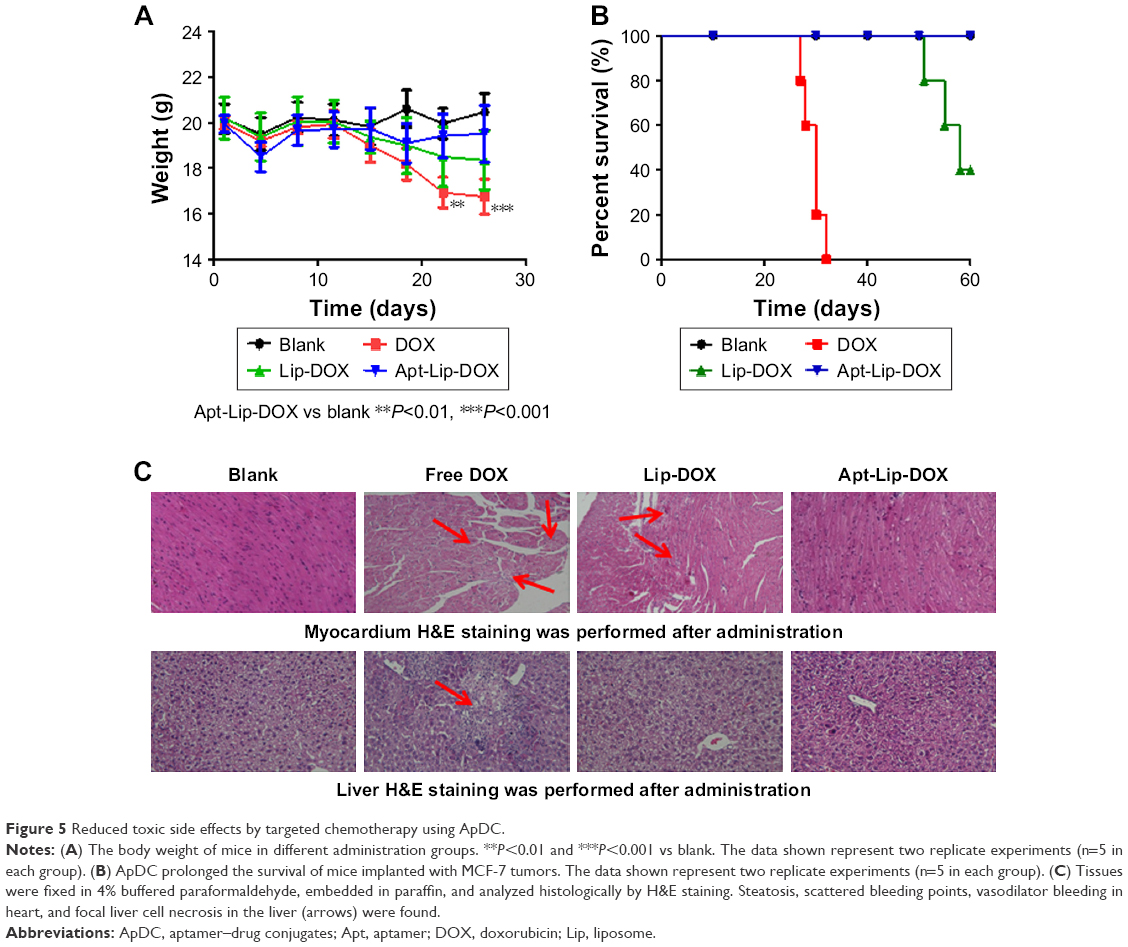 | Figure 5 Reduced toxic side effects by targeted chemotherapy using ApDC. |
Mice were sacrificed and several tissues (heart, liver, kidney, and stomach) were harvested. H&E staining of the tissue sections from formalin-fixed heart and liver showed dramatic change, as indicated in Figure 5C. NaCl-treated hearts and livers were normal, and similar results were also observed in the Apt-Lip-DOX treatment group. In the free DOX group, there were signs of steatosis and scattered bleeding points in the heart and focal liver cell necrosis in the liver. Though cardio and liver toxicity were reduced in Lip-DOX group compared with free DOX group, scattered damage points could still be observed.
To support the enhanced efficacy and reduced toxicity, a preliminary PK and biodistribution study was performed after the administration of single dosage of Lip-DOX and Apt-Lip-DOX in MCF-7 tumor-bearing mice. Plasma concentration–time curves for DOX showed that the addition of aptamers did not show rapid clearance properties of DOX, but increased accumulation of DOX was observed in the tumor tissues in Apt-Lip-DOX group compared with Lip-DOX group, which is consistent with the in vivo imaging results. In other tissues, the presence of aptamers seemed to reduce the biodistribution of DOX in heart, liver, and kidney to different degrees (Figure S5).
Discussion
An aptamer is a kind of single-stranded DNA or RNA oligonucleotide which alters the protein function through shape recognition. Generally, we refer to these biologically active, G-rich oligonucleotides as aptamers because their activities arise from binding to protein targets via shape-specific recognition (analogous to antibody–antigen binding). For example, quadruplex oligonucleotides are non-immunogenic, heat stable, have increased resistance to serum nucleases, and show enhanced cellular uptake compared to unstructured sequences.17 In our study, we observed the co-location of HER3 protein and aptamer in the cytoplasm and nucleus.18,19 The aptamer diffused strongly into the nucleus, which may be due to either the relatively small molecular weight or the specific cellular entry structures. We also found that Apt #13 could bind to the EGFR family (EGFR, HER2, and HER3), while Apt #7 showed no such phenomenon (as shown in Figure 1B). We supposed that Apt #13 might bind to the conserved three-dimensional space structure of the EGFR members based on the space docking mimic.20 Of course, the characteristic and function of Apt #13 would need to be fully investigated if necessary. Apt#13 was used for targeted chemotherapy in this study due to its high affinity and specificity during selection process.
Due to its high-affinity binding within the heart, DOX can cause severe dose-dependent cardiotoxicity.21 The risk of congestive heart failure restricts the clinical use of DOX to cumulative doses of 450–550 mg/m2.22 Pegylated liposomal DOX (PLD) is a stable formulation encapsulating DOX in a “stealth” (ie, pegylated) liposome, which further enhances the sustained release effect and also avoids recognition and phagocytosis by the body’s immune system (Figure S3B). Although PLD has the benefit of low cardiac toxicity, it also has the shortcoming of high skin toxicity.23 The PEG-modified DOX tends to aggregate on the skin, which means that a small portion of the drug exudates through the capillaries of the hands and feet, resulting in HFS characterized by numbness, paresthesia, skin swelling or erythema, scaling, chapping, and induration blisters.24 Half of the gynecologic oncology patients who received PLD at 40 mg/m2 for a maximum of six cycles had HFS.25 The HFS is partly caused by extravasation of the liposomes due to long circulation time.5 The presence of targeting aptamer in our ApDC system could probably reduce the nonspecific circulatory time and enhance the tumor distribution and permeation, making it possible to reduce HFS symptoms compared to pegylated liposome-encapsulated DOX.
Although current evidence suggests that liposomal DOX (Figure S3A) has lower cardiotoxicity than free DOX, R-CDOP (D is a liposomal DOX) is classified as associated with “left ventricular function incomplete or very weak in patients with first-line treatment” in the US National Comprehensive Cancer Network Non-Hodgkin Guidelines.5,6 However, so far, the replacement test of PLD in CHOP regimens has focused on patients with senile decay, and clinical trials are still under way for general patient trials.26,27 In these clinical trials of elderly patients and patients with cardiac dysfunction, a very noteworthy situation is that the doses used are lower than the conventional dose of DOX, ranging from 25 to 40 mg/m2. The conventional dose for DOX is 50 mg/m2. When 50 mg/m2 of liposomal DOX was compared with ordinary DOX, the NHL-14 (liposomal DOX) test showed that liposomal DOX does not have a strong advantage.2
In this study, we used the Apt-Lip-DOX delivery system, with PEG involved as a coupling agent rather than on the outer layer as with PLD (Figure S3C) coupled with aptamer. Generally, the addition of molecular weight and targeting molecule could improve the PK parameters. For example, PEG and cholesterol covalently conjugated to aptamer is a common strategy to improve the circulating half-time due to increase in molecular weight.28,29 For antibody–drug conjugates, the presence of targeting antibody has been well demonstrated as it can increase the tumor distribution and decrease the circulating half-time.30 The PK behaviors of ApDC aptamers have been rarely reported. In our preliminary study, the addition of aptamers did not show rapid clearance properties of DOX, but increased accumulation of DOX was observed in tumor tissues and decreased distribution in other major organs in the Apt-Lip-DOX group compared with Lip-DOX group, as observed in in vivo imaging and distribution results. Considering the PK and biodistribution results, whether ApDC can overcome the HFS caused by liposomal extravasation due to long circulation time is unclear. However, considering the in vivo efficacy and toxic results, we speculated that ApDC might reduce the potential cardiac toxicity and HFS due to the reduced administration doses and frequency. Of course, the conclusion needs to be supported by solid PK and pharmacodynamics data.
The dosage herein is equal to 25 mg/m2 in humans, which is far from 50 mg/m2; however, unlike regular chemotherapy, targeted chemotherapy could increase the accumulation of DOX in the tumor by increasing the targeting ability and reducing accumulation in non-target organs. In addition, low toxicity is obvious in this study, which is indicated by no loss in the body weight, prolonged survival rate, and reduced toxicity in heart and liver.
Conclusion
In conclusion, the results herein indicate that targeted chemotherapy using the ApDC format could provide better tolerability and efficacy compared with the current chemotherapy strategy. This proof-of-concept study for targeted chemotherapy may change the dose and regimen of conventional chemotherapy.
Acknowledgments
This study was supported by grants from the national science and technology major project “Non-clinical PK, PD, and immunogenicity evaluation of biosimilar” (2015ZX09501008-006); “Core technology-based non-clinical evaluation of biological macromolecular drugs” (grant number 2015ZX09501007-002). We thank Editage for linguistic assistance during the preparation of this manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Singal PK, Liiskovic NS, Hill M, Thomas TP, Li T. Combination therapy with probucol prevents adriamycininduced cariomyopathy. Mol Cell Cardiol. 1995;27:1055–1063. | ||
Fridrik MA, Jaeger U, Petzer A, et al. Cardiotoxicity with rituximab, cyclophosphamide, non-pegylated liposomal doxorubicin, vincristine and prednisolone compared to rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisolone in frontline treatment of patients with diffuse large B-cell lymphoma: a randomised phase-III study from the Austrian Cancer Drug Therapy Working Group [Arbeitsgemeinschaft Medikamentöse Tumortherapie AGMT](NHL-14). Eur J Cancer. 2016;58:112–121. | ||
Harrington KJ, Rowlinson-Busza G, Syrigos KN, Vile RG, Uster PS, Peters AM, Stewart JS. Pegylated liposome-encapsulated doxorubicin and cisplatin in the treatment of head and neck xenograft tumours. Clin Cancer Res. 2000;6(12):4939–4949. | ||
Yang J, Qiao L, Zeng Z, et al. The role of the ATM/Chk/P53 pathway in mediating DNA damage in hand-foot syndrome induced by PLD. Toxicol Lett. 2017;4(265):131–139. | ||
Yokomichi N, Nagasawa T, Coler-Reilly A, et al. Pathogenesis of hand-foot syndrome induced by PEG-modified liposomal doxorubicin. Hum Cell. 2013;26(1):8–18. | ||
Rohlfing S, Aurich M, Schöning T, Ho AD, Witzens-Harig M. Nonpegylated liposomal Doxorubicin as a component of R-CHOP is an effective and safe alternative to conventional Doxorubicin in the treatment of patients with diffuse large B-cell lymphoma and preexisting cardiac diseases. Clin Lymphoma Myeloma Leuk. 2015;15(8):458–463. | ||
Callahan R. Ado-trastuzumab emtansine in metastatic HER2-positive breast cancer. J Adv Pract Oncol. 2014;5(2):134–139. | ||
Uppal H, Doudement E, Mahapatra K, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21(1):123–133. | ||
Chen K, Liu B, Yu B, et al. Advances in the development of aptamer drug conjugates for targeted drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(3):Epub Oct 31, 2016. | ||
Zhao N, Pei SN, Qi J, et al. Oligonucleotide aptamer-drug conjugates for targeted therapy of acute myeloid leukemia. Biomaterials. 2015;67:42–51. | ||
Zhu CL, Song XY, Zhou WH, Yang HH, Wen YH, Wang XR. An efficient cell-targeting and intracellular controlled-release drug delivery system based on MSN-PEM-aptamer conjugates. J Mater Chem. 2009;19:7765–7770. | ||
Gala K, Chandarlapaty S. Molecular pathways: HER3 targeted therapy. Clin Cancer Res. 2014;20(6):1410–1416. | ||
Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancertherapy. Clin Cancer Res. 2010;16(5):1373–1383. | ||
Zhang J, Zhang X, Liu Q, et al. Mammalian cell display for rapid screening scFv antibody therapy. Acta Biochim Biophys Sin. 2014;46(10):859–866. | ||
Liu YJ, Dou XQ, Wang F, et al. IL-4RJ aptamer-liposome-CpG oligodeoxynucleotides suppress tumour growth by targeting the tumour microenvironment. J Drug Target. 2016;1:1–9. | ||
Dicheva BM, Seynhaeve AL, Soulie T, Eggermont AM, Ten Hagen TL, Koning GA. Pharmacokinetics, tissue distribution and therapeutic effect of cationic thermosensitive liposomal doxorubicin upon mild hyperthermia. Pharm Res. 2016;33(3):627–638. | ||
Bates PJ, Laber DA, Miller DM, Thomas SD, Trent JO. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol. 2009;86(3):151–164. | ||
Kim JH, Im KS, Kim NH, Yhee JY, Nho WG, Sur JH. Expression of HER-2 and nuclear localization of HER-3 protein in canine mammary tumors: histopathological and immunohistochemical study. Vet J. 2011;189(3):318–322. | ||
Trocmé E, Mougiakakos D, Johansson CC, et al. Nuclear HER3 is associated with favorable overall survival in uveal melanoma. Int J Cancer. 2012;130(5):1120–1127. | ||
Little field P, Liu L, Mysore V, Shan Y, Shaw DE, Jura N. Structural analysis of the EGFR/HER3 heterodimer reveals the molecular basis for activating HER3 mutations. Sci Signal. 2014;7(354):ra114. | ||
Bielack SS, Erttmann R, Winkler K, Landbeck G. Doxorubicin: effect of different schedules on toxicity and anti-tumor efficacy. Eur J Cancer Clin Oncol. 1989;25(5):873–882. | ||
Yildirim Y, Gultekin E, Avci ME, Inal MM, Yunus S, Tinar S. Cardiac safety profile of pegylated liposomal doxorubicin reaching or exceeding lifetime cumulative doses of 550 mg/m2 in patients with recurrent ovarian and peritoneal cancer. Int J Gynecol Cancer. 2008;18(2):223–227. | ||
Yuan Y, Orlow SJ, Curtin J, Downey A, Muggia F. Pegylated liposomal doxorubicin (PLD): enhanced skin toxicity in areas of vitiligo. Ecancermedicalscience. 2008;2:111. | ||
Shimada M, Sato S, Otsuki T, et al. Supportive care for hand-foot syndrome and stomatitis in relapsed ovarian cancer patients receiving pegylated liposomal doxorubicin. J Clin Oncol. 2011;29:e19722. | ||
von Gruenigen V, Frasure H, Fusco N, DeBernardo R, Eldermire E, Eaton S, Waggoner S. A double-blind, randomized trial of pyridoxine versus placebo for the prevention of pegylated liposomal doxorubicin-related hand-foot syndrome in gynecologic oncology patients. Cancer. 2010;116(20):4735–4743. | ||
Zaja F, Tomadini V, Zaccaria A, et al. CHOP-rituximab with pegylated liposomal doxorubicin for the treatment of elderly patientswith diffuse large B-cell lymphoma. Leuk Lymphoma. 2006;47(10):2174–2180. | ||
Oki Y, Ewer MS, Lenihan DJ, et al. Pegylated liposomal doxorubicin replacing conventional doxorubicin in standard R-CHOP chemotherapy for elderly patients with diffuse large B-cell lymphoma: an open label, single arm, phase II trial. Clin Lymphoma Myeloma Leuk. 2015;15(3):152–158. | ||
Bulbake U, Doppalapudi S, Kommineni N, Khan W. Liposomal formulations in clinical use: an updated review. Pharmaceutics. 2017;9(2):12. | ||
Lee CH, Lee SH, Kim JH, Noh YH, Noh GJ, Lee SW. Pharmacokinetics of a cholesterol-conjugated aptamer against the hepatitis C virus (HCV) NS5B protein. Mol Ther Nucleic Acids. 2015;4(10):e254. | ||
Leal M, Wentland J, Han X, et al. Preclinical development of an anti-5T4 antibody-drug conjugate: pharmacokinetics in mice, rats, and NHP and tumor/tissue distribution in mice. Bioconjug Chem. 2015;26(11):2223–2232. |
Supplementary materials
 | Figure S1 Reduced toxic side effects and enhanced therapeutic efficacy by targeted chemotherapy using ApDC. |
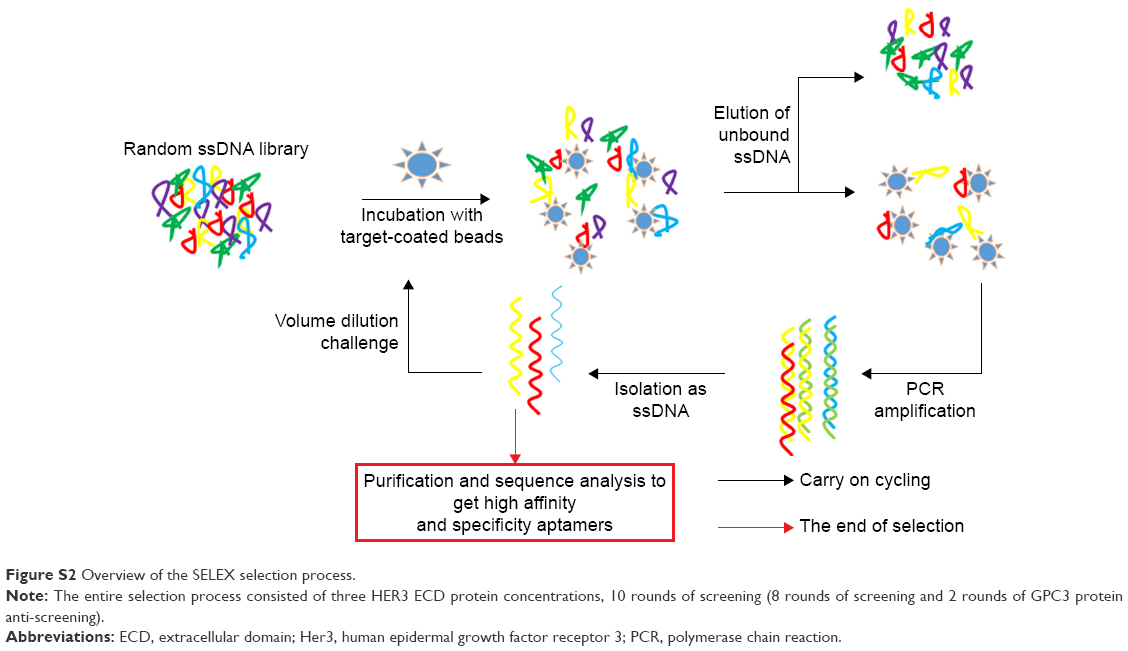 | Figure S2 Overview of the SELEX selection process. |
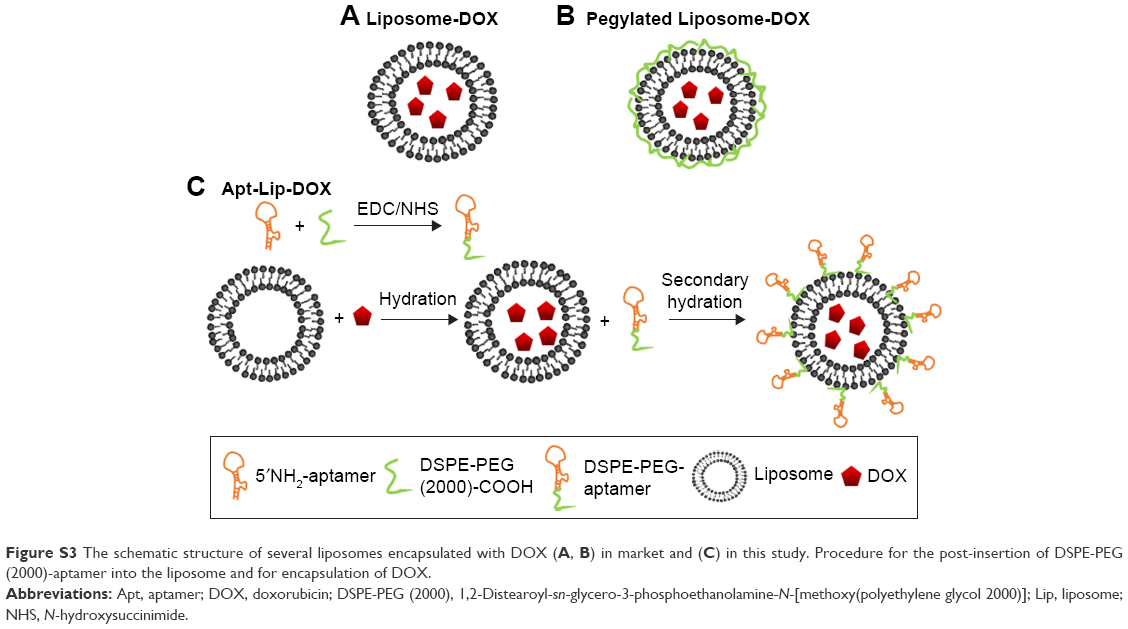 | Figure S3 The schematic structure of several liposomes encapsulated with DOX (A, B) in market and (C) in this study. Procedure for the post-insertion of DSPE-PEG (2000)-aptamer into the liposome and for encapsulation of DOX. |
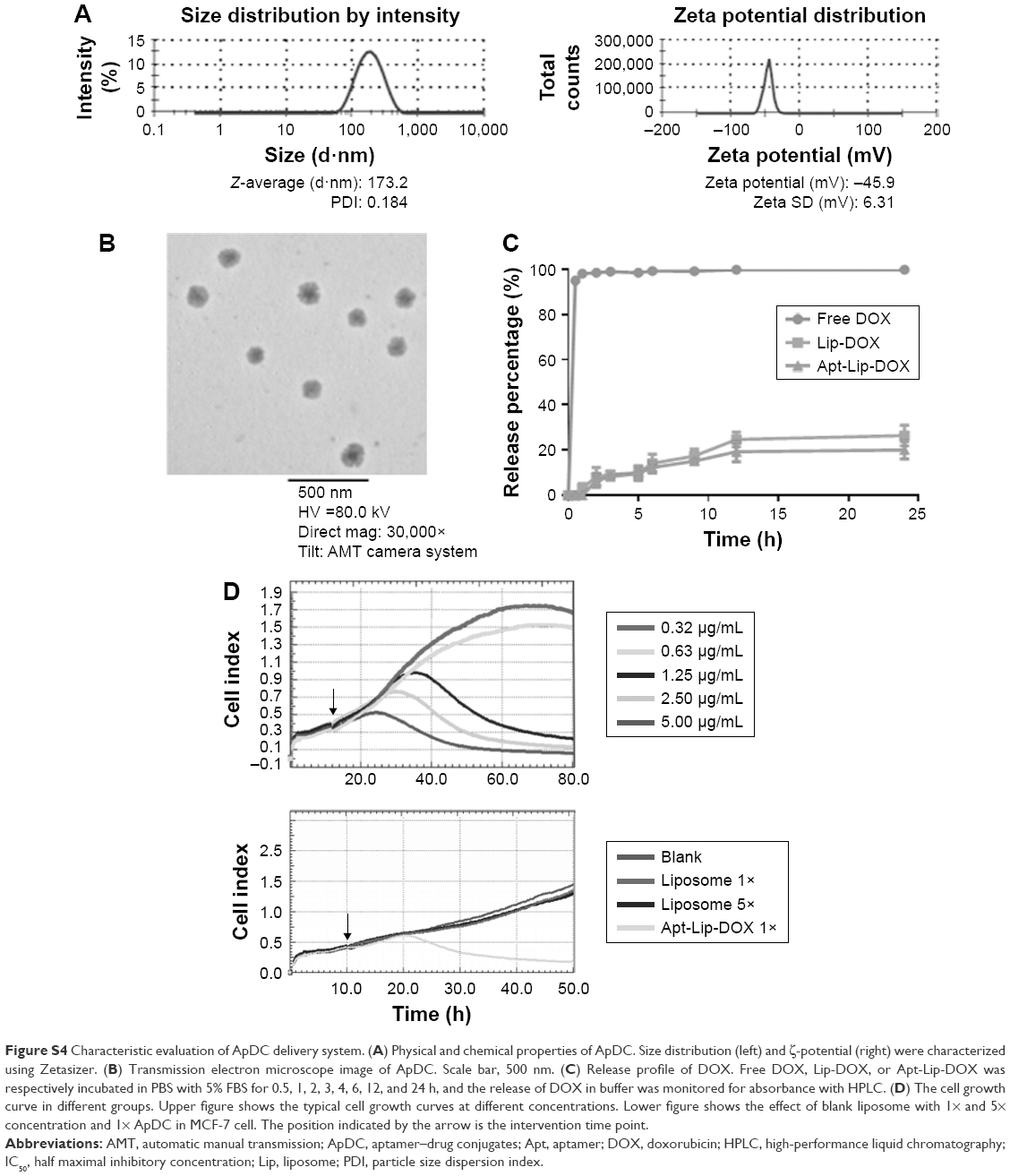 | Figure S4 Characteristic evaluation of ApDC delivery system. (A) Physical and chemical properties of ApDC. Size distribution (left) and ζ-potential (right) were characterized using Zetasizer. (B) Transmission electron microscope image of ApDC. Scale bar, 500 nm. (C) Release profile of DOX. Free DOX, Lip-DOX, or Apt-Lip-DOX was respectively incubated in PBS with 5% FBS for 0.5, 1, 2, 3, 4, 6, 12, and 24 h, and the release of DOX in buffer was monitored for absorbance with HPLC. (D) The cell growth curve in different groups. Upper figure shows the typical cell growth curves at different concentrations. Lower figure shows the effect of blank liposome with 1× and 5× concentration and 1× ApDC in MCF-7 cell. The position indicated by the arrow is the intervention time point. |
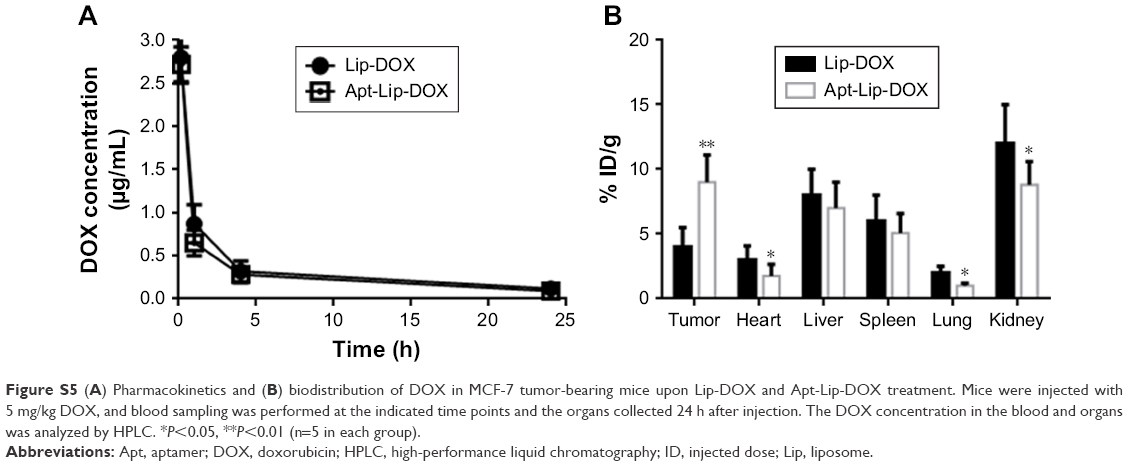 | Figure S5 (A) Pharmacokinetics and (B) biodistribution of DOX in MCF-7 tumor-bearing mice upon Lip-DOX and Apt-Lip-DOX treatment. Mice were injected with 5 mg/kg DOX, and blood sampling was performed at the indicated time points and the organs collected 24 h after injection. The DOX concentration in the blood and organs was analyzed by HPLC. *P<0.05, **P<0.01 (n=5 in each group). |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.