")
Back to Journals » OncoTargets and Therapy » Volume 12
Application Of Adoptive Immunotherapy In Ovarian Cancer
Authors Yang S, Yin X, Yue Y, Wang S
Received 3 July 2019
Accepted for publication 18 September 2019
Published 27 September 2019 Volume 2019:12 Pages 7975—7991
DOI https://doi.org/10.2147/OTT.S221773
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Carlos E Vigil
Siyu Yang,1 Xiaojiao Yin,1 Ying Yue,1 Siqing Wang2
1Department of Gynecological Oncology, The First Hospital of Jilin University, Changchun 130061, China; 2Department of Cancer Immunology, The First Hospital of Jilin University, Changchun 130061, China
Correspondence: Ying Yue
Department of Gynecological Oncology, The First Hospital of Jilin University, Changchun 130061, People’s Republic of China
Tel +8643113756661254
Email [email protected]
Siqing Wang
Department of Cancer Immunology, The First Hospital of Jilin University, Changchun 130061, People’s Republic of China
Tel +8643115943073518
Email [email protected]
Abstract: Ovarian cancer (OC) has been the most fatal gynecological disease that threatens women’s health. Surgery and platinum-based chemotherapy are the basic ovarian cancer treatments that can improve survival, but the five-year survival rate has not improved because of delayed diagnosis, drug resistance, and recurrence. Novel treatments are needed to improve the prognosis and survival rate of ovarian cancer patients. In recent years, adoptive cell therapy (ACT) has received increasing attention as an emerging therapeutic strategy in the treatment of solid tumors including OC. ACT has shown promising results in many preclinical and clinical trials of OC. The application of ACT depends on different effector cells, such as lymphokine-activated killer (LAK) cells, tumor-infiltrating lymphocytes (TILs), and genetically modified T cells. In this review, we focus on adoptive immunotherapies in ovarian cancer and summarize completed and ongoing preclinical/clinical trials. The future development directions and obstacles for ACT in OC treatment are discussed.
Keywords: ovarian cancer, adoptive cell therapy, cancer immunotherapy, immune cells
Introduction
Ovarian cancer (OC) is the primary gynecological causes of death in women. Worldwide, there are about 230, 000 cases of OC each year, with more than 150, 000 deaths.1 Surgery and chemotherapy are currently the main treatments for OC. Cytoreductive surgery is used to remove all visible tumor masses. However, most patients are diagnosed in the advanced stage of the tumor and need to receive postoperative adjuvant chemotherapy. In addition, patients with extensive tumor metastasis will receive neoadjuvant chemotherapy to shrink the tumor and destroy metastatic cells, so as to facilitate subsequent surgery and other treatments.2–4 Although radical surgery and adjuvant chemotherapy are performed to remove macroscopic tumors and improve outcomes, most patients with ovarian cancer will have recurrence and tumor resistance, which is usually fatal5 and widely studied anti-vascular endothelial growth factor (VEGF) therapy is also difficult to reverse this situation6 [Table 1]. Thus, there is a great need for more effective OC therapies to improve the long-term clinical prognosis.
 |
Table 1 Comparison Of Clinical Effects Of Four Ovarian Cancer Treatment Methods |
With the improved understanding of the relationship between the immune system and tumor development, immunotherapy is becoming a promising treatment for lung cancer,15 melanoma,16 liver cancer,17 and breast cancer.18 In recent years, increasing evidence has shown that immunotherapy is also a promising treatment in ovarian cancer since ovarian cancer is an immunogenic tumor that can be recognized and attacked by immune system.19–21 Recent immune therapies mainly include immune checkpoint inhibitors, cancer vaccine, and adoptive cell therapy (ACT).22–24 Among them, ACT has attracted increasing attention because a large number of specific effector cells against tumor cells results in a quick therapeutic effect and minimizes impact on the internal environment than other therapies.
ACT relies on intravenous infusion of autologous immune cells after stimulation/modification and expansion in vitro to improve autologous antitumor response in tumor patients [Figure 1]. In 1965, Mathé et al confirmed that adoptive immunotherapy had an obvious effect on acute leukemia in a murine experiment and clinical trial.25 Research on ACT for the treatment of hematological malignancies is constantly evolving and developing.26,27 In 2002, a clinical trial showed that adoptive cell immunotherapy was effective for solid tumors (metastatic melanoma)28 and ongoing clinical trials have confirmed this.29,30 Since OC was not originally considered to be an immunogenic tumor, adoptive immunotherapy for OC did not initially receive much attention. However, in 2003, OC was shown to be an immunogenic tumor that may be treat by immunotherapy.19,31 Adoptive immunotherapy is based on different cell types [Figure 2]: MHC-independent cells (e.g., lymphokine-activated killer (LAK) cells, natural killer (NK) cells, and cytokine-induced killer (CIK) cells) or MHC-dependent cells (tumor-infiltrating lymphocytes (TILs)). There are also two special and rapidly developing cell types: chimeric antigen receptor (CAR) T cells and T cell receptor (TCR) T cells.32 In this review, we discuss the application of adoptive immunotherapy of LAK cells, NK cells, CIK cells, TILs, CAR-T cells, and TCR-T cells in OC and outline the disadvantages and future development directions of ACT in OC treatment.
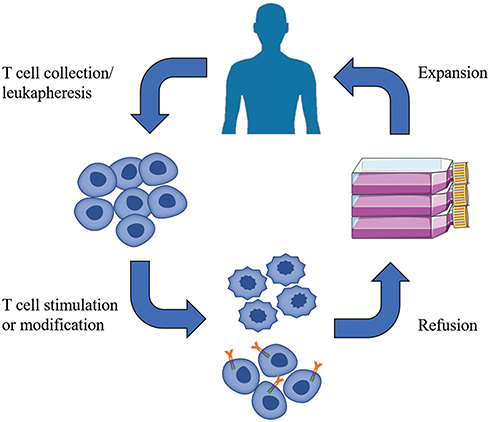 |
Figure 1 Adoptive cell immunotherapy (ACT) approaches: After obtaining immune cells from the patient, leukapheresis is performed. Immune cells are activated after stimulation or genetical modification. Effective immune cells are expanded and then refused to the patient. |
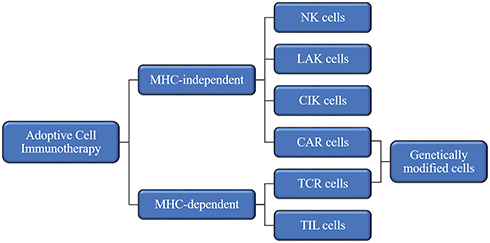 |
Figure 2 Effector cells for adoptive cell immunotherapy. Abbreviations: MHC, major histocompatibility complex; NK, natural killer; LAK, lymphokine-activated killer; CIK, cytokine-induced killer; CAR, chimeric-antigen receptors; TCR, T cell receptors; TIL, tumor-infiltrating lymphocytes. |
Major Histocompatibility Complex (MHC)-Independent Adoptive Immunotherapy
LAK Cells
LAK cells are induced by NK cells or T cells through adding high-dose IL-2 and other cytokines when cultured in vitro, rather than an independent lymphoid or subgroup.33 LAK cells can kill NK-instant tumor cells and have achieved certain therapeutic effects in cancer treatment. In 1985, Rosenberg et al suggested that LAK cells and IL-2 adoptive immunotherapy have therapeutic effects on metastatic tumors for which many traditional treatments are ineffective. Although the number of clinical patients in this study was limited, the same authors had also shown promising therapeutic effects in their previous in vivo experiments in mice;34 however, they also mentioned that high-dose IL-2 promotes the secretion of toxic cytokines by helper T cells, which is harmful to patients. Grimm et al also described this problem.35 Although LAK cell adoptive immunotherapy has a very broad-spectrum anti-tumor effect, the safe application of high-dose IL-2 is still problematic.
NK Cells
NK cells are part of the innate immune response and are key effectors in cancer immunosurveillance.36 They can defend against cancer development and metastasis without restricting the expression of MHC molecules.37–39 NK cells represent about 5–15% of human circulating lymphocytes and comprise CD56hiCD16− NK cells and CD56loCD16+ NK cells.40 CD56hiCD16− NK cells with high cytotoxic potential can produce plenty of cytokines, while CD56loCD16+ NK cells are highly cytotoxic and mediate antibody-dependent cellular cytotoxicity (ADCC) responses.36 NK cells can effectively treat tumors such as leukemia,41 but the therapeutic effect of NK cell adoptive immunotherapy in OC is still being explored. The promise of NK cell adoptive immunotherapy for OC was recognized in 2007, and it was shown that resting NK cells rely on DNAM-1 signaling with complementary contributions of NKG2D and NCR receptors to recognize and kill freshly isolated OC cells in vitro.42 In 2006, a new mouse model was established that will be helpful for further exploration of NK cell adoptive immunotherapy for OC.43
CIK Cells
CIK cells were first discovered in 1991 by Schmidt-Wolf et al.44 They were heterogeneous CD8+ T cells produced by human peripheral blood lymphocytes (PBLs) and induced by addition anti-CD3 antibody, interferon-γ (IFN-γ), and interleukin-2 (IL-2) ex vivo.44 CIK cells can be characterized by the presence of CD3+ CD56+ phenotype, which is mainly responsible for the antitumor activity of CIK cells and CD3+ CD56− phenotype, which is more similar to conventional T lymphocytes.45
Several studies have confirmed the feasibility, effectivity, and safety of CIKs for the treatment of malignant tumors.46,47 Leemhuis et al confirmed the effectiveness of CIK cells in the treatment of malignant tumors, and then conducted a clinical phase I trial of CIK cell therapy in patients with hematologic malignancies.46 Clinical phase I trials of patients with hematologic malignancies by Introna et al also demonstrated that it was feasible and well tolerated to produce allogeneic CIK cells under clinical conditions.47 Another study analyzed the efficacy of CIK cells in the treatment of OC.48 In a phase II study, Liu et al49 tested the effectivity of CIK cell therapy following first-line treatment in advanced OC. Further experiments are required to determine whether CIK cell maintenance immunotherapy can help improve overall survival, but CIK cell therapy does improve progression-free survival in patients with advanced OC after first-line treatment with slight toxicity. A retrospective analysis by Zhou et al further validated the effectiveness of CIK cell therapy as a therapeutic approach to prolonging the survival of OC patients.49 A clinical phase II trial to determine whether radiofrequency ablation (RFA) and cytokine-induced killer cell (CIK) infusion can prolong survival in patients with OC is also underway (NCT02487693).
MHC-Dependent Adoptive Immunotherapy
TILs
TILs are endogenous autologous T cells isolated from tumor tissues with certain tumor specificity and MHC restriction. Adoptive immunotherapy with TILs has a response rate of about 50% in melanoma patients.30,50 In 1991, Aoki et al showed that TIL cell adoptive therapy had a promising future in OC, although the experiment lacks randomness.51 Subsequently, Fujita et al52 found that TILs adoptive immunotherapy after chemotherapy was helpful for the prognosis of patients with OC; patients who received TILs had a 3-year survival rate of 100%, compared with 67.5% for patients who did not receive TILs.52 In recent years, with a deep understanding of OC and cellular immunotherapy, the application of TILs in OC has been further developed. Westergaard et al53 obtained 34 tumor specimens from 33 OC patients and analyzed the phenotype, antigen specificity, and function of TILs. It was found that TILs obtained from OC can be effectively expanded and exert anti-tumor effect in vitro, which supported the hypothesis that OC patients could benefit from ACT of TILs, and the relevant Phase I clinical trial ended in 2017.53,54 Owens et al55 recently discovered an effective method for isolating and expanding TILs from OC. Surprisingly, the expanded TILs retain the ability to recognize autologous tumor cells in vitro.55 Although numerous studies have indicated the promise of TILs adoptive immunotherapy for OC, problems remain that limit its development. For example, the anti-tumor effect of TILs is limited by the fact that unselected TILs in OC usually contain only a small number of tumor-reactive T cells. In addition, the greater financial support required for isolation and expansion of tumor-specific TILs and reperfusion into patient limits their clinical application.
CAR-T Cells And TCR-T Cells
Although TIL cell adoptive immunotherapy is a promising treatment for OC, the method of isolating and manufacturing TILs is labor intensive and successful in only a subset of patients, which limits its therapeutic effect and clinical application.56,57 In order to improve the therapeutic potential, genetically modified peripheral blood lymphocytes that exhibit tumor antigen specificity have received more and more attention. Genetically modified T cells can express a chimeric antigen receptor (CAR) or a tumor-antigen specific T-cell receptor (TCR).
CAR-T Cell Therapy
CAR-T cells are T lymphocytes that have been genetically modified to express an engineered T cell receptor that is able to recognize tumor-specific antigens MHC unrestrictedly and activate the immune response.
The CAR comprises four main parts: the extracellular antigen-binding domain, the spacer domain (hinge domain), the transmembrane domain, and the intracellular T cell activation/signaling domain. The extracellular antigen-binding domain, also known as the ectodomain, is derived from the light and heavy chains of the antibody and is a single-chain variable fragment (scFv) that can recognize tumor-specific antigens on cell surface in an MHC-unrestricted manner.58–60 The CAR-T cell structure design has been constantly updated, through four generations, producing CAR-T cells that survive longer in vivo and have stronger killing ability [Figure 3]. The first generation of CAR-T cells only contained the extracellular scFv antigen recognition region and the intracellular CD3ζ chain signal region. To improve the proliferation and persistence of CAR-T cells, the costimulatory molecules such as CD28 and OX-40/4-1BB (CD134/CD137) were added in the second and third generation. The fourth generation is characterized by releasing cytokines like IL-12 and IL-15, which enhance immune response.61,62
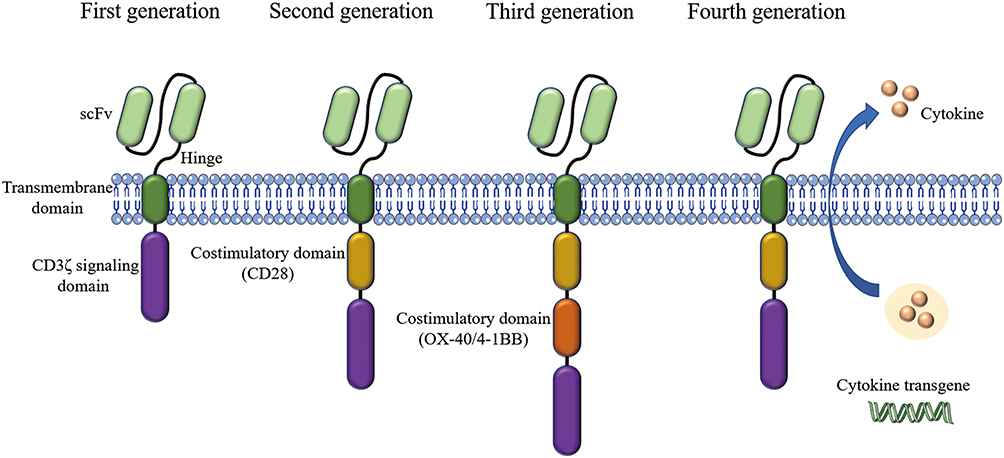 |
Figure 3 Four generation of CARs: The first generation contains a single-chain fragment of variable region (scFv) and CD3ζ signaling domain. Costimulatory molecules such as CD28 are added in the second generation. Third-generation CARs include more signaling domains. The fourth generation are characterized by addition of cytokine transgenes like IL-12 and IL-15. |
The most exciting results with CAR-T cell therapy have been achieved in hematological tumors. In a phase I clinical trial conducted by Park et al,63 53 patients with relapsed B-cell acute lymphoblastic leukemia (ALL) received an infusion of autologous T cells expressing the 19-28z CAR. Long-term follow-up of outcomes and safety indicated that 19-28z CAR-T cells have potent anti-tumor capability with many patients achieving long-term relief and possibly cure.63 Currently, anti-CD19 CAR-T cells are approved by the FDA for the treatment of diffuse large B-cell lymphoma. Brudno et al’s study of CAR-BCMA (B-cell maturation antigen) T cells in the treatment of multiple myeloma also confirmed the enormous potential of CAR-T cells in hematological tumor treatment.64 Research on CAR-T cell therapy continues to extend to solid tumors including OC.
The main targets for CAR-T cells in OC include MUC16, mesothelin and folate receptor-α. Pre-clinical studies of CAR-T cell therapy targeting MUC16 in murine models have shown promising results suggesting that this approach may be effective for the treatment of OC, and a parallel clinical trial is ongoing.65 MUC16 plays an important role in the progression and metastasis of OC and has become a key target for OC treatment.66 Mesothelin is an antigen target that is overexpressed in OC.67 The expression of mesothelin is associated with the prognosis of OC; hence, mesothelin is both a therapeutic target for OC and a prognostic marker.68 The results of preliminary trials are promising, and more clinical trials on CAR-T cell therapy targeting mesothelin are still ongoing, such as NCT03692637 and NCT03814447. Folate receptor-α (FRα) is a glycosylphosphatidylinositol-anchored protein that expressed on the surface of normal ovarian cells. FRα is also overexpressed on the surface of malignant epithelial cancer cells, making it a potential target for the treatment of OC.69–71 The safety and efficacy of CAR-T cell therapy targeting FRα are supported by preclinical studies and phase I clinical trials.72,73 Another clinical trial (NCT03585764) is also underway. Other antigens such as HER2 and CD133 have also been tested in preclinical animal models for OC therapy (NCT01935843, NCT02541370).
Although CAR-T cell therapy has shown great potential in the treatment of OC, it still faces many problems that remain to be solved. The CAR target antigen is also expressed in some normal tissue, resulting in immune-mediated rejection that is known as an “on-target, off-tumor” response. This rejection can even cause damage to vital organs such as the liver and lungs.74,75 Also, the potentially immunosuppressive environment in OC, including the highly immunosuppressive ascites, will prevent T cells from effectively infiltrating into tumor cells and dysfunctional T cells. CAR-T cell adoptive immunotherapy also has some common problems of ACT treatment, such as cytokine release syndrome and a more effective T cell transport pathway. In conclusion, there are both hopes and challenges in the treatment of OC by CAR. Further research is necessary to improve the safety and efficacy of CAR-T cell therapy in OC.
TCR-T Cell Therapy
TCRs are characteristic markers on the surface of T cells that recognize specific antigens.76,77 TCR-T cells are T cells that express a genetically engineered TCR alpha and beta chain pair that can recognize tumor-specific antigens. TCR-T cell treatment has been successful in patients with malignant cancer such as colorectal carcinoma,78 metastatic melanoma,79 and multiple myeloma.80,81 Genetically modified TCR-T cells are also considered as a potentially promising treatment for OC patients. In OC, the TCR target antigens include MAGE-A4, WT1, and NY-ESO-1, with NY-ESO-1 being widely studied.
New York esophageal squamous cell carcinoma-1 (NY-ESO-1) is an 18 kDa protein that can be detected in normal testis, fetal ovary, and placenta.82,83 However, NY-ESO-1 antibodies can also be detected in the serum of ovarian cancer, lung cancer, breast cancer, bladder cancer, esophageal cancer, as well as melanoma patients.82,83 Odunsi et al84 detected the expression of NY-ESO-1 by reverse transcription PCR (RT-PCR) and immunohistochemistry (IHC) in OC tissues and cell lines, confirming its expression and persistence in OC. Thus, NY-ESO-1 is an attractive target for antigen-specific adoptive immunotherapy in OC.84 Several phase I/II clinical trials with TCR-T cells are currently ongoing in OC patients (NCT01567891, NCT03691376, NCT02457650, NCT02869217, NCT03159585). Like other types of ACT, there are barriers to the application of TCR-T cells to ACT. Tumor antigen-specific T cells of autoantigens isolated from cancer patients usually have low affinity due to central tolerance.85 As observed with CAR-T cells, the “on-target, off-tumor” response occurs when TCR-T cells are infused, due to the expression of the same antigen on normal tissue, and the infused TCR-T cells also induce the occurrence of cytokine release syndrome (CRS).
Although both TCR-T cells and CAR-T cells are genetically modified, they have several differences. Since TCRs can recognize epitopes from both intracellular and cell surface antigens of tumor cells via TAA/MHC complex, tumor-specific TCRs can only be used in patients with the specific MHC or HLA allele. In contrast, CAR recognition does not rely on peptide processing or MHC molecules; hence, CARs can only recognize tumor surface antigens; however, all surface target molecules may become potential CAR trigger epitopes.60
Obstacles For ACT In OC
Immunosuppressive Tumor Microenvironment
Immune suppression in tumor microenvironment (TME) is mediated by three main factors:86 1) immunosuppressive cells in the TME; 2) cytokines and enzymes released from the tumor or myeloid cells; and 3) barriers. An immunosuppressive TME seriously affects the therapeutic effect of ACT on tumors, especially in OC,87 which can build a highly suppressive environment in the peritoneal cavity.
T-regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) are both important immunosuppressive cells in the OC TME.88,89 Tregs can induce immunosuppressive cytokines such as IL-10 and TGF-β to suppress the function of tumor-infiltrating cytotoxic T cells in the OC environment.90,91 MDSCs express high levels of substances such as arginase, inducible nitric oxide synthase (iNOS), and reactive oxygen species (ROS) to inhibit T-cell function.92,93 In order to reduce immunosuppression in TME, non-myeloablative chemotherapy can be used to decrease the number of Tregs and suppressive cellular cytokines.94 This can also be achieved by combinations of ACT with immune checkpoint inhibitors and targeted therapies.95
Some cytokines and enzymes released from the tumor or myeloid cells also can suppress the immune response. High expression of vascular endothelial growth factor (VEGF), which is expressed in most OC,96,97 can recruit MDSCs to the tumor site and inhibited tumor immunity in ovarian carcinoma.98 In tumor cells, the high expression of TGF-β will inhibit the function of human memory CD8+ T cells and tumor-infiltrating lymphocytes.99 Indoleamine 2,3-dioxygenase (IDO) is an enzyme that catalyzes the degradation of tryptophan, which in turn inhibits T cell proliferation. In addition, IDO can directly inhibit T cells and enhance local Treg-mediated immunosuppression.100 To solve the immunosuppression caused by these cytokines and enzymes, we can combine antagonists and blocking antibodies for different targets with ACT treatment.
Tumor vasculature and tumor stroma are both common barriers that prevent homing of effector T cells.101,102 Leung et al suggested that cancer-associated fibroblasts (CAFs) can upregulate the lipoma-preferred partner (LPP) and this was correlated with survival and chemoresistance in patients with OC.102 They demonstrate the importance of CAF–endothelial cell crosstalk signaling in cancer treatment and the strategy of using LPP targeting siRNA in combination with cytotoxic drugs to improve treatment efficacy. Antiangiogenic agents are also being used to normalize the tumor vasculature and reduce the tumor metastasis in OC.6 A recent study showed that losartan has the effect of reducing collagen content as well as improving perfusion and relieving tumor hypoxia, and this is helpful to enhance chemotherapeutic efficacy in OC models.103
Cytokine Release Syndrome (CRS)
CRS is a potentially life-threatening acute inflammation that occurs after ACT infusion in hematologic and solid tumor patients.104–106 The typical clinical manifestations of CRS include constitutional symptoms like fever, malaise, anorexia, and myalgias,107,108 and organic damage like cardiac dysfunction, respiratory distress syndrome, and renal/hepatic/neurologic toxicity.107 CRS is caused by a massive cytokine release by the infused T cells or other immune cells that are activated as they recognize tumor antigens. IL-6 is a crucial mediator in CRS and is therefore an important target for the treatment of CRS.109 An antagonist of IL-6 receptor, tocilizumab, can alleviate the toxicity of CRS without affecting the therapeutic effect of ACT.109,110
On-Target, Off-Tumor Toxicity
“Tumor-specific” antigens are also commonly expressed in normal tissues, which results in an on-target, off-tumor immune response after stimulation of T cells in ACT. If these antigens are expressed in vital organs, such as heart, liver, and kidney, they will cause fatal damage.74,75 The fundamental solution to this problem is to select tumor-specific antigens that are not expressed in normal tissues, but this is difficult. To overcome this, Kloss et al proposed a method where T cells are transduced with both a CAR, offering suboptimal activation upon binding of one antigen, and a chimeric costimulatory receptor (CCR) that recognizes a second antigen.111 This technology has made progress in OC. Lanitis et al generated trans-signaling T cells with two distinct CARs: anti-Meso scFv-CD3ζ and anti-FRα scFv-CD28.112 Both FRα and mesothelin show high expression in OC tissue, compared to much lower expression in normal tissues. The trans-signaling CAR strategy can more accurately identify tumor cells and diminish damage to normal tissues.
Future Perspective
Many clinical trials of ACT for ovarian cancer are ongoing [Table 2]. However, owing to the complexity of the OC tumor microenvironment and the human immune system, there are still many problems to be solved in the ACT treatment of OC. For example, how to reduce costs while ensuring efficient production of effector cells; and how infused T cells could home in on the tumor site and infiltrate it more accurately. Furthermore, many of the current clinical and preclinical experiments lack randomness, and more randomized trials are needed to confirm that ACT can improve overall survival (OS) or progression-free survival (PFS) in patients with ovarian cancer.
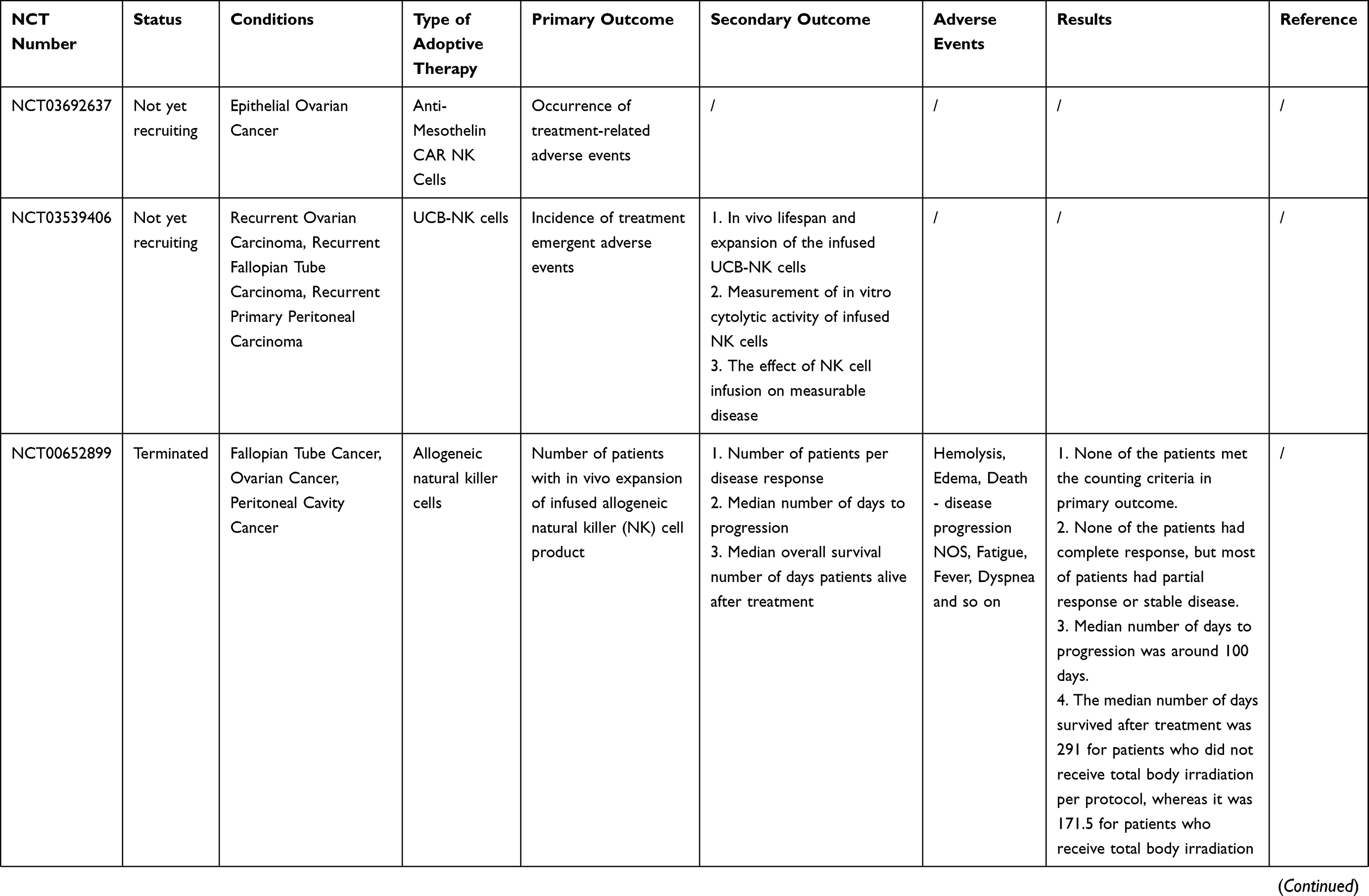 | 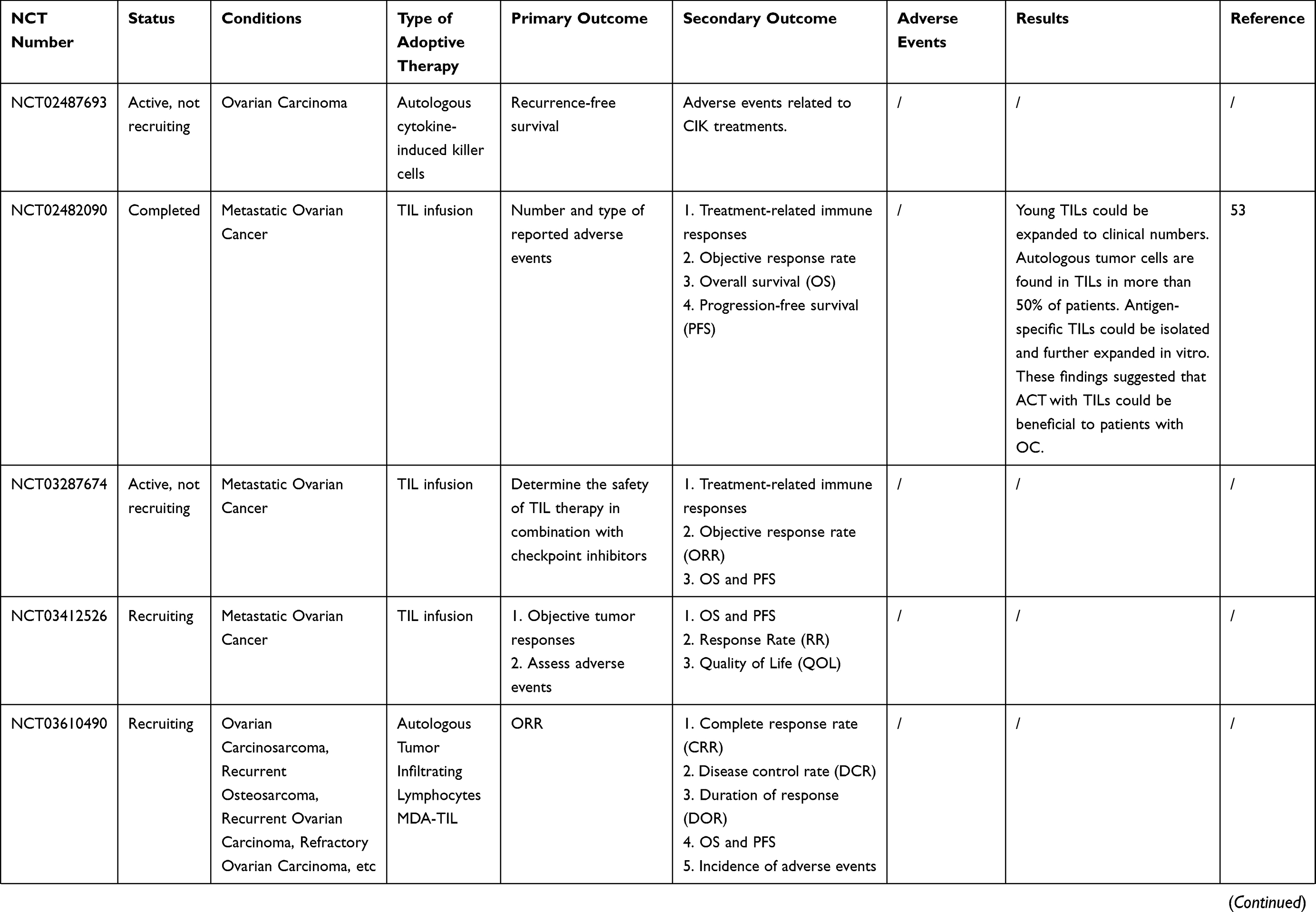 | 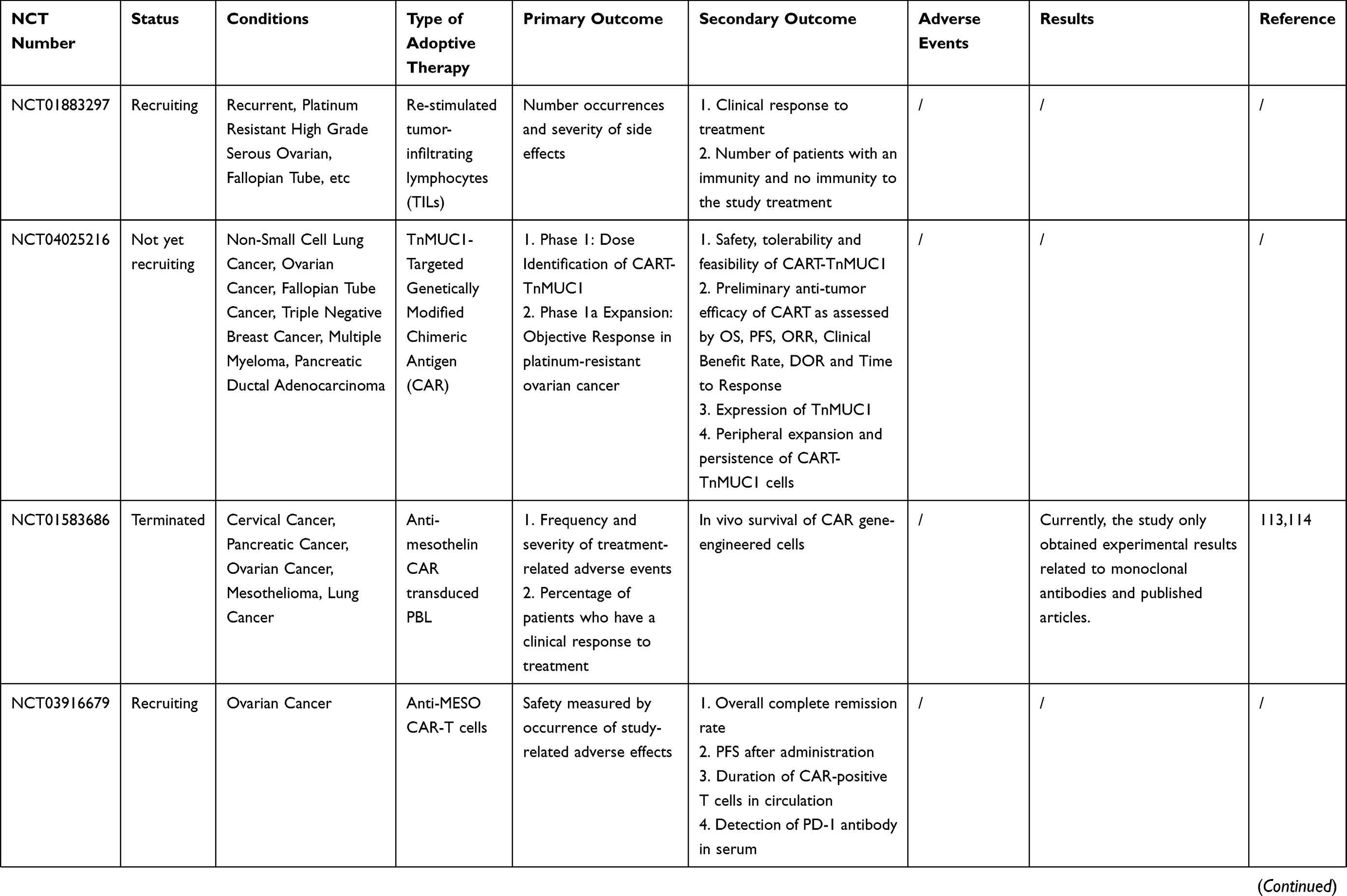 | 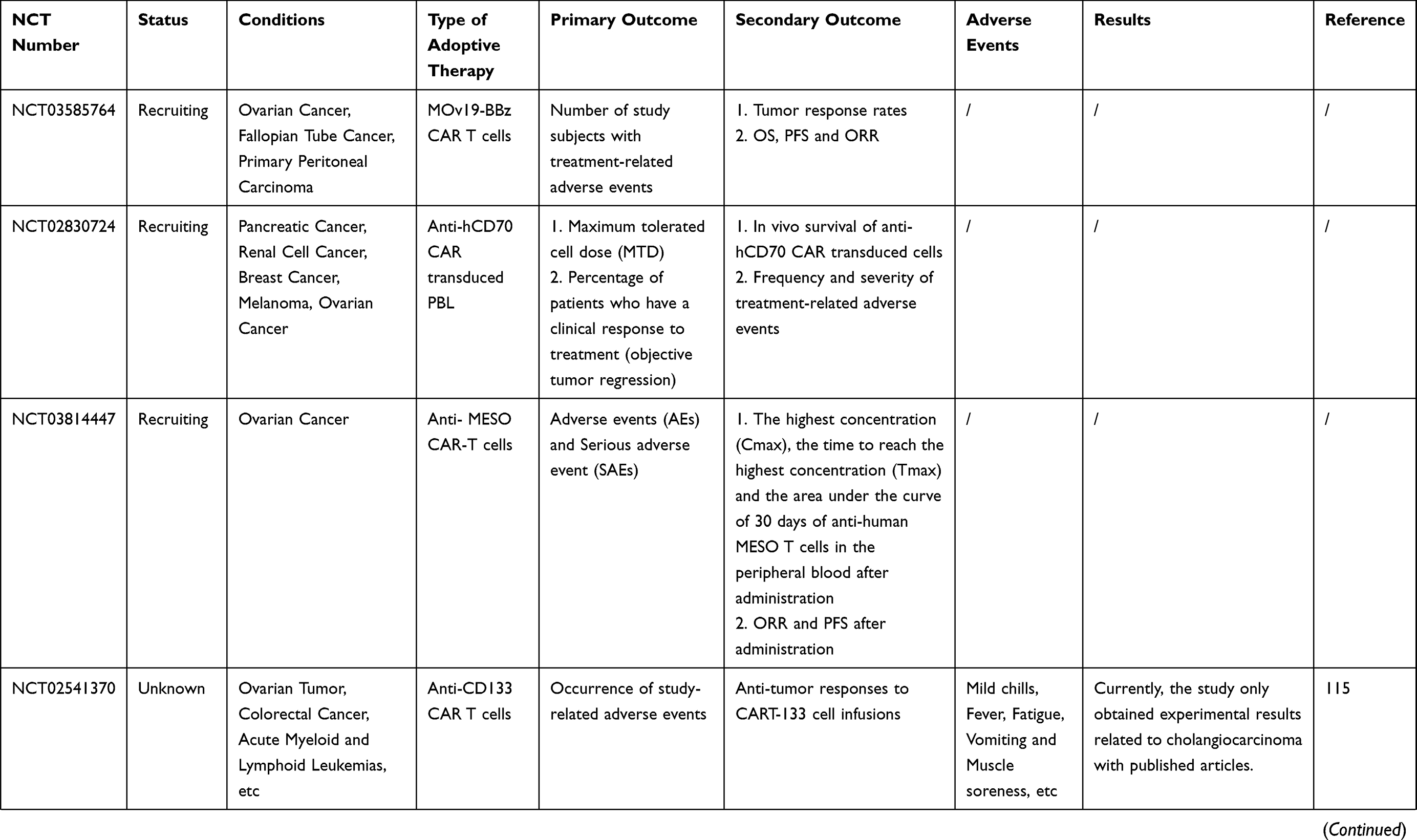 | 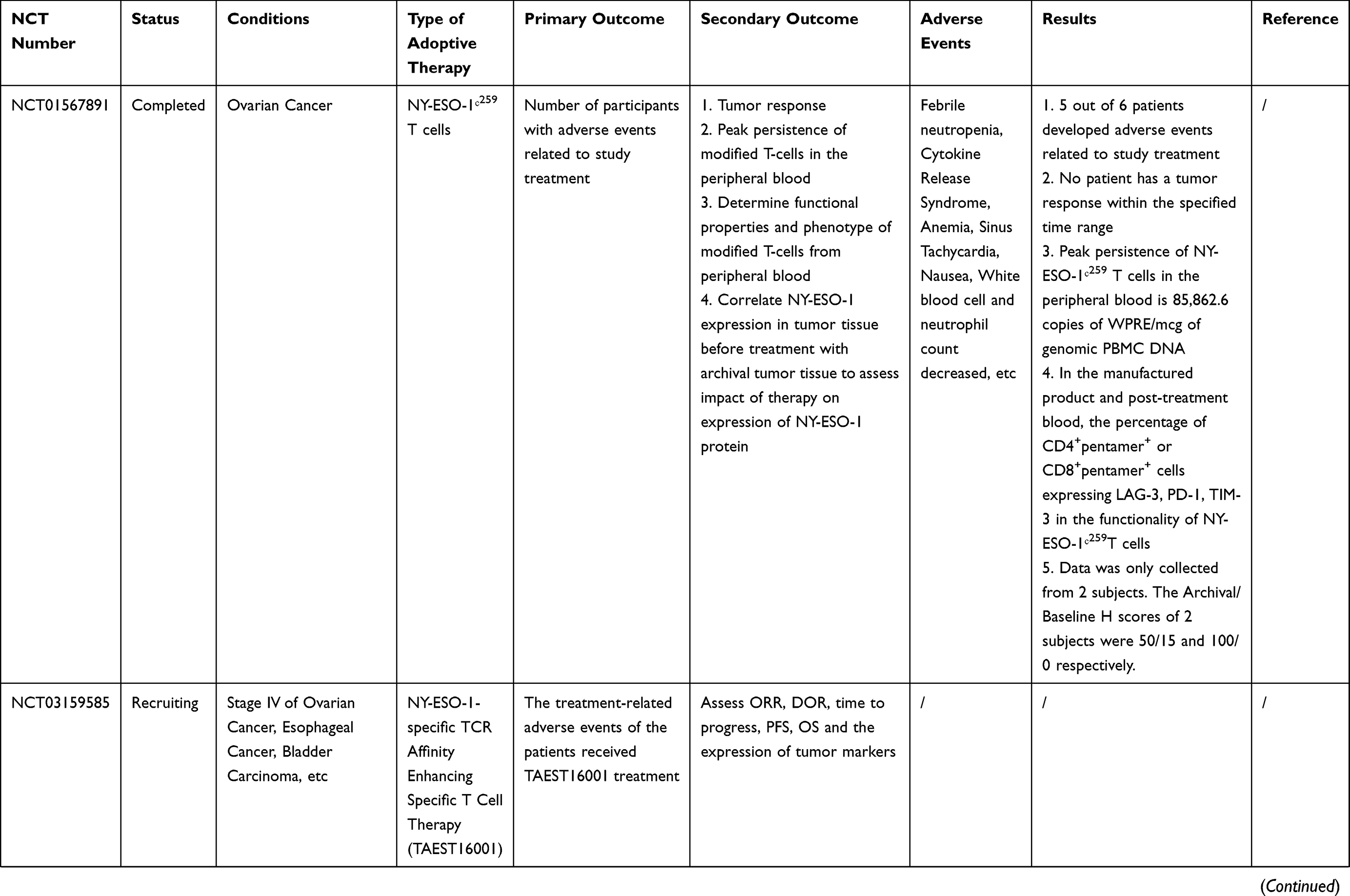 | 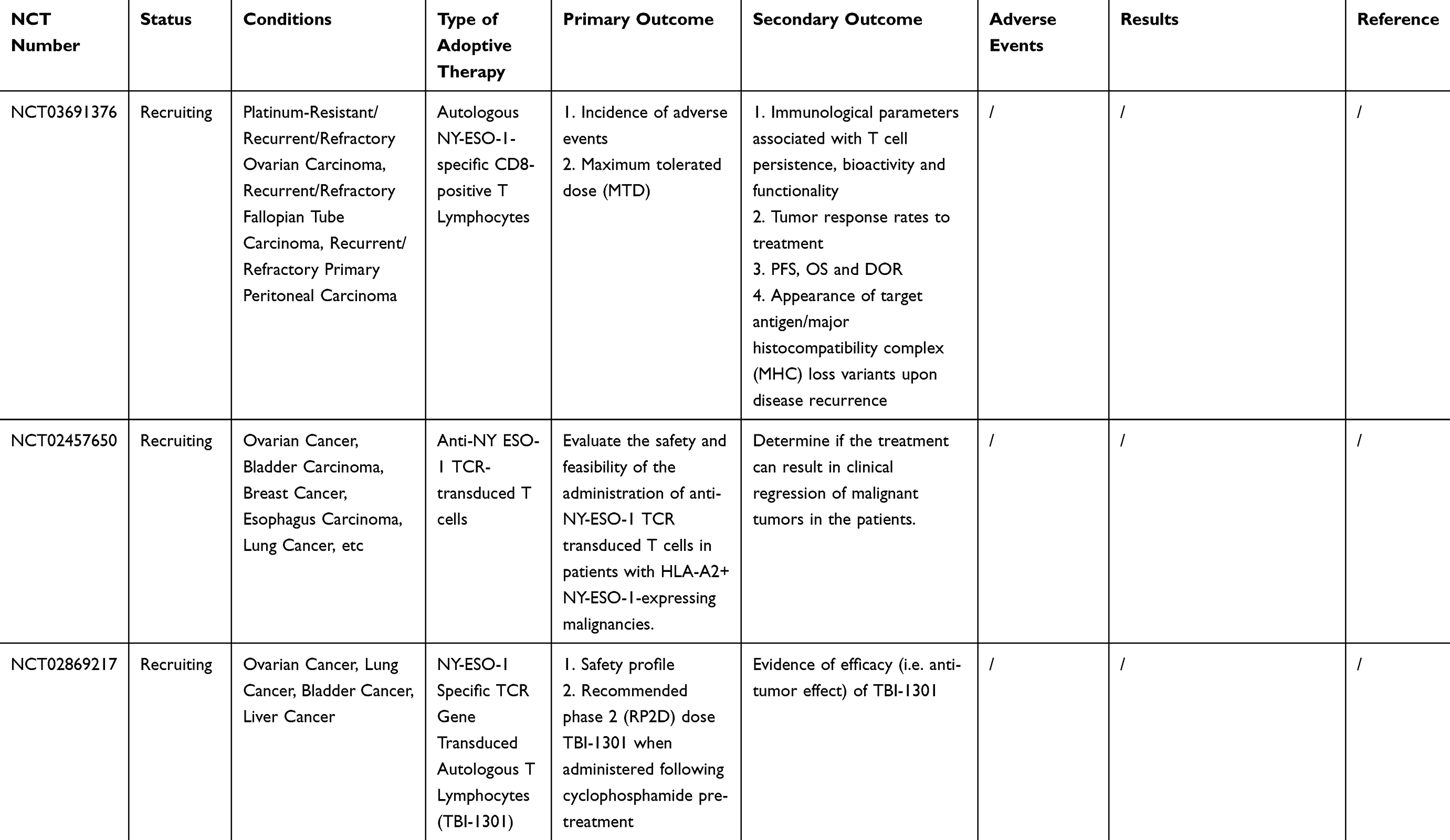 |
Table 2 Current On-Going And Completed Clinical Trials On ACT In Ovarian Cancer |
Nonetheless, in preclinical and clinical trials, the efficacy of ACT in OC is promising. Therefore, ACT still shows potential to become the new effective therapy for OC if we can address the abovementioned obstacles to reduce toxicity and improve the efficiency of ACT. The immune response inhibition caused by the tumor microenvironment can be reduced by combining the application of immunological checkpoint inhibitors or anti-angiogenesis agents, enabling improved effector cell function. In addition, new technology is evolving that utilizes two different CARs on T cells, enabling effector cells to more accurately identify tumor cells and improving their antitumor efficacy. Cell metabolism plays an important role in the anti-tumor effect of immune cells. Increasingly, studies have shown that the regulation of immune cell metabolism can affect the immune response;116–118 therefore, regulation of the effector cell metabolism process may be another approach to improve the anti-tumor effect and efficacy of ACT. Further refinement of technologies will hopefully generate more successful treatment methods for ovarian cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi:10.1002/ijc.29210
2. Elies A, Riviere S, Pouget N, et al. The role of neoadjuvant chemotherapy in ovarian cancer. Expert Rev Anticancer Ther. 2018;18(6):555–566. doi:10.1080/14737140.2018.1458614
3. Gorodnova TV, Sokolenko AP, Kuligina E, Berlev IV, Imyanitov EN. Principles of clinical management of ovarian cancer. Chin Clin Oncol. 2018;7(6):56. doi:10.21037/cco
4. Cho JH, Kim S, Song YS. Neoadjuvant chemotherapy in advanced ovarian cancer: optimal patient selection and response evaluation. Chin Clin Oncol. 2018;7(6):58. doi:10.21037/cco
5. Vargas-Hernandez VM, Moreno-Eutimio MA, Acosta-Altamirano G, Vargas-Aguilar VM. Management of recurrent epithelial ovarian cancer. Gland Surg. 2014;3(3):198–202. doi:10.3978/j.issn.2227-684X.2013.10.01
6. Monk BJ, Minion LE, Coleman RL. Anti-angiogenic agents in ovarian cancer: past, present, and future. Ann Oncol. 2016;27(Suppl 1):i33–i39. doi:10.1093/annonc/mdw093
7. du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer. 2009;115(6):1234–1244. doi:10.1002/cncr.24149
8. Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: evolution of management in the era of precision medicine. CA Cancer J Clin. 2019;69(4):280–304. doi:10.3322/caac.21559
9. Burger RA, Brady MF, Bookman MA, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. doi:10.1056/NEJMoa1104390
10. Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. doi:10.1056/NEJMoa1103799
11. Han ES, Monk BJ. What is the risk of bowel perforation associated with bevacizumab therapy in ovarian cancer? Gynecol Oncol. 2007;105(1):3–6. doi:10.1016/j.ygyno.2007.01.038
12. Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. doi:10.1056/NEJMoa1200694
13. Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–144. doi:10.1056/NEJMoa1305133
14. Hamanishi J, Mandai M, Ikeda T, et al. Safety and antitumor activity of Anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol. 2015;33(34):4015–4022. doi:10.1200/JCO.2015.62.3397
15. Proto C, Ferrara R, Signorelli D, et al. Choosing wisely first line immunotherapy in non-small cell lung cancer (NSCLC): what to add and what to leave out. Cancer Treat Rev. 2019;75:39–51. doi:10.1016/j.ctrv.2019.03.004
16. Rodriguez-Cerdeira C, Carnero Gregorio M, Lopez-Barcenas A, et al. Advances in immunotherapy for melanoma: a comprehensive review. Mediators Inflamm. 2017;2017:3264217. doi:10.1155/2017/3264217
17. Floudas CS, Brar G, Greten TF. Immunotherapy: current status and future perspectives. Dig Dis Sci. 2019;64(4):1030–1040. doi:10.1007/s10620-019-05516-7
18. Adams S, Gatti-Mays ME, Kalinsky K, et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 2019. doi:10.1001/jamaoncol.2018.7147
19. Zhang L, Conejo-Garcia JR, Katsaros D, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–213. doi:10.1056/NEJMoa020177
20. Coukos G, Tanyi J, Kandalaft LE. Opportunities in immunotherapy of ovarian cancer. Ann Oncol. 2016;27(Suppl 1):i11–i15. doi:10.1093/annonc/mdw084
21. Wefers C, Lambert LJ, Torensma R, Hato SV. Cellular immunotherapy in ovarian cancer: targeting the stem of recurrence. Gynecol Oncol. 2015;137(2):335–342. doi:10.1016/j.ygyno.2015.02.019
22. Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol. 2017;28(suppl_8):viii1–viii7. doi:10.1093/annonc/mdx444
23. Fan CA, Reader J, Roque DM. Review of immune therapies targeting ovarian cancer. Curr Treat Options Oncol. 2018;19(12):74. doi:10.1007/s11864-018-0584-3
24. Pakish JB, Jazaeri AA. Immunotherapy in gynecologic cancers: are we there yet? Curr Treat Options Oncol. 2017;18(10):59. doi:10.1007/s11864-017-0504-y
25. Zhang M, Zhang DB, Shi H. Application of chimeric antigen receptor-engineered T cells in ovarian cancer therapy. Immunotherapy. 2017;9(10):851–861. doi:10.2217/imt-2017-0039
26. Bortin MM, Rimm AA, Saltzstein EC. Graft versus leukemia: quantification of adoptive immunotherapy in murine leukemia. Science. 1973;179(4075):811–813. doi:10.1126/science.179.4075.811
27. Fraietta JA, Lacey SF, Orlando EJ, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563–571. doi:10.1038/s41591-018-0010-1
28. Dudley ME, Wunderlich JR, Robbins PF, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi:10.1126/science.1076514
29. Dudley ME, Yang JC, Sherry R, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi:10.1200/JCO.2008.16.5449
30. Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23(10):2346–2357. doi:10.1200/JCO.2005.00.240
31. Liu YL, Zamarin D. Combination immune checkpoint blockade strategies to maximize immune response in gynecological cancers. Curr Oncol Rep. 2018;20(12):94. doi:10.1007/s11912-018-0740-8
32. Perica K, Varela JC, Oelke M, Schneck J. Adoptive T cell immunotherapy for cancer. Rambam Maimonides Med J. 2015;6(1):e0004. doi:10.5041/RMMJ.20769172
33. Phillips JH, Lanier LL. Dissection of the lymphokine-activated killer phenomenon. Relative contribution of peripheral blood natural killer cells and T lymphocytes to cytolysis. J Exp Med. 1986;164(3):814–825. doi:10.1084/jem.164.3.814
34. Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313(23):1485–1492. doi:10.1056/NEJM198512053132327
35. Grimm EA. Human lymphokine-activated killer cells (LAK cells) as a potential immunotherapeutic modality. Biochim Biophys Acta. 1986;865(3):267–279. doi:10.1016/0304-419x(86)90017-x
36. Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9(5):503–510. doi:10.1038/ni1582
37. Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–154. doi:10.1016/j.ccell.2017.06.009
38. Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 1975;16(2):230–239. doi:10.1002/ijc.2910160205
39. Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5(2):117–121. doi:10.1002/eji.1830050209
40. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17(9):1025–1036. doi:10.1038/ni.3518
41. Baragano Raneros A, Lopez-Larrea C, Suarez-Alvarez B. Acute myeloid leukemia and NK cells: two warriors confront each other. Oncoimmunology. 2019;8(2):e1539617. doi:10.1080/2162402X.2018.1539617
42. Carlsten M, Bjorkstrom NK, Norell H, et al. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007;67(3):1317–1325. doi:10.1158/0008-5472.CAN-06-2264
43. Hermanson DL, Bendzick L, Kaufman DS. Mouse xenograft model for intraperitoneal administration of NK cell immunotherapy for ovarian cancer. Methods Mol Biol. 2016;1441:277–284. doi:10.1007/978-1-4939-3684-7_23
44. Schmidt-Wolf IG, Negrin RS, Kiem HP, Blume KG, Weissman IL. Use of a SCID mouse/human lymphoma model to evaluate cytokine-induced killer cells with potent antitumor cell activity. J Exp Med. 1991;174(1):139–149. doi:10.1084/jem.174.1.139
45. Linn YC, Lau LC, Hui KM. Generation of cytokine-induced killer cells from leukaemic samples with in vitro cytotoxicity against autologous and allogeneic leukaemic blasts. Br J Haematol. 2002;116(1):78–86. doi:10.1046/j.1365-2141.2002.03247.x
46. Leemhuis T, Wells S, Scheffold C, Edinger M, Negrin RS. A phase I trial of autologous cytokine-induced killer cells for the treatment of relapsed hodgkin disease and non-hodgkin lymphoma. Biol Blood Marrow Transplant. 2005;11(3):181–187. doi:10.1016/j.bbmt.2004.11.019
47. Introna M, Borleri G, Conti E, et al. Repeated infusions of donor-derived cytokine-induced killer cells in patients relapsing after allogeneic stem cell transplantation: a phase I study. Haematologica. 2007;92(7):952–959. doi:10.3324/haematol.11132
48. Liu J, Li H, Cao S, et al. Maintenance therapy with autologous cytokine-induced killer cells in patients with advanced epithelial ovarian cancer after first-line treatment. J Immunother. 2014;37(2):115–122. doi:10.1097/CJI.0000000000000021
49. Zhou Y, Chen CL, Jiang SW, et al. Retrospective analysis of the efficacy of adjuvant CIK cell therapy in epithelial ovarian cancer patients who received postoperative chemotherapy. Oncoimmunology. 2019;8(2):e1528411. doi:10.1080/2162402X.2018.1528411
50. Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi:10.1158/1078-0432.CCR-11-0116
51. Aoki Y, Takakuwa K, Kodama S, et al. Use of adoptive transfer of tumor-infiltrating lymphocytes alone or in combination with cisplatin-containing chemotherapy in patients with epithelial ovarian cancer. Cancer Res. 1991;51(7):1934–1939.
52. Fujita K, Ikarashi H, Takakuwa K, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1(5):501–507.
53. Westergaard MCW, Andersen R, Chong C, et al. Tumour-reactive T cell subsets in the microenvironment of ovarian cancer. Br J Cancer. 2019;120(4):424–434. doi:10.1038/s41416-019-0384-y
54. Radestad E, Klynning C, Stikvoort A, et al. Immune profiling and identification of prognostic immune-related risk factors in human ovarian cancer. Oncoimmunology. 2019;8(2):e1535730. doi:10.1080/2162402X.2018.1535730
55. Owens GL, Price MJ, Cheadle EJ, Hawkins RE, Gilham DE, Edmondson RJ. Ex vivo expanded tumour-infiltrating lymphocytes from ovarian cancer patients release anti-tumour cytokines in response to autologous primary ovarian cancer cells. Cancer Immunol Immunother. 2018;67(10):1519–1531. doi:10.1007/s00262-018-2211-3
56. Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26(4):332–342.
57. Junker N, Andersen MH, Wenandy L, et al. Bimodal ex vivo expansion of T cells from patients with head and neck squamous cell carcinoma: a prerequisite for adoptive cell transfer. Cytotherapy. 2011;13(7):822–834. doi:10.3109/14653249.2011.563291
58. Priceman SJ, Forman SJ, Brown CE. Smart CARs engineered for cancer immunotherapy. Curr Opin Oncol. 2015;27(6):466–474. doi:10.1097/CCO.0000000000000232
59. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422–442. doi:10.1038/s41571-018-0003-5
60. Met O, Jensen KM, Chamberlain CA, Donia M, Svane IM. Principles of adoptive T cell therapy in cancer. Semin Immunopathol. 2019;41(1):49–58. doi:10.1007/s00281-018-0703-z
61. Wang L, Xu T, Cui M. Are ovarian cancer stem cells the target for innovative immunotherapy? Onco Targets Ther. 2018;11:2615–2626. doi:10.2147/OTT.S155458
62. Jindal V, Arora E, Gupta S, Lal A, Masab M, Potdar R. Prospects of chimeric antigen receptor T cell therapy in ovarian cancer. Med Oncol. 2018;35(5):70. doi:10.1007/s12032-018-1131-6
63. Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi:10.1056/NEJMoa1709919
64. Brudno JN, Maric I, Hartman SD, et al. T cells genetically modified to express an Anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018;36(22):2267–2280. doi:10.1200/JCO.2018.77.8084
65. Chekmasova AA, Rao TD, Nikhamin Y, et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res. 2010;16(14):3594–3606. doi:10.1158/1078-0432.CCR-10-0192
66. Felder M, Kapur A, Gonzalez-Bosquet J, et al. MUC16 (CA125): tumor biomarker to cancer therapy, a work in progress. Mol Cancer. 2014;13:129. doi:10.1186/1476-4598-13-129
67. Hassan R, Kreitman RJ, Pastan I, Willingham MC. Localization of mesothelin in epithelial ovarian cancer. Appl Immunohistochem Mol Morphol. 2005;13(3):243–247.
68. Yen MJ, Hsu CY, Mao TL, et al. Diffuse mesothelin expression correlates with prolonged patient survival in ovarian serous carcinoma. Clin Cancer Res. 2006;12(3 Pt 1):827–831. doi:10.1158/1078-0432.CCR-05-1397
69. Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108(3):619–626. doi:10.1016/j.ygyno.2007.11.020
70. Toffoli G, Cernigoi C, Russo A, Gallo A, Bagnoli M, Boiocchi M. Overexpression of folate binding protein in ovarian cancers. Int J Cancer. 1997;74(2):193–198. doi:10.1002/(sici)1097-0215(19970422)74:2<193::aid-ijc10>3.0.co;2-f
71. Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer. 2006;119(2):243–250. doi:10.1002/ijc.21712
72. Song DG, Ye Q, Carpenito C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71(13):4617–4627. doi:10.1158/0008-5472.CAN-11-0422
73. Kandalaft LE, Powell DJ
74. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi:10.1038/mt.2010.24
75. Cai B, Guo M, Wang Y, et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J Hematol Oncol. 2016;9(1):131. doi:10.1186/s13045-016-0357-z
76. Germain RN, Stefanova I. The dynamics of T cell receptor signaling: complex orchestration and the key roles of tempo and cooperation. Annu Rev Immunol. 1999;17:467–522. doi:10.1146/annurev.immunol.17.1.467
77. van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi:10.1146/annurev.immunol.21.120601.141036
78. Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi:10.1038/mt.2010.272
79. Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. doi:10.1126/science.1129003
80. Robbins PF, Kassim SH, Tran TL, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015;21(5):1019–1027. doi:10.1158/1078-0432.CCR-14-2708
81. San Miguel JF, Paiva B, Lasarte JJ. Engineering Anti-myeloma responses using affinity-enhanced TCR-engineered T cells. Cancer Cell. 2015;28(3):281–283. doi:10.1016/j.ccell.2015.08.009
82. Matsuda T, Leisegang M, Park JH, et al. Induction of neoantigen-specific cytotoxic T cells and construction of T-cell receptor-engineered T cells for ovarian cancer. Clin Cancer Res. 2018;24(21):5357–5367. doi:10.1158/1078-0432.CCR-18-0142
83. Jungbluth AA, Chen YT, Stockert E, et al. Immunohistochemical analysis of NY-ESO-1 antigen expression in normal and malignant human tissues. Int J Cancer. 2001;92(6):856–860. doi:10.1002/ijc.1282
84. Odunsi K, Jungbluth AA, Stockert E, et al. NY-ESO-1 and LAGE-1 cancer-testis antigens are potential targets for immunotherapy in epithelial ovarian cancer. Cancer Res. 2003;63(18):6076–6083.
85. Schmitt TM, Aggen DH, Ishida-Tsubota K, Ochsenreither S, Kranz DM, Greenberg PD. Generation of higher affinity T cell receptors by antigen-driven differentiation of progenitor T cells in vitro. Nat Biotechnol. 2017;35(12):1188–1195. doi:10.1038/nbt.4004
86. Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80. doi:10.1126/science.aaa6204
87. Stone ML, Chiappinelli KB, Li H, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A. 2017;114(51):E10981–E10990. doi:10.1073/pnas.1712514114
88. Li L, Wang L, Li J, et al. Metformin-induced reduction of CD39 and CD73 blocks myeloid-derived suppressor cell activity in patients with ovarian cancer. Cancer Res. 2018;78(7):1779–1791. doi:10.1158/0008-5472.CAN-17-2460
89. Curiel TJ, Coukos G, Zou L, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi:10.1038/nm1093
90. Woo EY, Chu CS, Goletz TJ, et al. Regulatory CD4(+) CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61(12):4766–4772.
91. Chen ML, Pittet MJ, Gorelik L, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. 2005;102(2):419–424. doi:10.1073/pnas.0408197102
92. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
93. Marvel D, Gabrilovich DI. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest. 2015;125(9):3356–3364. doi:10.1172/JCI80005
94. Gattinoni L, Finkelstein SE, Klebanoff CA, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202(7):907–912. doi:10.1084/jem.20050732
95. Mitsui J, Nishikawa H, Muraoka D, et al. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res. 2010;16(10):2781–2791. doi:10.1158/1078-0432.CCR-09-3243
96. Cascinu S, Staccioli MP, Gasparini G, et al. Expression of vascular endothelial growth factor can predict event-free survival in stage II colon cancer. Clin Cancer Res. 2000;6(7):2803–2807.
97. Brustmann H, Naude S. Vascular endothelial growth factor expression in serous ovarian carcinoma: relationship with high mitotic activity and high FIGO stage. Gynecol Oncol. 2002;84(1):47–52. doi:10.1006/gyno.2001.6467
98. Horikawa N, Abiko K, Matsumura N, et al. Expression of vascular endothelial growth factor in ovarian cancer inhibits tumor immunity through the accumulation of myeloid-derived suppressor cells. Clin Cancer Res. 2017;23(2):587–599. doi:10.1158/1078-0432.CCR-16-0387
99. Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174(9):5215–5223. doi:10.4049/jimmunol.174.9.5215
100. Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–1154. doi:10.1172/JCI31178
101. Natarajan S, Foreman KM, Soriano MI, et al. Collagen remodeling in the hypoxic tumor-mesothelial niche promotes ovarian cancer metastasis. Cancer Res. 2019;79(9):2271–2284. doi:10.1158/0008-5472.CAN-18-2616
102. Leung CS, Yeung TL, Yip KP, et al. Cancer-associated fibroblasts regulate endothelial adhesion protein LPP to promote ovarian cancer chemoresistance. J Clin Invest. 2018;128(2):589–606. doi:10.1172/JCI95200
103. Zhao Y, Cao J, Melamed A, et al. Losartan treatment enhances chemotherapy efficacy and reduces ascites in ovarian cancer models by normalizing the tumor stroma. Proc Natl Acad Sci U S A. 2019;116(6):2210–2219. doi:10.1073/pnas.1818357116
104. Godel P, Shimabukuro-Vornhagen A, von Bergwelt-Baildon M. Understanding cytokine release syndrome. Intensive Care Med. 2018;44(3):371–373. doi:10.1007/s00134-017-4943-5
105. Park JH, Geyer MB, Brentjens RJ. CD19-targeted CAR T-cell therapeutics for hematologic malignancies: interpreting clinical outcomes to date. Blood. 2016;127(26):3312–3320. doi:10.1182/blood-2016-02-629063
106. Tanyi JL, Stashwick C, Plesa G, et al. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T cells. J Immunother. 2017;40(3):104–107. doi:10.1097/CJI.0000000000000160
107. Neelapu SS, Tummala S, Kebriaei P, et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47–62. doi:10.1038/nrclinonc.2017.148
108. Lee DW, Gardner R, Porter DL, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi:10.1182/blood-2014-05-552729
109. Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–748. doi:10.1038/s41591-018-0036-4
110. Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–528. doi:10.1016/S0140-6736(14)61403-3
111. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi:10.1038/nbt.2459
112. Lanitis E, Poussin M, Klattenhoff AW, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res. 2013;1(1):43–53. doi:10.1158/2326-6066.CIR-13-0008
113. Hassan R, Bullock S, Premkumar A, et al. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin Cancer Res. 2007;13(17):5144–5149. doi:10.1158/1078-0432.CCR-07-0869
114. Hassan R, Cohen SJ, Phillips M, et al. Phase I clinical trial of the chimeric anti-mesothelin monoclonal antibody MORAb-009 in patients with mesothelin-expressing cancers. Clin Cancer Res. 2010;16(24):6132–6138. doi:10.1158/1078-0432.CCR-10-2275
115. Feng KC, Guo YL, Liu Y, et al. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J Hematol Oncol. 2017;10(1):4. doi:10.1186/s13045-016-0378-7
116. Hu ZL, Qu GJ, Yu XY, et al. Acylglycerol kinase maintains metabolic state and immune responses of CD8(+) T cells. Cell Metab. 2019;30(2):
117. Gubser PM, Bantug GR, Razik L, et al. Rapid effector function of memory CD8(+) T cells requires an immediate-early glycolytic switch. Nat Immunol. 2013;14(10):
118. Zhou PP, Chi HB. AGK unleashes CD8(+) T cell glycolysis to combat tumor growth. Cell Metab. 2019;30(2):233–234. doi:10.1016/j.cmet.2019.07.008
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.