")
Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Apoptotic membrane dynamics in health and disease
Received 15 April 2015
Accepted for publication 14 May 2015
Published 14 July 2015 Volume 2015:7 Pages 133—142
DOI https://doi.org/10.2147/CHC.S57893
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Linda Julian, Michael F Olson
Cancer Research UK Beatson Institute, Glasgow, UK
Abstract: Apoptosis, also known as programmed cell death, is a fundamental homeostatic mechanism essential for normal embryonic development and the maintenance of healthy adult tissues. The processes that affect cell membrane dynamics during the tightly regulated apoptotic program have attracted considerable interest over the years. Distinct biochemical and structural alterations to plasma membrane composition and topography contribute to the efficient removal of cellular corpses. In this review, we will discuss these membrane alterations and their importance in maintaining cell and tissue homeostasis.
Keywords: apoptosis, blebbing, membrane dynamics, homeostasis, apoptotic cell clearance
Introduction
The term “apoptosis” was first used in 1972 by Kerr et al1 to describe a distinct form of programmed cell death that contributes to the elimination of excess, unwanted, or damaged cells during embryonic development and normal cell turnover in healthy adult tissues. The distinct morphological manifestations of a cell undergoing apoptosis include cell shrinkage, membrane blebbing, nuclear and cytoplasmic condensation, and ultimately cell fragmentation into membrane bound apoptotic bodies.2
Although this programmed cell death mechanism was described earlier, it was in the 1980s and 1990s that most of the molecular components governing this highly efficient process came to light. Genetic studies on the nematode Caenorhabditis elegans were critically important for the elucidation of the apoptotic pathway, revealing the ced3, ced4, and ced9 genes as being centrally involved in developmental cell death.3 It was soon discovered that proteases known as cysteine-dependent aspartate specific proteases or caspases were key drivers of the apoptotic program.4–7 Caspases involved in apoptosis may be divided into initiator or effector caspases. Intracellular and extracellular apoptotic stimuli activate the initiator caspases 2, 8, 9, and 10, which in turn signal via a caspase activation cascade to propagate the death stimulus by activating the downstream effector caspases 3, 6, and 7. Activated effector caspases orchestrate the destruction of diverse cellular structures by selectively cleaving target proteins to elicit the morphological and biochemical features associated with apoptosis (Figure 1). Subsequent studies involving targeted genetic disruptions in mice revealed differential requirements for specific caspases in mammalian development, some of them displaying heart and brain development abnormalities, or even resulting in embryonic and perinatal lethality.8
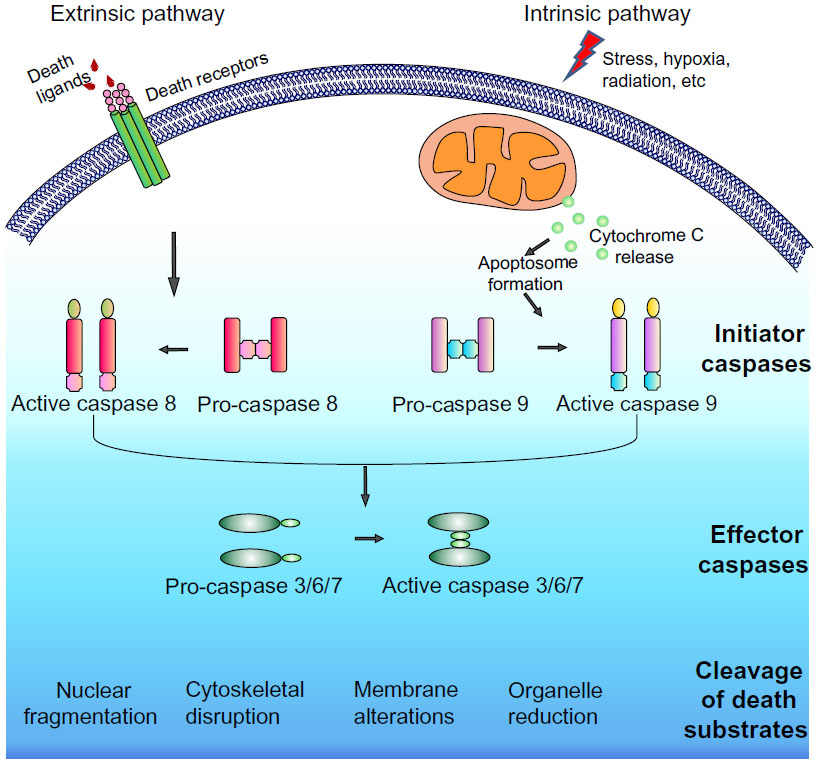 | Figure 1 Caspase cascade. |
Apoptosis in development and maintenance of tissue homeostasis
A central function of apoptosis in vertebrate development is digit formation, since impaired apoptosis leads to ineffective removal of cells between developing digits resulting in retention of interdigital webbing.9 Similarly, the immune system uses apoptosis to eliminate developing lymphocytes lacking functional antigen receptors.10,11 The role of apoptosis in nervous system development has also been widely recognized, where superfluous neurons and neurons that fail to establish functional synaptic connections are eliminated.12 Apoptosis during embryonic development also helps give tissues and organs their form and structure. For example, during gastrulation, the conversion of solid embryonic ectoderm into hollow closed tubes of columnar epithelial cells is made possible by apoptosis of inner ectodermal cells.13,14 In addition, mechanical force generated by apoptotic cells has recently been shown to be vital for inducing tension in developing Drosophila melanogaster tissues to induce epithelial folding, thus establishing proper topography.15
In addition to its roles in development, apoptosis is also important for the maintenance of tissue homeostasis through adult life. Cell turnover in adult tissues is highly variable across tissue types, and heavily relies on the apoptotic machinery to remove damaged and superfluous cells. In adults, cell death also plays a key role in tissue remodeling, such as mammary gland involution16 and follicular atresia of the postovulatory follicle.17 Along with the tightly controlled apoptotic program, highly efficient phagocytic mechanisms ensure prompt removal of apoptotic debris, thereby reducing the potential for apoptotic cells to become necrotic (secondary necrosis), resulting in inflammation.18 In contrast to the immunologically silent apoptotic process, necrosis may stimulate inflammatory responses that could be harmful to tissue health. Despite the large number of cells dying by apoptosis in adult tissues, it is difficult to detect apoptotic cells even in tissues with high cell turnover such as the thymus.19 Defects in clearance mechanisms have been linked to inflammation and autoimmunity,20 reflecting the importance and efficiency of apoptotic cell clearance under normal circumstances.
Membrane dynamics and changes in membrane properties during apoptosis
The process of apoptosis includes important changes in apoptotic cell membranes that aid in efficient recognition and removal. For the purpose of this review, membrane alterations are broadly divided into biochemical and structural.
Biochemical alterations
A common feature of plasma membranes in eukaryotic cells is the asymmetric distribution of lipids between the inner and outer leaflets, with phosphatidylcholine and sphingomyelin predominantly present on the outer leaflet, whereas phosphatidylserine (PS), phosphatidylinositol, and phosphatidylethanolamine are confined to the cytoplasmic face of membranes (Figure 2).21 The maintenance of lipid asymmetry under homeostatic conditions is under the control of transporters called flippases and floppases.22 A third type of enzyme known as scramblases contributes to the loss of lipid asymmetry and increased PS extracellular exposure when activated.23 Loss of membrane asymmetry and PS externalization on the outer leaflet of the plasma membrane are common features of apoptotic cells, independent of the form of apoptotic stimulus.24,25 However, PS exposure is not restricted to apoptotic cells alone, but may also be seen on activated platelets.26,27 The events responsible for PS externalization have been widely debated over the years. Recently, Transmembrane Protein 16F, which has a Ca2+-dependent phospholipid scramblase activity, was identified as a mediator of PS externalization in activated platelets.28,29 In the case of apoptotic cells, another scramblase called XK-related protein 8 that gets activated by caspase cleavage was identified as a Ca2+-independent mediator of PS externalization.30 Recent work by Segawa et al identified adenosine triphosphate type 11C (ATP11C) and cell division cycle protein 50A (CDC50A) as being involved in apoptotic PS exposure.31 ATP11C, a flippase responsible for redirecting PS from the outer to the inner leaflet, is inactivated by caspase-mediated cleavage.31 CDC50A is required for localization of ATP11C to the plasma membrane, and knockout of CDC50A results in constitutive PS exposure.31 These data strongly indicate that during apoptosis, caspase-mediated activation of Xkr8 and caspase-mediated inactivation of ATP11C work in a coordinated fashion to permit PS exposure (Figure 2).
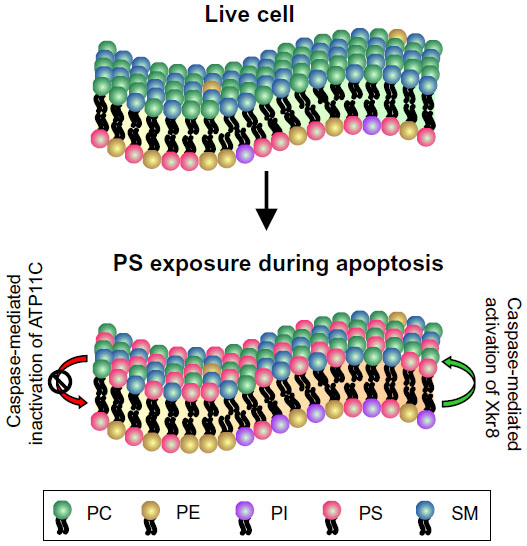 | Figure 2 Phosphatidylserine exposure during apoptosis. |
Early studies confirmed that PS externalization was not just an indicator of loss of membrane asymmetry during apoptosis, but actually has an important active function to play in the clearance of apoptotic cells by acting as a recognition signal for phagocytic cells.24,32 When receptors on phagocytes engage with PS, a signaling cascade is initiated within both professional and non-professional phagocytes that lead to cytoskeletal rearrangements to facilitate apoptotic cell internalization.33,34 A repertoire of PS receptors on macrophages and monocytes has been identified over the years, including Stabilin-2, Tim-4, and Bai1.35–37 PS-dependent recognition of apoptotic cells may also occur via bridging molecules such as milk fat globulin-EGF-factor 8 (MFG-E8) and growth arrest-specific gene 6, which tether externalized PS on apoptotic cells to the phagocytic receptors.38,39 MFG-E8 is secreted by activated macrophages to promote tethering of PS on apoptotic cells to integrins on phagocytes.38 Growth arrest-specific gene 6, which is a ligand for receptor tyrosine kinases Mer, also binds to PS to bridge apoptotic cells to phagocytes for clearance.39 Although PS externalization has been considered to be an important “eat me” signal, it is worth noting that since PS trans-bilayer asymmetry is also seen in some viable, non-apoptotic cells such as activated platelets, which suggested that apoptotic cell engulfment requires exceeding a sensitivity threshold for PS exposure.27,40 However, this theory was challenged when studies showed that the levels of PS exposure on viable or apoptotic cells were not critical for engulfment by macrophages, suggesting that additional molecules may be required to co-operate with PS to aid efficient phagocytosis.41 Interestingly, oxidized PS in conjunction with non-oxidized PS was also found to be required for efficient apoptotic cell engulfment.42 Over the years, a number of molecular determinants and bridging ligands have been identified on apoptotic cells including C1q, a member of the complement pathway,43 and mannose-binding lectin, a collectin protein.44
Structural alterations
Apart from these biochemical changes that occur in apoptotic cells, several dramatic morphological alterations are also key events when cells die by apoptosis, as described by Kerr et al in their seminal report.1 Broadly termed as the execution phase of apoptosis, these structural modifications include cell shrinkage, chromatin condensation, nuclear fragmentation, and formation of dynamic plasma membrane protrusions called blebs, which eventually detach, culminating in fragmentation into apoptotic bodies.1 These membrane protrusions and apoptotic bodies serve as concentration sites for phagocytic markers allowing efficient detection and engulfment by patrolling phagocytes and/or neighboring cells.2,44–46 Packaging of intracellular contents into membrane bound apoptotic bodies is believed to be crucial in avoiding an unwanted inflammatory response by preventing the release of potential self-antigens into the surrounding milieu. Nuclear fragments that are not sequestered within apoptotic bodies are a source of antigens in autoimmune diseases such as systemic lupus erythematosus (SLE).47,48 Until the early 1990s, comprehensive characterization of the execution phase of apoptosis remained elusive because of the asynchronous nature of apoptotic cell death.49,50
Caspases play major roles both in the commitment phase and in the execution phase of apoptosis through their cleavage of numerous protein substrates. Adherent cells retract from neighboring cells and from extracellular substrates via caspase cleavage of β-catenin, a critical regulator of adherens junctions,51 and focal adhesion proteins such as tensin.52 Forces generated by the actin–myosin cytoskeleton induced by the phosphorylation of myosin light chains (MLCs) by caspase-cleaved Rho-associated coiled coil kinase (ROCK1) drive cell contraction and dynamic membrane blebbing during apoptosis.53–55 Two phases of plasma membrane blebbing were reported in apoptotic cells, with the first phase occurring concomitant with cell detachment.56 Several non-adherent cell lines fail to display this first phase of blebbing, suggesting a role for the initial phase in maintaining detachment from substrate and/or neighboring cells.56 The second phase, observed in both adherent and non-adherent cells, results in the formation of blebs that contain chromatin and other cellular contents.56 Caspase-mediated cleavage and weakening of nuclear structural lamin proteins, as well as contractile force generated by the actin–myosin cytoskeleton, result in nuclear fragmentation and redistribution of chromatin to the blebs and apoptotic bodies.57–60
Cytoskeletal dynamics during membrane blebbing
Blebs are blister-like protrusions that occur when cell membranes delaminate from cortical cytoskeletal structures, which may appear and disappear on a timescale of minutes. Although most often considered in the context of cell death, blebbing is also linked to other processes, including locomotion and cytokinesis.61,62 Due to their transient nature, little was known about blebbing until the discovery of the human melanoma cell line M2, deficient in actin–binding protein of 280 kDa (ABP-280; filamin A), that exhibits prolonged periods of extensive plasma membrane blebbing.63,64 Over the years, the physical process of blebbing has been carefully investigated using various theoretical predictions and biophysical experiments. Broadly, the lifecycle of blebs can be subdivided into three steps: nucleation, expansion, and retraction (Figure 3).
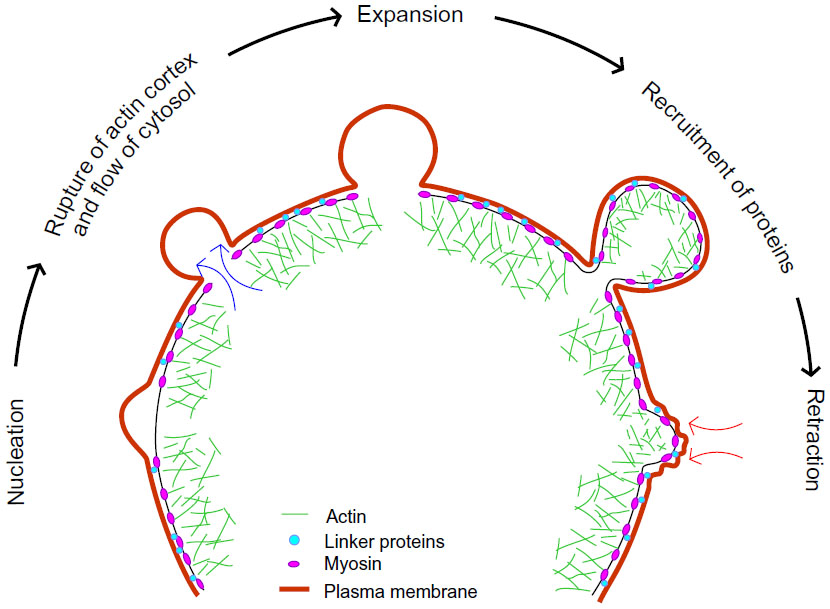 | Figure 3 Life cycle of a bleb. |
Nucleation
Nucleation occurs when the cell membrane delaminates from the cortical cytoskeleton, an event that reflects a change in the balance between cytoplasmic hydrostatic pressure pushing outward and the opposing forces of membrane attachment to cytoskeletal proteins. Bleb protrusion may be initiated when intracellular pressure produced by contraction of cortical actin–myosin structures leads to localized compression of the cytoplasm. Consistent with a role for actin–myosin contraction, inhibition of myosin II activity resulted in abrogation of bleb protrusion.53,61,65 Conversely, laser ablation of the actin–myosin cortex led to bleb formation in L929 fibroblasts, indicating that bleb size and growth are restricted by cortical tension.66 Alteration of ezrin expression in A375 melanoma cells by knockdown or overexpression to change the strength of plasma membrane coupling to the submembrane cortex resulted in corresponding increased or decreased blebbing, respectively.67 This supports the conclusion that bleb protrusion results from changes in the strength of membrane–cortex coupling.
Expansion
Once blebs have been nucleated, they are further expanded by pressure generated by actomyosin contraction and the flow of cytosol into the newly formed bleb. Several possible mechanisms of bleb expansion to increase surface area has been postulated68 including membrane tear from the actin cortex69 and unfolding of membrane folds.70 Bleb expansion eventually ceases due to transient decreases in local pressure, as well as reassembly and/or contraction of the actin–myosin cortex,71 at which point retraction may begin.
Retraction
Retraction has been reported to occur approximately 30 seconds after bleb initiation and takes approximately 90 seconds for full bleb retraction.71,72 It was reported that initially, expanding blebs in filamin-deficient M2 melanoma cells are devoid of cortical actin–myosin; however, there is a sequential recruitment of the membrane-cortex linker protein ezrin, actin, actin-bundling proteins such as α-actinin, and contractile proteins including MLC II during cortex reassembly.71 Concomitant with the regrowth of cortical actin under bleb membranes, the actin–myosin cortex at the base of the bleb disappears, which may serve to supply material for reassembly within blebs. These observations have recently been challenged using refined image analysis of blebbing induced by vaccinia infection of HeLa cells.72 In contrast to previous reports, ezrin was detected within blebs prior to the onset of retraction while filamentous actin continued to accumulate after the onset of retraction. In addition, inhibition of Arp2/3 activity, which contributes to nucleating cortical actin, reduced bleb retraction speeds, decreased the rate of myosin II regulatory light chain recruitment, and increased filamentous actin stability, which indicate that filamentous actin flux combined with actin–myosin contraction both contribute to bleb retraction.
Although the trigger of bleb retraction is still unclear, it has been shown that myosin-driven contraction plays a role in powering retraction.71 What remains unresolved is whether material from the retracted bleb integrates back into the cell cortex. It has been observed that occasionally secondary blebs appear on existing blebs, possibly because of increased pressure within the bleb during retraction that might cause membrane to delaminate from the newly formed cortex.73 In addition, new blebs may occur at the site of a retracted bleb,72 possibly due to instability of the recently perturbed actin cortex and/or relatively weak attachments between membrane and cortical structures.
Inducers of membrane blebbing
Although blebbing may be observed during several cellular processes including cytokinesis, migration, and spreading, arguably its most totemic manifestation is during the execution phase of apoptosis.24 Despite being one of the hallmark features of apoptosis, little was known about the mechanistic drivers underlying this phenomenon until the 1990s. Various studies showed that reorganization of cytoskeletal structures drives the formation of membrane blebs and apoptotic bodies.53 Disruption of filamentous actin inhibited the formation of apoptotic bodies without affecting DNA fragmentation, indicating a requirement for polymerized actin during the later stages of apoptosis.74 One early study implicating myosin contraction in bleb formation showed that microinjecting an active form of MLC kinase resulted in active surface blebbing during mitosis.75 Subsequently, it was shown that blebbing cells had increased levels of myosin II regulatory light chain phosphorylation and that inhibition of MLC kinase activity decreased blebbing.53 Actin–myosin contractile force generation results from the phosphorylation of the regulatory MLCs, which promotes myosin binding to filamentous actin, coupling of actin–myosin filaments to linker proteins at the plasma membrane, and ATP-dependent actin–myosin filament contraction.76,77 Inhibition of myosin contractility with the myosin II ATPase blocker blebbistatin reduced apoptotic blebbing.78
Two homologues of the serine/threonine kinase ROCKs (ROCK1 and ROCK2) have been identified as downstream effectors of Rho.79 ROCKs have dual roles in the regulation of MLC phosphorylation in that they directly phosphorylate MLC and decrease dephosphorylation indirectly by phosphorylating and inhibiting the MLC-binding MYPT1 subunit of the phosphatase complex.80–82 Apoptotic membrane blebbing was found to be dependent on ROCK activity and independent of the function of its upstream regulator, RhoA.54,55 During apoptosis, activated caspases cleave ROCK1 after a aspartate-glutamate-threonine-aspartate (amino acids 1,110–1,113) sequence, thus removing the autoinhibitory C terminal region and generating a truncated kinase fragment with increased intrinsic activity. The ROCK kinase fragment generated can phosphorylate downstream targets including MLC, leading to actin–myosin contractile force generation, cell contractility, and consequent membrane blebbing and apoptotic body formation. The significance of ROCK1 for apoptotic blebbing has been shown in other cell types, including cardiac myocytes, lymphoma cells, and non-small-cell lung carcinoma cells.83–85 Of notable importance was the relocalization of fragmented DNA into blebs and apoptotic bodies upon caspase-activated ROCK1 cleavage.54 In addition to these morphological responses, ROCK1 activity was also required for nuclear envelope tearing and disintegration.60 During the early stages of rapid membrane blebbing and apoptotic body release, there may be leakage of damage-associated molecular pattern proteins, such as nucleosomal histones, highlighting a possible role for blebbing cells and apoptotic bodies to alert the immune cells for efficient phagocytic uptake prior to secondary necrosis.86 In the specialized case of cell death induced by natural killer cells, Granzyme B cleaves ROCK2 at a homologous position to the caspase-cleavage site on ROCK1, leading to constitutive ROCK2 activation that is sufficient to promote caspase-independent membrane blebbing.55 However, given that Granzyme B also activates caspases leading to ROCK1 activation, there are no obvious situations where ROCK2 would be activated without concomitant ROCK1 activation in apoptotic cells.
Pathological consequences of dysfunction in apoptotic membrane dynamics
The final phase during the active apoptotic program is the clearance of cell corpses and the associated debris, a process termed efferocytosis, derived from the Latin word “effero” meaning “to bury”.87 The fact that it is difficult to detect apoptotic cells in situ, even in tissues with high cellular turnover, indicates that clearance mechanisms are highly efficient and rapid. Several elegant studies have shown that apoptotic cells release “find me” signals including lysophosphatidylcholine, sphingosine-1-phosphate, fractalkine, and the nucleotides ATP and UTP to “advertise” their presence to local and distant phagocytes, as reviewed previously.88,89 Once alerted, phagocytes must be capable of distinguishing live cells from the dead, and this is facilitated by different biochemical and structural alterations that occur on the plasma membrane of apoptotic cells as discussed above. Since the apoptotic program is immunologically silent during development and in adult steady-state tissues, largely due to the efficient removal of apoptotic cells, it is not surprising that disturbances in the rapid recognition and removal of the apoptotic debris may lead to pathological consequences.
Autoimmunity
Apoptotic cells that linger in tissues due to inefficient clearance mechanisms may undergo secondary necrosis, allowing intracellular antigens to be recognized as non-self, thus leading to autoantibody production and consequent autoimmunity development.90–92 One of the most widely studied pathological consequences of failed clearance of apoptotic debris is SLE, which is a chronic autoimmune disorder affecting multiple organs.93,94 Evidence gleaned from several genetic mouse models bearing defective PS-mediated recognition of apoptotic cells suggests a strong link between inefficient apoptotic debris clearance and development of autoimmunity.92,95–98 Given the fact that there are multiple PS recognition mechanisms, it is intriguing that these mechanisms are not redundant in suppressing the autoimmune phenotype in these mouse models. In accordance with the mouse data, accumulation of apoptotic cells was observed in SLE patients, which was attributed to impaired efferocytosis rather than an aberrant apoptotic program.99–101 Defects in structural alterations of apoptotic cells can be attributed to the lack of a proper display of membrane modifications. C1q has been shown to bind to apoptotic blebs of keratinocytes45 and vascular endothelial cells,46 suggesting a possible role for apoptotic blebs as focal points for “eat me” signals, to enable efficient efferocytosis.2 C1q-deficient mice exhibited the autoimmune phenotype of glomerulonephritis, as well as an increased number of uncleared apoptotic bodies in the glomeruli, implying defective clearance.90
Nevertheless, animal model studies have revealed that not all defects in apoptotic clearance mechanisms lead to autoimmunity. One example is the non-development of autoimmunity in animals lacking mannose-binding lectin, a collectin that shares functional similarities with C1q, binds to apoptotic cells, and plays a major role in clearance of apoptotic cells in vivo.102 It is possible that the uncleared apoptotic debris has an immunosuppressive effect that helps prevent autoimmunity in these animals, which would require further investigation to validate.
Cancer
In addition to evasion of cell death as a hallmark feature of cancer cells, escaping programmed cell removal has also surfaced as a determinant in cancer development.103,104 Whether apoptotic membrane defects or aberrant clearance mechanisms have beneficial or detrimental effects in the context of tumor progression has been ardently debated over many years. Expression levels of “eat me” and “don’t eat me” are altered in tumor cells as revealed by several animal model studies and human genetic studies. Pro-phagocytic markers such as annexin 1 have been found to be reduced at the mRNA and protein levels in several human cancers including prostate cancer, esophageal cancer, and head and neck cancer.105,106 In addition, “don’t eat me” signals such as CD47 and CD200 were upregulated in lung, breast, and prostate and bladder cancer.107 Although these studies imply that tumors adopt evasion of apoptotic clearance as a cancer promoting strategy, several studies reported upregulation of different apoptotic recognition molecules such as PS108,109 and calreticulin104 in tumor cells, raising questions regarding the role of efficient apoptotic cell clearance in the context of tumorigenesis.
Chronic inflammation is a key driver of tumorigenesis.110–112 Efficient clearance mechanisms are accompanied by the production of anti-inflammatory mediators, resulting in an immunosuppressive environment. Given the high levels of cell death that can occur within a tumor microenvironment, clearance defects could trigger antitumor immune responses, as demonstrated by the inhibition of PS recognition of lymphoma cells that impaired immunosuppressive clearance and heightened tumor immunogenicity.113 Conversely, inflammation caused by delayed or inefficient clearance may favor tumor growth by providing a suitable environment, facilitating genomic instability and promoting angiogenesis.114 Hence, both possibilities have to be carefully deliberated while interpreting the role of dysfunctional clearance mechanisms in tumors.
Conclusion
Alterations in the plasma membrane during apoptosis, both structural and biochemical, play crucial roles in discriminating dying cells from their healthy counterparts. These morphological features ensure efficient recognition and clearance of apoptotic debris by phagocytes to maintain homeostatic equilibrium. Hence, it is not surprising that defects in these alterations may have significant implications in diseases including autoimmunity and cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. | |
Wickman G, Julian L, Olson MF. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012;19(5):735–742. | |
Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44(6):817–829. | |
Cerretti DP, Kozlosky CJ, Mosley B, et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science. 1992;256(5053):97–100. | |
Nicholson DW, Ali A, Thornberry NA, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376(6535):37–43. | |
Alnemri ES, Livingston DJ, Nicholson DW, et al. Human ICE/CED-3 protease nomenclature. Cell. 1996;87(2):171. | |
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. | |
Wang J, Lenardo MJ. Roles of caspases in apoptosis, development, and cytokine maturation revealed by homozygous gene deficiencies. J Cell Sci. 2000;113(Pt 5):753–757. | |
Milligan CE, Prevette D, Yaginuma H, et al. Peptide inhibitors of the ICE protease family arrest programmed cell death of motoneurons in vivo and in vitro. Neuron. 1995;15(2):385–393. | |
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–676. | |
Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4(5):410–415. | |
Nijhawan D, Honarpour N, Wang X. Apoptosis in neural development and disease. Annu Rev Neurosci. 2000;23:73–87. | |
Coucouvanis E, Martin GR. Signals for death and survival: a two- step mechanism for cavitation in the vertebrate embryo. Cell. 1995; 83(2):279–287. | |
Coucouvanis EC, Martin GR, Nadeau JH. Genetic approaches for studying programmed cell death during development of the laboratory mouse. Methods Cell Biol. 1995;46:387–440. | |
Monier B, Gettings M, Gay G, et al. Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature. 2015;518(7538):245–248. | |
Lund LR, Romer J, Thomasset N, et al. Two distinct phases of apoptosis in mammary gland involution: proteinase-independent and -dependent pathways. Development. 1996;122(1):181–193. | |
Tilly JL, Kowalski KI, Johnson AL, Hsueh AJ. Involvement of apoptosis in ovarian follicular atresia and postovulatory regression. Endocrinology. 1991;129(5):2799–2801. | |
Franz S, Gaipl US, Munoz LE, et al. Apoptosis and autoimmunity: when apoptotic cells break their silence. Curr Rheumatol Rep. 2006;8(4):245–247. | |
Platt N, Suzuki H, Kodama T, Gordon S. Apoptotic thymocyte clearance in scavenger receptor class A-deficient mice is apparently normal. J Immunol. 2000;164(9):4861–4867. | |
Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–630. | |
Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev. Biophys. 2010;39:407–427. | |
Contreras FX, Sanchez-Magraner L, Alonso A, Goni FM. Transbilayer (flip–flop) lipid motion and lipid scrambling in membranes. FEBS Lett. 2010;584(9):1779–1786. | |
Bevers EM, Williamson PL. Phospholipid scramblase: an update. FEBS Lett. 2010;584(13):2724–2730. | |
Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216. | |
Rimon G, Bazenet CE, Philpott KL, Rubin LL. Increased surface phosphatidylserine is an early marker of neuronal apoptosis. J Neurosci Res. 1997;48(6):563–570. | |
Zwaal RF, Comfurius P, Bevers EM. Lipid–protein interactions in blood coagulation. Biochim Biophys Acta. 1998;1376(3):433–453. | |
Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42(5):423–438. | |
Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–838. | |
Yang H, Kim A, David T, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151(1):111–122. | |
Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403–406. | |
Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164–1168. | |
Fadok VA, Savill JS, Haslett C, et al. Different populations of macrophages use either the vitronectin receptor or the phosphatidylserine receptor to recognize and remove apoptotic cells. J Immunol. 1992; 149(12):4029–4035. | |
Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2(12):899–905. | |
Gumienny TL, Hengartner MO. How the worm removes corpses: the nematode C. elegans as a model system to study engulfment. Cell Death Differ. 2001;8(6):564–568. | |
Park SY, Jung MY, Kim HJ, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192–201. | |
Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. | |
Park D, Tosello-Trampont AC, Elliott MR, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–434. | |
Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. | |
Nakano T, Ishimoto Y, Kishino J, et al. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J Biol Chem. 1997;272(47):29411–29414. | |
Borisenko GG, Matsura T, Liu SX, et al. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells – existence of a threshold. Arch Biochem Biophys. 2003;413(1):41–52. | |
Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A. 2011;108(48):19246–19251. | |
Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 2004;11(8):943–945. | |
Paidassi H, Tacnet-Delorme P, Verneret M, et al. Investigations on the C1q-calreticulin–phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol. 2011;408(2):277–290. | |
Ogden CA, deCathelineau A, Hoffmann PR, et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194(6):781–795. | |
Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. 1997;158(10):4525–4528. | |
Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol. 2001;166(5):3231–3239. | |
Rosen A, Casciola-Rosen L. Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell Death Differ. 1999;6(1):6–12. | |
Stollar BD, Stephenson F. Apoptosis and nucleosomes. Lupus. 2002;11(12):787–789. | |
Casciola-Rosen La, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–1330. | |
Lazebnik YA, Cole S, Cooke CA, Nelson WG, Earnshaw WC. Nuclear events of apoptosis in vitro in cell-free mitotic extracts: a model system for analysis of the active phase of apoptosis. J Cell Biol. 1993;123(1):7–22. | |
Brancolini C, Lazarevic D, Rodriguez J, Schneider C. Dismantling cell–cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. J Cell Biol. 1997;139(3):759–771. | |
Kook S, Kim DH, Shim SR, Kim W, Chun JS, Song WK. Caspase-dependent cleavage of tensin induces disruption of actin cytoskeleton during apoptosis. Biochem Biophys Res Commun. 2003;303(1):37–45. | |
Mills JC, Stone NL, Erhardt J, Pittman RN. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 1998;140(3):627–636. | |
Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3(4):339–345. | |
Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3(4):346–352. | |
Lane JD, Allan VJ, Woodman PG. Active relocation of chromatin and endoplasmic reticulum into blebs in late apoptotic cells. J Cell Sci. 2005;118(Pt 17):4059–4071. | |
Neamati N, Fernandez A, Wright S, Kiefer J, McConkey DJ. Degradation of lamin B1 precedes oligonucleosomal DNA fragmentation in apoptotic thymocytes and isolated thymocyte nuclei. J Immunol. 1995;154(8):3788–3795. | |
Rao L, Perez D, White E. Lamin proteolysis facilitates nuclear events during apoptosis. J Cell Biol. 1996;135(6 Pt 1):1441–1455. | |
Broers JL, Bronnenberg NM, Kuijpers HJ, Schutte B, Hutchison CJ, Ramaekers FC. Partial cleavage of A-type lamins concurs with their total disintegration from the nuclear lamina during apoptosis. Eur J Cell Biol. 2002;81(12):677–691. | |
Croft DR, Coleman ML, Li S, et al. Actin-myosin-based contraction is responsible for apoptotic nuclear disintegration. J Cell Biol. 2005;168(2):245–255. | |
Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5(8):711–719. | |
Hickson GR, Echard A, O’Farrell PH. Rho-kinase controls cell shape changes during cytokinesis. Curr Biol. 2006;16(4):359–370. | |
Cunningham CC, Gorlin JB, Kwiatkowski DJ, et al. Actin-binding protein requirement for cortical stability and efficient locomotion. Science. 1992;255(5042):325–327. | |
Cunningham CC. Actin polymerization and intracellular solvent flow in cell surface blebbing. J Cell Biol. 1995;129(6):1589–1599. | |
Yoshida K, Soldati T. Dissection of amoeboid movement into two mechanically distinct modes. J Cell Sci. 2006;119(Pt 18):3833–3844. | |
Tinevez JY, Schulze U, Salbreux G, Roensch J, Joanny JF, Paluch E. Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A. 2009;106(44):18581–18586. | |
Lorentzen A, Bamber J, Sadok A, Elson-Schwab I, Marshall CJ. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J Cell Sci. 2011;124(Pt 8):1256–1267. | |
Bovellan M, Fritzsche M, Stevens C, Charras G. Death-associated protein kinase (DAPK) and signal transduction: blebbing in programmed cell death. FEBS J. 2010;277(1):58–65. | |
Charras GT, Coughlin M, Mitchison TJ, Mahadevan L. Life and times of a cellular bleb. Biophys J. 2008;94(5):1836–1853. | |
Hamill OP, Martinac B. Molecular basis of mechanotransduction in living cells. Physiol Rev. 2001;81(2):685–740. | |
Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175(3):477–490. | |
Barry DJ, Durkin CH, Abella JV, Way M. Open source software for quantification of cell migration, protrusions, and fluorescence intensities. J Cell Biol. 2015;209(1):163–180. | |
Charras GT. A short history of blebbing. J Microsc. 2008;231(3):466–478. | |
Cotter TG, Lennon SV, Glynn JM, Green DR. Microfilament-disrupting agents prevent the formation of apoptotic bodies in tumor cells undergoing apoptosis. Cancer Res. 1992;52(4):997–1005. | |
Fishkind DJ, Cao LG, Wang YL. Microinjection of the catalytic fragment of myosin light chain kinase into dividing cells: effects on mitosis and cytokinesis. J Cell Biol. 1991;114(5):967–975. | |
Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372(6503):231–236. | |
Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358. | |
Orlando KA, Stone NL, Pittman RN. Rho kinase regulates fragmentation and phagocytosis of apoptotic cells. Exp Cell Res. 2006;312(1):5–15. | |
Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases. 2014;5(2):e29846. | |
Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273(5272):245–248. | |
Amano M, Ito M, Kimura K, et al. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem. 1996; 271(34):20246–20249. | |
Noda M, Yasuda-Fukazawa C, Moriishi K, et al. Involvement of rho in GTP gamma S-induced enhancement of phosphorylation of 20 kDa myosin light chain in vascular smooth muscle cells: inhibition of phosphatase activity. FEBS Lett. 1995;367(3):246–250. | |
Chang J, Xie M, Shah VR, et al. Activation of Rho-associated coiled-coil protein kinase 1 (ROCK-1) by caspase-3 cleavage plays an essential role in cardiac myocyte apoptosis. Proc Natl Acad Sci U S A. 2006;103(39):14495–14500. | |
Parent N, Sané A-T, Droin N, Bertrand R. Procaspase-2S inhibits procaspase-3 processing and activation, preventing ROCK-1-mediated apoptotic blebbing and body formation in human B lymphoma Namalwa cells. Apoptosis. 2005;10(2):313–322. | |
Zihni C, Mitsopoulos C, Tavares IA, Baum B, Ridley AJ, Morris JD. Prostate-derived sterile 20-like kinase 1-alpha induces apoptosis. JNK- and caspase-dependent nuclear localization is a requirement for membrane blebbing. J Biol Chem. 2007;282(9):6484–6493. | |
Wickman GR, Julian L, Mardilovich K, et al. Blebs produced by actin–myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013;20(10):1293–1305. | |
Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129(6):1673–1682. | |
Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189(7):1059–1070. | |
Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14(3):166–180. | |
Botto M, Dell’ Agnola C, Bygrave A, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19(1):56–59. | |
Cohen PL, Caricchio R, Abraham V, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–140. | |
Hanayama R, Tanaka M, Miyasaka K, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (New York, NY). 2004;304(5674):1147–1150. | |
Kalden JR. Defective phagocytosis of apoptotic cells: possible explanation for the induction of autoantibodies in SLE. Lupus. 1997;6(3):326–327. | |
Gaipl US, Munoz LE, Grossmayer G, et al. Clearance deficiency and systemic lupus erythematosus (SLE). J Autoimmun. 2007;28(2–3):114–121. | |
Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8(4):279–289. | |
Cohen PL, Caricchio R, Abraham V, et al. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J Exp Med. 2002;196(1):135–140. | |
Scott RS, McMahon EJ, Pop SM, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–211. | |
Rodriguez-Manzanet R, Sanjuan MA, Wu HY, et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci U S A. 2010;107(19):8706–8711. | |
Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41(7):1241–1250. | |
Ren Y, Tang J, Mok MY, Chan AW, Wu A, Lau CS. Increased apoptotic neutrophils and macrophages and impaired macrophage phagocytic clearance of apoptotic neutrophils in systemic lupus erythematosus. Arthritis Rheum. 2003;48(10):2888–2897. | |
Gaipl US, Franz S, Voll RE, Sheriff A, Kalden JR, Herrmann M. Defects in the disposal of dying cells lead to autoimmunity. Curr Rheumatol Rep. 2004;6(6):401–407. | |
Stuart LM, Takahashi K, Shi L, Savill J, Ezekowitz RA. Mannose-binding lectin-deficient mice display defective apoptotic cell clearance but no autoimmune phenotype. J Immunol. 2005;174(6):3220–3226. | |
Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. | |
Chao MP, Jaiswal S, Weissman-Tsukamoto R, et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med. 2010;2(63):63ra94. | |
Garcia Pedrero JM, Fernandez MP, Morgan RO, et al. Annexin A1 down-regulation in head and neck cancer is associated with epithelial differentiation status. Am J Pathol. 2004;164(1):73–79. | |
Paweletz CP, Ornstein DK, Roth MJ, et al. Loss of annexin 1 correlates with early onset of tumorigenesis in esophageal and prostate carcinoma. Cancer Res. 2000;60(22):6293–6297. | |
Moreaux J, Veyrune JL, Reme T, De Vos J, Klein B. CD200: a putative therapeutic target in cancer. Biochem Biophys Res Commun. 2008; 366(1):117–122. | |
Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51(11):3062–3066. | |
Woehlecke H, Pohl A, Alder-Baerens N, Lage H, Herrmann A. Enhanced exposure of phosphatidylserine in human gastric carcinoma cells overexpressing the half-size ABC transporter BCRP (ABCG2). Biochem J. 2003;376(Pt 2):489–495. | |
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. | |
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420(6917):860–867. | |
Philip M, Rowley DA, Schreiber H. Inflammation as a tumor promoter in cancer induction. Semin Cancer Biol. 2004;14(6):433–439. | |
Bondanza A, Zimmermann VS, Rovere-Querini P, et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med. 2004;200(9):1157–1165. | |
Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res. 2006;4(4):221–233. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.