")
Back to Journals » Biologics: Targets and Therapy » Volume 8
Apoptosis and the FLIP and NF-kappa B proteins as pharmacodynamic criteria for biosimilar TNF-alpha antagonists
Authors Urbano PCM, Soccol V, Azevedo V
Received 8 November 2013
Accepted for publication 21 November 2013
Published 31 July 2014 Volume 2014:8 Pages 211—220
DOI https://doi.org/10.2147/BTT.S57253
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Paulo César Martins Urbano,1 Vanete Thomaz Soccol,1 Valderilio Feijó Azevedo2
1Biotechnology and Bioprocess Engineering Program, Federal University of Parana, Curitiba, Parana, Brazil; 2Hospital de Clínicas, Federal University of Parana, Curitiba, Parana, Brazil
Abstract: Various criteria are necessary to assess the efficacy and safety of biological medications in order to grant companies the right to register these medications with the appropriate bodies that regulate their sale. The imminent expiration of the patents on reference biological products which block the cytokine TNF-α (tumor necrosis factor-α) raises the possibility of bringing so-called biosimilars to the market (similar to the biologicals of reference products). This occurrence is inevitable, but criteria to adequately evaluate these medications are now needed. Even among controversy, there is a demand from publications correlating the pro-apoptotic mechanism of the original TNF-α antagonists (etanercept, infliximab, adalimumab, golimumab, and certolizumab pegol) in the treatment of rheumatoid arthritis and other diseases. In this article, the authors discuss the possibility of utilizing the pro-apoptotic effect correlated with the regulation of the anti-apoptotic proteins FLIP and NF-κB as new criteria for analyzing the pharmacodynamics of possible biosimilar TNF-α antagonists which should be submitted to regulatory agencies for evaluation.
Keywords: anti-TNF drugs, rheumatoid arthritis, apoptosis, NF-κB, FLIP
Introduction
In the mid-80s and the early 90s Tumor necrosis factor-alpha (TNF-α), a soluble or transmembranal cytokine, became a promising candidate for the treatment of rheumatoid arthritis (RA).1 A direct relation was determined between this cytokine and pro-inflammatory signaling, which results in the migration of immune system cells to the synovial region, producing chronic synovial inflammation, bone erosion, and cartilage damage.2
Preclinical and clinical studies show that blocking the cytokine TNF-α reduced not only the inflammation of RA, but also the progression of intestinal inflammation characteristic of Crohn’s Disease (CD).3–5 As a result, the industry proposed development of medications which function as TNF-α antagonists. Now these biologicals, which belong to the class of disease-modifying anti-rheumatic drugs (DMARDs), are part of the therapeutic arsenal against RA and spondyloarthritis. Currently, the main TNF-α antagonists available are etanercept (ENBREL®; Amgen and Wyeth, Thousand Oask, CA, USA), infliximab (REMICADE®; Janssen Biotech, Mitsubishi Tanabe Pharma and Merck & Co, Inc., Whitehouse Station, NJ, USA), adalimumab (HUMIRA®; Abbott Laboratories, Abbott Park, IL, USA), certolizumab pegol (CIMZIA®; Union chimique belge, Brussels, Belgium) and golimumab (SIMPONI®; Centocor Ortho Biotech Inc., Horsham, PA, USA).6
Due to the expiration of the patents on TNF-α antagonist medications, an opportunity has arisen for various manufacturers to produce so-called biosimilar versions of these molecules. These are, in theory, similar to the standard biopharmaceuticals, but not identical. In order to protect consumers from possible damage caused by these copies, strong criteria are required for analyzing these molecules and conducting studies that demonstrate their effectiveness and safety.
Assessing the bioequivalence and biocomparability of a protein produced by two different manufacturers can be a challenge for regulating agencies. The criteria for analyzing each case are crucial to the perceived efficacy and safety of the medication.7
Currently, various clinical and non-clinical assessments are used to evaluate the efficacy of these TNF-α blockers. While the mechanism by which TNF-α antagonists act is not yet completely clear, there are studies correlating these medications with the occurrence of cellular apoptosis. The action of these antagonists with receptors of cell death prohibits the expression of some anti-apoptotic proteins in RA.
Most notable among these inhibitory proteins is FLIP (FLICE-like inhibitory protein, where FLICE is a FADD-like interleukin-1 beta-converting enzyme, FADD is a Fas-associated death domain protein and Fas is a death receptor [also known as CD95 and APO-1]). FLIP is a protein which inhibits caspase-8, impeding the uptake of this caspase for the death-inducing signaling complex (DISC) and thereby inhibiting apoptosis. Another important inhibitory protein, NF-κB (nuclear factor kappa-light-chain- enhancer of activated B-cells), promotes the expression of anti-apoptotic genes, resulting in the production of the proteins TRAF1, TRAF2, cIAP-1, cIAP2 and FLIPL.8–10
A potential correlation between the apoptotic mechanisms of the TNF-α antagonists which are already on the market and their respective biosimilars would allow the criteria for evaluating the copies to be expanded. In the meantime, as will be discussed in this article, apoptosis may have distinct repercussions on each TNF-α antagonist under various experimental conditions.
The objective of this article is to discuss the importance of the apoptosis mechanism and of the proteins FLIP and NF-κB as criteria for analyzing the pharmacodynamics of possible biosimilar TNF-α antagonists.
Biosimilars: perspectives and methods of registration
It is well-known that biological medications are tested more often (eg, preclinical studies and clinical trials) than chemical medications (produced by chemical synthesis). These molecules are more complex because they are produced by biotechnological processes based on living organisms. The majority are proteins with high molecular weights, unstable, and are more likely to cause adverse immunological reactions.11 The complexity is such that the same protein produced by one particular company may have structural variations from one batch to the next.
After a patent expires, specific information such as the production line, and principally, the master cell line are the exclusive property of the company that holds the patent. Consequently, it is impossible to consider biological medications as “generics”, but instead “biosimilars” which are similar in structure, functionality, efficacy, and safety to the patented medications.
The need for and importance of these two classes of medication are indisputable. The original or reference biologicals are the trailblazers of innovation in the pharmaceutical industry which should be constant. Yet, at the same time, biosimilars are a path to high-quality treatment at a low cost in order to increase the access to biologics, especially in developing countries.12
A reference biological, as a rule, undergoes strict assessment. Its efficacy and safety are demonstrated in a full dossier that contains preclinical tests such as molecular quality, pharmacokinetics (absorption, distribution, biotransformation and excretion), pharmacodynamics (mechanism of drug action), and acute and chronic toxicity studies in animals in the first stage. In the next step, phase I, II, III, and IV clinical trials are conducted; immunogenicity and clinical safety are assessed, and a pharmacovigilance plan is developed.13,14
In terms of biocomparability, evaluation criteria should establish the correlation, within an acceptable margin, between quality, safety, and efficacy in order to demonstrate the similarity between a reference molecule and a biosimilar.
The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have determined the following basic regulatory principles for approval of biosimilar molecules: a dossier describing the manufacture and quality of the molecule, preclinical studies, clinical studies, and immunogenicity and pharmacovigilance studies.15,16
The planning of clinical studies to detect differences between the reference molecule and the biosimilar is a crucial factor in measuring comparability, especially studies of equivalency and non-inferiority.17
Among the variables explored by comparability studies, the pharmacodynamics of the reference molecule are important in evaluating similarity; as a result, it is necessary to understand the mechanism of action.
In the context of TNF-α antagonists, the basic pharmacodynamics are characterized by the neutralization of soluble or transmembranal TNF, which results in the anti-inflammatory action that demonstrates the benefits of this treatment. The interaction between these biologicals and the complement system is also assessed, by recognition of the Fc portion (fragment crystallizable, in the cases of the monoclonal antibodies infliximab, adalimumab and golimumab).
The increase in apoptosis in RA and CD correlated with inhibition of FLIP and NF-κB proteins in RA can be considered as criteria for defining comparability studies between the reference molecule and its biosimilar. This proposal is based on references in the literature which affirm that apoptotic action can be a specific effect of some TNF-α antagonists, and its possible means of action could be through neutralization of FLIP and NF-κB proteins by a reverse signal.3,15–21
TNF-α antagonists and apoptosis
To better understand the process which results in apoptosis, Micheau and Tschopp22 described the two main pathways for the occurrence of cellular death or survival (Figure 1).
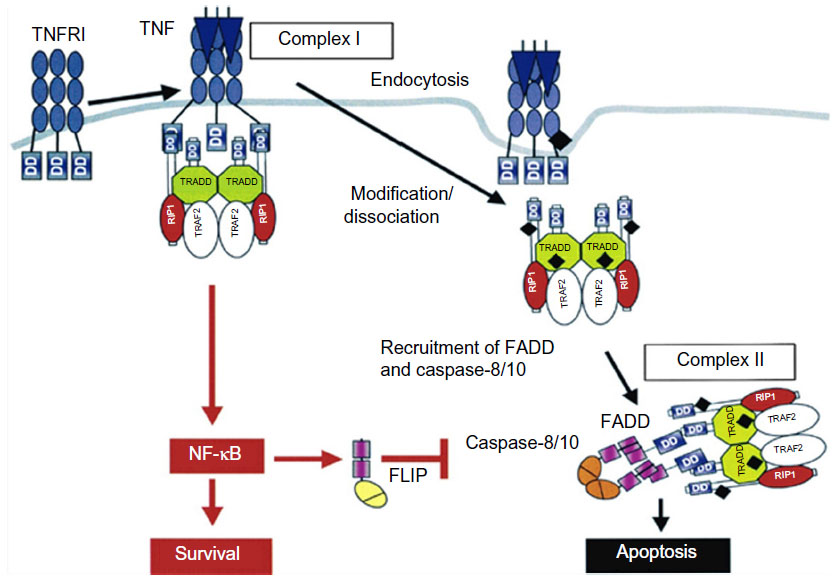 | Figure 1 Signaling pathways of the cytokine TNF-α. |
During the inflammatory response, the cytokine TNF-α can act by means of binding to transmembranal TNFR1 and 2 receptors in some cells in the immune system, transmitting survival signals (apoptosis inhibitors), the route of inflammation, and routes promoting mitosis. As a result, it is well-known that in RA, low levels of cellular apoptosis occur in the joints and synovial fluid.23 Studies reveal that the interaction of soluble TNF-α is more accentuated with TNFR2 which, more than TNFR1, possesses an ambiguous characteristic24 (Figure 1), activating the apoptosis pathway or activating the cellular survival pathway depending on the external signal, on the adaptation proteins present in the cytosol, and on other factors which have not yet been determined.25
During the past decade, various studies related to TNF-α antagonists demonstrated that the use of etanercept, infliximab, adalimumab and certolizumab pegol not only reduces serum levels of TNF-α, the immune response promoter, but also promotes the death of some inflammatory cells. This death occurs by reactivation of the apoptotic pathway, providing greater efficacy in treatment. However, each TNF-α antagonist responds differently with relation to apoptosis. Table 1 shows the specifics of each TNF-α inhibitor, highlighting the potential of each to promote cellular apoptosis.
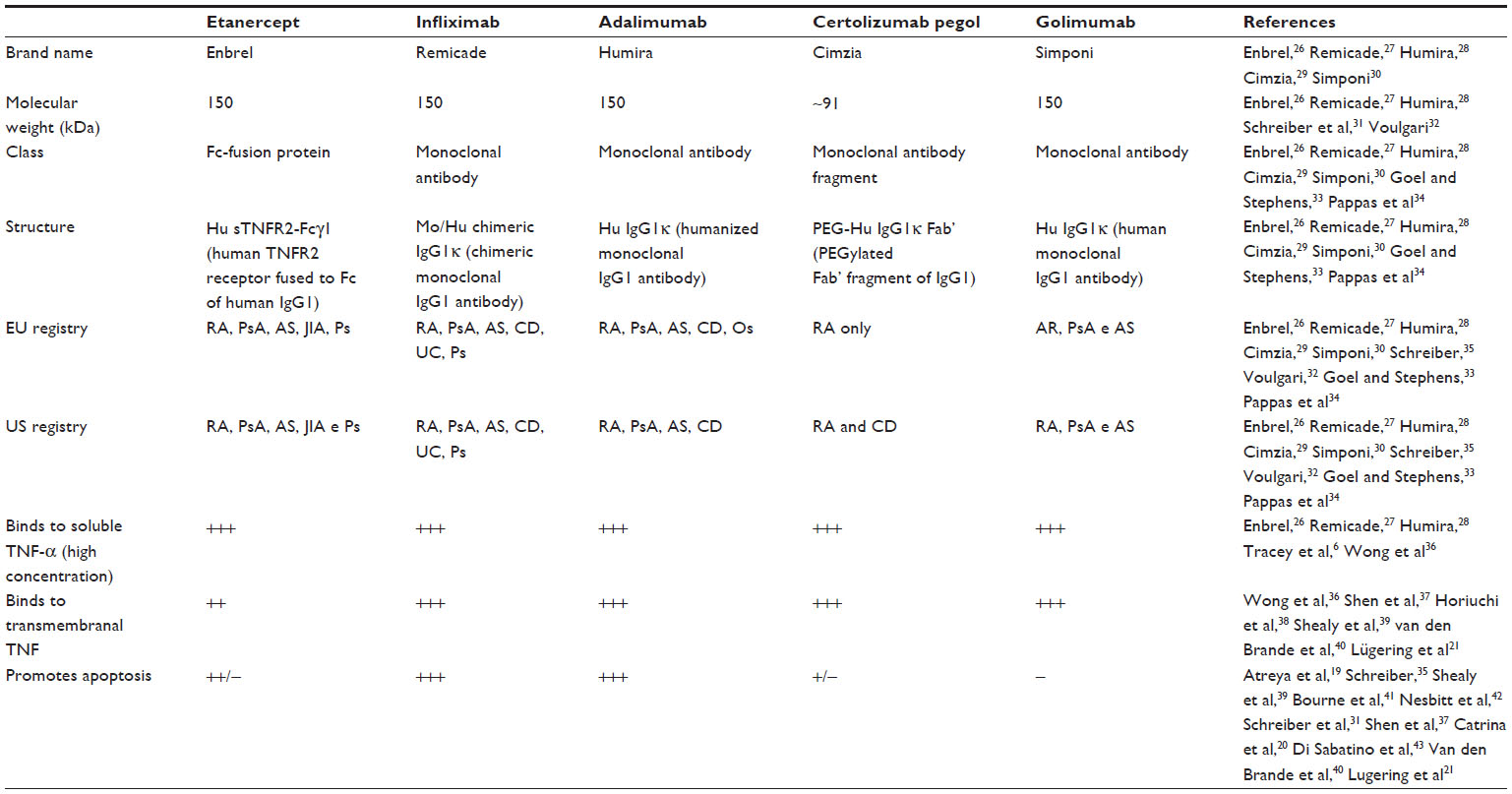 | Table 1 Profile of TNF-α antagonists |
Lügering et al21 in turn have identified a possible relation between the action of infliximab on cellular apoptosis in patients suffering from CD. They observed that 4 hours after administration of infliximab, monocyte apoptosis occurred, as determined by assessing the activation of caspases 8, 9, and 3, which act independently of signaling from CD95/95L (CD95 and ligand) receptors.
Di Sabatino et al43 conducted experiments in which they administered infliximab to patients with CD. The patients received the medication over 10 weeks at a concentration of 5 mg/kg. After treatment, it was verified that infliximab promoted apoptosis by increasing the susceptibility of lamina propria cells to peripheral blood T-cells. In in vivo and in vitro studies, the results also indicated that the mechanism of apoptosis is initiated by dependent caspase and not by the interaction receptor Fas-Fas in CD.
Ohshima et al44 assessed the action of TNF-α antagonists based on studies of treatment of synovial hyperplasia (an event characteristic of RA). The authors demonstrated that treatment can promote the reactivation of CD95 receptors (death receptors), which are crucial in the apoptotic process as they facilitate cellular apoptosis.
The authors demonstrated that the chimeric monoclonal antibody (infliximab) activated another death pathway through other receptors such as TNFR1. Meusch et al45 studied the blocking of TNF-α expression and of cellular apoptosis by means of TNF-α antagonists in in vitro studies using samples of peripheral blood monocytes taken from patients with RA. They demonstrated a significant increase in cellular apoptosis, reduction of TNF-α, and a possible drug intervention by means of interaction with the TNFR receptors.
Clinical tests of infliximab and etanercept in patients with CD demonstrated that these molecules can attach to the transmembranal TNF (tmTNF) of some inflammatory cells, mainly in monocytes found in tissue and synovial fluid. After attaching, they transmit an intracellular signal; apoptosis is one of the possible results of this interaction.46,47
Pattacini et al48 also demonstrated the correlation between the effectiveness of TNF-α antagonists and apoptosis and concluded that etanercept has a stronger pro-apoptotic effect. In vitro experiments were conducted, in which TNF-α antagonists were administered into cells cultured from fibroblasts (synovioctyes) and peripheral blood monocytes collected from patients with RA. The results demonstrated activation of cellular apoptosis in these cell types, which are crucial in the progression of RA.
Catrina et al20 evaluated patients with RA with regards to the relation between the TNF-α antagonists etanercept and infliximab and apoptosis. The authors concluded that these medications promote an increase in apoptosis in CD14 cells such as macrophages, dendritic cells and synovial fluid mononuclear cells (SFMCs). Furthermore, they stated that the possible cause of apoptosis could be due to the regulation of the proteins FLIP and NF-κB.8 However, it was stressed that etanercept does not induce apoptosis in lymphocytes, only in macrophages. In other studies, high doses of infliximab promoted apoptosis in both lymphocytes and macrophages, but doses of 3 mg/kg resulted in the decrease of macrophages only.49
Shen et al37 in turn concluded that adalimumab and infliximab affect the production of monocyte cytokines and induce apoptosis in activated monocytes. It was also demonstrated that adalimumab can activate the caspase 3 pathway, resulting in a higher level of cellular apoptosis in treated chimeric rats. The results finally show that cellular apoptosis can be blocked by the pan-caspase inhibitor, assuring that adalimumab’s apoptosis mechanism is independent of caspase.
Herman et al50 verified the pro-apoptotic action of TNF-α antagonists in a clinical study. Fifteen patients with RA were administered TNF-α antagonists together with methotrexate, 20 patients received only methotrexate, and eleven patients did not receive any treatment. A significant increase in cellular apoptosis in T-helper 1 and T-helper 2 (Th1/Th2) lymphocytes was noted; this amplified inflammation, resulting in significant reduction of joint inflammation and restoration of the balance of Th1/Th2 lymphocytes.
Experiments conducted by Aravena et al51 demonstrated that treatment with the TNF-α antagonist adalimumab over a period of 16 weeks in patients with RA promoted a population balance of the CD4 Th 1, and Th 17 lymphocytes population. There was also a reduction in secretion of interferon gamma (IFN-γ) by CD8, indicating that it is not apoptosis itself that promotes reduction of the inflammatory process, but instead the apoptosis of specific cells which play an important role in the process of chronic inflammation.
The important connection between the Fc fragment of the monoclonal antibodies infliximab, adalimumab and golimumab and the cellular depletion pathways has also been determined. It has been proposed that there is a certain dependence on this fragment in the pro-apoptotic process of these molecules. The exception is golimumab, which despite possessing the Fc fragment has not yet shown a relationship with the apoptotic process.52 These data have been confirmed by Bourne et al,41 who have proposed the hypothesis that apoptosis is not a clinically effective action in CD, considering that etanercept promotes apoptosis but is not effective in treating CD. While certolizumab pegol does not have Fc, it does not promote apoptosis, but is effective in the treatment of CD. Yet etanercept, even with Fc, does not promote the activation of the complement system,53 and certolizumab pegol promoted apoptosis in a study by Atreya et al.19 As a result, dependency on the Fc fragment in this mechanism is questionable.
Van den Brande et al,40 in turn, stressed that etanercept did not induce apoptosis in in vitro experiments with T lymphocytes in patients with CD. In another study, Fries et al54 administered etanercept and infliximab to rats with colitis and determined that both of these TNF-α antagonists reduced the population of enterocytes by apoptosis, probably as a result of the reduction of circulating TNF-α.
In a recent study, Eder et al55 demonstrated in an in vitro study with mononuclears and enterocytes of patients with CD, that infliximab and adalimumab exert a pro-apoptotic influence, producing an increase in the pro-apoptotic proteins Bax/Bcl-2 which assist in the process of apoptosis. The authors also stress that the efficiency of the TNF-α antagonist depends, at least in part, on this process. This study corroborates data from Van den Brande et al.40 There are drawbacks to the use of apoptosis such as clinical effectiveness of TNF-α antagonists in CD and in RA, but certainly the pharmacodynamics of TNF-α antagonists carry out apoptosis.
It should also be taken into account that the in vitro studies with RA and CD obtained more favorable results in comparing the action of TNF-α antagonists to the apoptotic mechanism. Nevertheless, Makrygiannakis and Catrina18 do not consider apoptosis as a criteria for the clinical effectiveness of TNF-α antagonists in RA, as all antagonists have similar efficacy but different apoptotic effects. This shows that in theory, apoptosis is not fundamental to the treatment of RA, but is a criteria for extrapolation of treatment and certainly for the pharmacodynamics of TNF antagonists. There are still many doubts and questions to be discussed in order to define apoptosis as a clinically effective treatment process; for this reason, more studies should be conducted in this area.
The pro-apoptotic effect, in the context of treating chronic inflammation, is not restricted to only TNF-α antagonists. Some non-biological medications classified as DMARDS promote apoptosis in specific cells by modulating actions on the expression of anti-apoptotic proteins such as FLIP, families of X-linked inhibitor of apoptosis protein (XIAP), inhibitors, execution caspases, and survivins. According to Smith et al,56 elevated levels of FLIP, XIAP, and survivins were detected in samples taken from patients with RA. However, 6 weeks after beginning treatment with DMARDS, analysis showed that the levels of these inhibitors fell steeply, causing a substantial increase in apoptosis in synovial cells. Other studies showed the pro-apoptotic action of DMARDS (non-biologicals) such as sulfasalazine,57 and hydroxichloroquine.58 Methotrexate, which in many cases is used in conjunction with TNF-α antagonists, and cyclophosphamide also promote cellular apoptosis in activated T CD4 and CD8 cells, but not in resting cells, through CD95 sensitivity.59,60
Research on the pro-apoptotic effect of TNF-α antagonists heavily focuses on RA and CD, but there are also studies with psoriatic arthritis (PsA) and psoriasis (Ps).61,62
FLIP and NF-κB proteins
Among the various possible routes, one probable pathway for the pro-apoptotic action of TNF-α antagonists in RA could be related to the inhibited expression of FLIP and NF-κB proteins, which are abundant in RA and fundamental to the process of inflammation.
Perlman et al63 have investigated the relation between FLIP in monocytes treated with lipopolysaccharide (LPS) and TNF-α. The authors showed that there was a significant increase in the expression of FLIP and a reduction in the expression of Fas-FasL (Fas and ligand), protecting the monocytes from apoptosis.
Further, they stated that after blocking FLIP through specific oligonucleotides, there was subsequently a return of sensitivity to apoptosis in the monocytes. The authors propose that the action of LPS and TNF-α in reducing apoptosis is associated with the FLIP protein.
Palao et al64 demonstrated that low regulation of the FLIP protein strongly affects the population of synovial fibroblasts through apoptosis, mediated by Fas receptors. They suggest the existence of a reverse pro-apoptotic signal after the inhibition of FLIP. Schedel et al65 also propose that the expression of FLIP in the synovial tissue in RA is related to the accumulation of inflammatory cells, indicating that FLIP may potentially be responsible for the extension of the life of synovial cells, and accordingly may contribute to the progression of articular damage. To arrive at this conclusion, the expression of mRNA for synthesizing the FLIP protein was analyzed in five samples of synovial tissue from patients with RA, two samples from patients with osteoarthritis (OA), and two samples from healthy volunteers. At the end of the study, the expression of mRNA was observed only in the tissue samples from patients with RA and OA, and was not detected in the samples from healthy volunteers; this demonstrated a relevant connection between the apoptotic action of the FLIP protein with the progression of RA.
Wu et al,66 in analyzing eleven samples from patients with juvenile idiopathic arthritis (JIA) and three samples from healthy volunteers using the RC-PCR technique (reverse transcriptase polymerase chain reaction), verified that in all patient samples the expression of mRNA FLIP was present, while in the normal samples there was no considerable expression. This demonstrated that an elevated incidence of this protein could promote a longer life for synovial cells, boosting inflammation.
Elevated levels of FLIP are directly related to the expression of NF-κB9 characterized by a protein complex which acts as a transcription factor in B-cells as RA progresses.67 A common characteristic of RA is the activation of NF-κB68 which functions to transcribe pro-inflammatory genes in immune responses, cellular growth and development processes, protecting monocytes and macrophages from apoptosis. In this way the reduction of NF-κB can consequently promote apoptosis in macrophages and in monocytes after treatment with TNF-α antagonists.69 Blocking NF-κB in RA synovial fluid cells lowered regulation of the FLIP-L genes,70 suggesting that reverse signaling could also promote apoptosis with dependent caspase.
There is evidence that pro-inflammatory cytokines such as TNF-α and IL-1 work by activating NF-κB dependent pathways which play an important role in synovial proliferation, leukocyte infiltration, and the synthesis of other pro-inflammatory mediators.71,72
The moment at which FLIP and NF-κB, as key factors in the pathology of RA, approach and encounter the pro-apoptotic mechanism of TNF-α antagonists is described by Makrygiannakis and Catrina.18 They stress that the modulation of NF-κB promotes the reduction in expression of FLIP, after utilization of the TNF-α antagonists, resulting in a reverse signal, which culminates in apoptosis (Figure 2). They also describe the possible relationship between the reverse signal and the modulation in expression of p53 (tumor suppressing protein) and pro-apoptotic proteins, such as Bcl-2 and cytochrome C, for the formation of the apoptosome and activation of apoptosis.
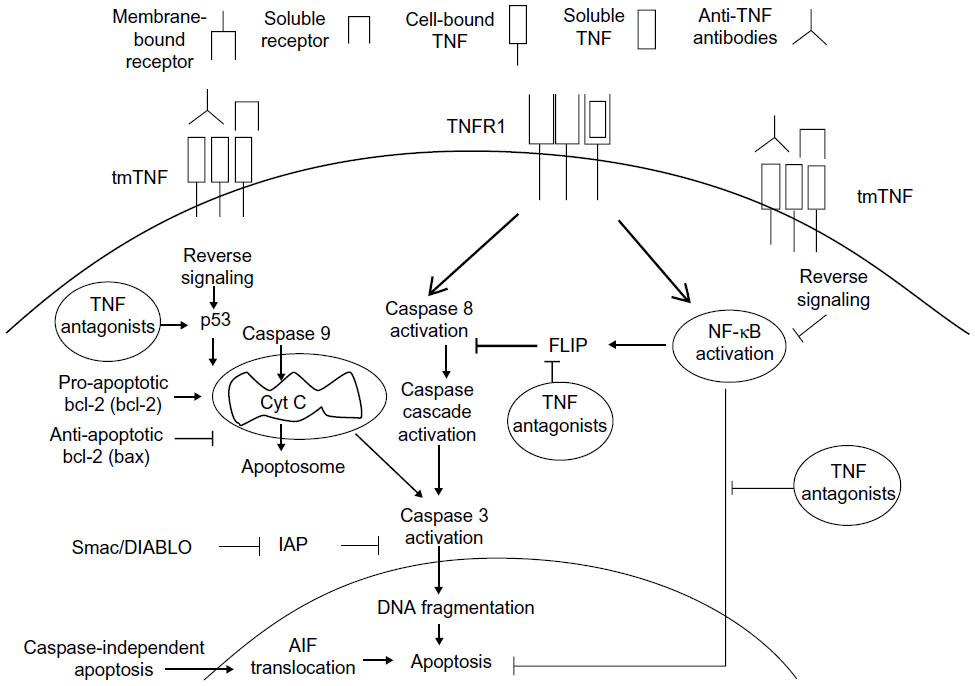 | Figure 2 Reverse signaling in the pharmacodynamics of TNF-α antagonists promotes apoptosis. |
Conclusion
In the context of studies comparing a reference molecule and a biosimilar, certainly the individual data regarding the promotion of apoptosis by etanercept, infliximab, adalimumab and certolizumab pegol (to a lesser extent), should be taken into consideration. However, there is a need to standardize experiments, keeping in mind that in the literature, the pro-apoptotic action of the antagonists varies in accordance with various factors such as the quantity of the biological administered, the length of treatment, and the type of disease. In this way, a more accurate analysis with regards to the apoptotic mechanism can be considered, as one of the main apoptosis pathways used by TNF-α antagonists is dependent on caspase 3, which in turn is strongly influenced by FLIP (expressed by NF-κB), the suppressor of caspase 8 (activator of caspase 3). The reverse signal promoted by the TNF-α antagonists could be an action mechanism that should be explored in comparability studies in the context of RA.
As the costs of biosimilar medications drop, there is an overall expectation, especially from governmental organizations in many countries, that there will be an increase in prescriptions of these medications compared to the reference biologicals. Nevertheless, it should be stressed that proof of the efficacy and safety of biosimilars is the sine qua non condition for their use and sale.
Acknowledgments
The authors thank the Department of Product Physician – Pfizer Brazil for all the support provided. This work is funded by Pfizer Brazil.
Disclosure
The authors declare that there are no conflicts of interest.
References
Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66(8):1403–1408. | |
Kaushansky K, Broudy VC, Harlan JM, Adamson JW. Tumor necrosis factor-alpha and tumor necrosis factor-beta (lymphotoxin) stimulate the production of granulocyte-macrophage colony-stimulating factor, macrophage colony-stimulating factor, and IL-1 in vivo. J Immunol. 1988;141(10):3410–3415. | |
Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M. Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet. 1989;2(8657):244–247. | |
Butler DM, Maini RN, Feldmann M, Brennan FM. Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw. 1995;6(4):225–230. | |
Van Dullemen HM, Van Deventer SJ, Hommes DW, et al. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology. 1995;109(1):129–135. | |
Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117(2):244–279. | |
Schellekens H. How similar do “biosimilars” need to be? Nat Biotechnol. 2004;22(11):1357–1359. | |
Catrina AI, Ulfgren AK, Lindblad S, Grondal L, Klareskog L. Low levels of apoptosis and high FLIP expression in early rheumatoid arthritis synovium. Ann Rheum Dis. 2002;61(10):934–936. | |
Micheau O, Lens S, Gaide O, Alevizopoulos K, Tschopp J. NF-kappaB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–5305. | |
Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281(5383):1680–1683. | |
Azevedo VF. Are we prepared to prescribe biosimilars? Rev Bras Reumatol. 2010;50(3):221–224. | |
McCamish M, Woollett G. The state of the art in the development of biosimilars. Clinical pharmacology and Therapeutics. 2012; 91(3):405–417. | |
Calvo B, Zúñiga L. Medicamentos Biotecnológicos: Requisitos Exigidos para el Desarrollo y Aprobación de Biosimilares. [Biological Medicinal Products: Requirements for the Development and Approval of Biosimilars]. Información tecnológica. 2010;21(6):125–132. Spanish. | |
Cai XY, Gouty D, Baughman S, Ramakrishnan M, Cullen C. Recommendations and requirements for the design of bioanalytical testing used in comparability studies for biosimilar drug development. Bioanalysis. 2011;3(5):535–540. | |
FDA Guidance for Industry Scientific Considerations in Demonstrating Biosimilarity to a Reference Product. | |
European Medicines Agency – Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: quality issues. | |
Pinto VF. Estudos clínicos de não-inferioridade: fundamentos e controvérsias. [Non-inferiority clinical trials: concepts and issues]. J Vasc Bras. 2010;9(3):145–151. Portuguese. | |
Makrygiannakis D, Catrina AI. Apoptosis as a mechanism of action of tumor necrosis factor antagonists in rheumatoid arthritis. J Rheumatol. 2012;39(4):679–685. | |
Atreya R, Zimmer M, Bartsch B, et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14+ macrophages. Gastroenterology. 2011;141(6):2026–2038. | |
Catrina AI, Trollmo C, af Klint E, et al. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum. 2005;52(1):61–72. | |
Lügering A, Schmidt M, Lügering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology. 2001;121(5):1145–1157. | |
Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. | |
Ottonello L. Synovial fluid from patients with rheumatoid arthritis inhibits neutrophil apoptosis: role of adenosine and proinflammatory cytokines. Rheumatology (Oxford). 2002;41(11):1249–1260. | |
Angermeier M, Eckardt-Schupp F, Moertl S. A novel function of Ubc13 in TNFR1 receptor activation. Cell Signal. 2010;22(9):1388–1396. | |
Bogliolo BFG: Patologia Geral. [General Pathology]. 4a edition. Rio de Janeiro: Guanabara Koogan; 2009:378. Portuguese. | |
Enbrel (Etanercept) [prescribing information]. Immunex Corporation; 2007. | |
Remicade (infliximab) [prescribing information]. Centocor I; 2006. | |
Humira (adalimumab) [prescribing information]. Laboratories A; 2007. | |
Cimzia (Certolizumab pegol) [prescribing information]. UCB I; 2009. | |
Simponi (Golimumab) [prescribing information]. Centocor I; 2009. | |
Schreiber S, Khaliq-Kareemi M, Lawrance IC, et al. Maintenance therapy with certolizumab pegol for Crohn’s disease. N Engl J Med. 2007;357(3):239–250. | |
Voulgari PV. Golimumab: a new anti-TNF-alpha agent for rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. Expert Rev Clin Immunol. 2010;6(5):721–733. | |
Goel N, Stephens S. Certolizumab pegol. MAbs. 2010;2(2):137–147. | |
Pappas DA, Bathon JM, Hanicq D, Yasothan U, Kirkpatrick P. Golimumab. Nature reviews Drug Discovery. 2009;8:695–696. | |
Schreiber S. Certolizumab pegol for the treatment of Crohn’s disease. Therap Adv Gastroenterol. 2011;4(6):375–389. | |
Wong M, Ziring D, Korin Y, et al. TNFalpha blockade in human diseases: mechanisms and future directions. Clin Immunol. 2008;126(2):121–136. | |
Shen C, Van Assche G, Rutgeerts P, Ceuppens JL. Caspase activation and apoptosis induction by adalimumab: demonstration in vitro and in vivo in a chimeric mouse model. Inflamm Bowel Dis. 2006;12(1):22–28. | |
Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford). 2010;49(7):1215–1228. | |
Shealy D, Cai A, Staquet K, et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor alpha. MAbs. Epub July 1, 2010;2(4). | |
Van den Brande JMH, Braat H, Van den Brink GR, et al. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology. 2003;124(7):1774–1785. | |
Bourne T, Fossati G, Nesbitt A. A PEGylated Fab’ fragment against tumor necrosis factor for the treatment of Crohn disease: exploring a new mechanism of action. BioDrugs. 2008;22(5):331–337. | |
Nesbitt A, Fossati G, Bergin M, et al. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm Bowel Dis. 2007;13(11):1323–1332. | |
Di Sabatino A, Ciccocioppo R, Cinque B, et al. Defective mucosal T cell death is sustainably reverted by infliximab in a caspase dependent pathway in Crohn’s disease. Gut. 2004;53(1):70–77. | |
Ohshima S, Mima T, Sasai M, et al. Tumour necrosis factor alpha (TNF-alpha) interferes with Fas-mediated apoptotic cell death on rheumatoid arthritis (RA) synovial cells: a possible mechanism of rheumatoid synovial hyperplasia and a clinical benefit of anti-TNF-alpha therapy for RA. Cytokine. 2000;12(3):281–288. | |
Meusch U, Rossol M, Baerwald C, Hauschildt S, Wagner U. Outside-to-inside signaling through transmembrane tumor necrosis factor reverses pathologic interleukin-1beta production and deficient apoptosis of rheumatoid arthritis monocytes. Arthritis Rheum. 2009;60(9):2612–2621. | |
Van Deventer SJ. Transmembrane TNF-alpha, induction of apoptosis, and the efficacy of TNF-targeting therapies in Crohn’s disease. Gastroenterology. 2001;121(5):1242–1246. | |
Mitoma H, Horiuchi T, Hatta N, et al. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology. 2005;128(2):376–392. | |
Pattacini L, Boiardi L, Casali B, Salvarani C. Differential effects of anti-TNF-alpha drugs on fibroblast-like synoviocyte apoptosis. Rheumatology (Oxford). 2010;49(3):480–489. | |
Taylor PC, Peters AM, Paleolog E, et al. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43(1):38–47. | |
Herman S, Zurgil N, Machlav S, et al. Distinct effects of anti-tumor necrosis factor combined therapy on TH1/TH2 balance in rheumatoid arthritis patients. Clin Vaccine Immunol. 2011;18(7):1077–1082. | |
Aravena O, Pesce B, Soto L, et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology. 2011;216(12):1256–1263. | |
Vos ACW, Wildenberg ME, Duijvestein M, Verhaar AP, Van den Brink GR, Hommes DW. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. 2011;140(1):221–230. | |
Kaymakcalan Z, Sakorafas P, Bose S, et al. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131(2):308–316. | |
Fries W, Muja C, Crisafulli C, et al. Infliximab and etanercept are equally effective in reducing enterocyte APOPTOSIS in experimental colitis. Int J Med Sci. 2008;5(4):169–180. | |
Eder P, Lykowska-Szuber L, Krela-Kazmierczak I, Stawczyk-Eder K, Zabel M, Linke K. The influence of infliximab and adalimumab on the expression of apoptosis-related proteins in lamina propria mononuclear cells and enterocytes in Crohn’s disease – an immunohistochemical study. J Crohns Colitis. 2013;7(9):706–716. | |
Smith MD, Weedon H, Papangelis V, Walker J, Roberts-Thomson PJ, Ahern MJ. Apoptosis in the rheumatoid arthritis synovial membrane: modulation by disease-modifying anti-rheumatic drug treatment. Rheumatology (Oxford). 2010;49(5):862–875. | |
Oakley F, Meso M, Iredale JP, et al. Inhibition of inhibitor of kappaB kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology. 2005;128(1):108–120. | |
Kim WU, Yoo S-A, Min S-Y, et al. Hydroxychloroquine potentiates Fas-mediated apoptosis of rheumatoid synoviocytes. Clin Exp Immunol. 2006;144(3):503–311. | |
Nielsen CH, Albertsen L, Bendtzen K, Baslund B. Methotrexate induces poly(ADP-ribose) polymerase-dependent, caspase 3-independent apoptosis in subsets of proliferating CD4+ T cells. Clin Exp Immunol. 2007;148(2):288–295. | |
Strauss G, Osen W, Debatin KM. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin Exp Immunol. 2002;128(2):255–266. | |
Malaviya R, Sun Y, Tan JK, et al. Etanercept induces apoptosis of dermal dendritic cells in psoriatic plaques of responding patients. J Am Acad Dermatol. 2006;55(4):590–597. | |
Goedkoop AY, Kraan MC, Picavet DI, et al. Deactivation of endothelium and reduction in angiogenesis in psoriatic skin and synovium by low dose infliximab therapy in combination with stable methotrexate therapy: a prospective single-centre study. Arthritis Research and Therapy. 2004;6(4):R326–R334. | |
Perlman H, Pagliari LJ, Nguyen N, Bradley K, Liu H, Pope RM. The Fas-FasL death receptor and PI3K pathways independently regulate monocyte homeostasis. Eur J Immunol. 2001;31(8):2421–2430. | |
Palao G, Santiago B, Galindo M, Payá M, Ramirez JC, Pablos JL. Down-regulation of FLIP sensitizes rheumatoid synovial fibroblasts to Fas-mediated apoptosis. Arthritis Rheum. 2004;50(9):2803–2810. | |
Schedel J, Gay RE, Kuenzler P, et al. FLICE-inhibitory protein expression in synovial fibroblasts and at sites of cartilage and bone erosion in rheumatoid arthritis. Arthritis Rheum. 2002;46(6):1512–1518. | |
Wu FX, Wu LJ, Luo XY, et al. Expression of FLICE-inhibitory protein in synovial tissue and its association with synovial inflammation in juvenile idiopathic arthritis. Chin Med Sci J. 2010;25(1):20–26. | |
Bai S, Liu H, Chen K-H, et al. NF-kappaB-regulated expression of cellular FLIP protects rheumatoid arthritis synovial fibroblasts from tumor necrosis factor alpha-mediated apoptosis. Arthritis Rheum. 2004;50(12):3844–3855. | |
Hammaker D, Sweeney S, Firestein GS. Signal transduction networks in rheumatoid arthritis. Ann Rheum Dis. 2003;62(Suppl 2):ii86–ii89. | |
Pagliari LJ, Perlman H, Liu H, Pope RM. Macrophages require constitutive NF-kappaB activation to maintain A1 expression and mitochondrial homeostasis. Mol Cell Biol. 2000;20(23):8855–8865. | |
Zhang HG, Hyde K, Page GP, et al. Novel tumor necrosis factor alpha-regulated genes in rheumatoid arthritis. Arthritis Rheum. 2004;50(2):420–431. | |
Collantes E, Valle Blázquez M, Mazorra V, Macho A, Aranda E, Muñoz E. Nuclear factor-kappa B activity in T cells from patients with rheumatic diseases: a preliminary report. Ann Rheum Dis. 1998;57(12):738–741. | |
Fujisawa K, Aono H, Hasunuma T, Yamamoto K, Mita S, Nishioka K. Activation of transcription factor NF-kappa B in human synovial cells in response to tumor necrosis factor alpha. Arthritis Rheum. 1996;39(2):197–203. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.