")
Back to Journals » Cancer Management and Research » Volume 12
Antitumor Efficacy of Oncolytic Herpes Virus Type 1 Armed with GM-CSF in Murine Uveal Melanoma Xenografts
Authors Liu S, Liu F , Zhao M, Zhang J
Received 3 August 2020
Accepted for publication 29 October 2020
Published 18 November 2020 Volume 2020:12 Pages 11803—11812
DOI https://doi.org/10.2147/CMAR.S274605
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xueqiong Zhu
Sisi Liu,1 Fusheng Liu,2 Mingwei Zhao,1 Junwen Zhang2
1Department of Ophthalmology, Peking University People’s Hospital, Eye Diseases and Optometry Institute, Beijing Key Laboratory of Diagnosis and Therapy of Retinal and Choroid Diseases, College of Optometry, Peking University Health Science Center, Beijing, People’s Republic of China; 2Brain Tumor Research Center, Beijing Neurosurgical Institute, Beijing Laboratory of Biomedical Materials, Beijing Tiantan Hospital Affiliated to Capital Medical University, Beijing 100070, People’s Republic of China
Correspondence: Junwen Zhang
Brain Tumor Research Center, Beijing Neurosurgical Institute, Beijing Laboratory of Biomedical Materials, Beijing Tiantan Hospital Affiliated to Capital Medical University, No. 115 Nansihuan West Road, Fengtai District, Beijing 100070, People’s Republic of China
Tel +86-10-59975626
Email [email protected]
Mingwei Zhao
Department of Ophthalmology, Peking University People’s Hospital, Eye Diseases and Optometry Institute, Beijing Key Laboratory of Diagnosis and Therapy of Retinal and Choroid Diseases, College of Optometry, Peking University Health Science Center, No. 11 Xizhimen South Street, Xi Cheng District, Beijing 100044, People’s Republic of China
Tel +86-10-8832666
Email [email protected]
Background: Uveal melanoma (UM) is the most common primary intraocular tumor in adults with a high incidence of metastasis. Standard care therapies for UM include enucleation and radiation, which are minimally effective in prolonging patient survival. Oncolytic virus treatment has become a new trend in cancer field. Of which, oncolytic herpes simplex virus type 1 (HSV-1) therapy is one of the most effective antitumor treatments. Here, we established an oncolytic HSV-1 encoding granulocyte-macrophage colony-stimulating factor (GM-CSF), tested its efficacy in UM therapy, and investigated the innate immune response induced by this virus.
Methods: Oncolytic HSV-1 expressing GM-CSF (HSV-GM-CSF) was constructed, then verified using qPCR and Western blot assays. Cell viability assays and transmission electron microscopy were conducted on three UM cell lines, MUM2B, 92.1, and MP41, to assess the cell-killing ability and virus infection of this virus. For in vivo experiments, BALB/c-nude mice in situ UM xenografts were established to testify the efficacy of the oncolytic virus, oncolytic HSV-1, and HSV-GM-CSF groups, respectively. IVIS images, ocular volumes, mice weights, and survivals were tracked to see the efficacy of the virus. Hematoxylin and eosin staining, immunohistochemistry, and flow cytometry analyses were conducted to demonstrate the immune activity after virus treatment.
Results: All three tested UM cell lines were sensitive to infection by HSV-GM-CSF. In vivo xenograft experiments revealed that oncolytic virus HSV-1 reduced UM tumor volume and that oncolytic virus HSV-1 armed with GM-CSF enhanced the antitumor effect compared with unmodified HSV-1. The bodyweights of untreated control group mice were significantly lower than those of mice in either virus-treated group (HSV-1 or HSV-GM-CSF). Follow-up survivals were prolonged in the virus-treated groups compared with the control group and were prolonged to a greater extent in the HSV-GM-CSF group than in the HSV-1 group. Macrophage stimulation was observed following HSV-GM-CSF treatment.
Conclusion: Our results indicate that the recombinant oncolytic virus HSV-GM-CSF is a potential therapeutic treatment for UM.
Keywords: uveal melanoma, oncolytic virus, HSV-1, GM-CSF, antitumor efficacy
Introduction
Uveal melanoma (UM) is the most common adult primary intraocular tumor. It arises from melanocytes of the choroid plexus, ciliary body, and iris of the eye.1 The incidence of UM is low, with 6–7 new cases per 1 million individuals.2 Despite the currently available treatments of enucleation and radiation, the prognosis for UM remains poor, leaving a 5-year survival rates range from 25% to 66%.3,4 The overall death rate of UM is mostly due to liver metastatic disease, which can begin several years before its diagnosis, complicating its detection and treatment. For uncontrolled metastasis, patients who are not treated in time survive only 2 to 6 months.5 Therefore, it is vital to develop new therapeutics for treating UM that can prolong patient survival.
Oncolytic virus therapy is currently under investigation for use in cancer treatment. This approach is regarded as promising due to its advantage of selectively targeting and replicating in tumor cells, subsequently causing tumor cell lysis. Recent reports have focused mainly on adenovirus, herpes virus, poxvirus, and reovirus.6,7 Oncolytic adenovirus H101 was the first oncolytic virus approved by the Chinese State Food and Drug Administration for clinical cancer treatment.8 Of the currently available oncolytic viruses, only adenovirus H101 and ICOVIR-5 have been investigated in UM cells.9–11 However, these viruses showed less efficacy towards UM cells. Hence, the highly efficient oncolytic herpes simplex virus type 1 (HSV-1) has been proposed as another candidate therapeutic for UM treatment because it has proven highly effective in a broad range of tumor cell types.12–16
The large DNA-based genome of oncolytic HSV-1 allows it to act as an ideal carrier for gene modification. HSV-1 can be genetically modified to express various molecules that enhance its efficacy as a cancer treatment, such as suicide genes (thymidine kinase [TK],17 cytosine deaminase [CD]18). One of the most promising transgenes for this purpose is granulocyte-macrophage colony-stimulating factor (GM-CSF), which was found to be effective for improving the therapy response rates in patients with advanced melanoma.16,19 GM-CSF is renowned for recruiting T-cells to a specific antigen-mediated antitumor response. Furthermore, GM-CSF can also recruit both dendritic cells (DCs) and natural killer (NK) cells, as well as increase macrophage activity.20,21 There is an increasing evidence that non-specific innate immune cells may aid in the treatment of UM, and clinical observations indicate that macrophage infiltration leads to prolonged survival in patients with UM.22 GM-CSF has not yet been implemented in UM treatment. We hypothesized that modifying HSV-1 by inserting GM-CSF would stimulate innate immune responses, thereby improving the antitumor efficacy of this oncolytic virus.
In this study, we developed an oncolytic HSV-1 virus encoding the human GM-CSF gene (HSV-GM-CSF) and verified its therapeutic efficacy towards UM cell lines. Through in vitro and in vivo experiments, we investigated the potential of applying HSV-GM-CSF to the treatment of UM and assessed the immunostimulatory efficacy of this virus in a murine UM xenograft.
Materials and Methods
Cell Lines
UM cell lines MUM2B, 92.1, and MP41 were used in the experiment. The 92.1 cells were a gift from Prof. Vavvas Demetrios and Efstathiou Nikolaos of Massachusetts General Hospital. 92.1 cell line was authenticated by STR profile and was approved by the Ethical Committee of Beijing Neurosurgical Institute. Operations about the cell line were according to the guidelines of laboratory cells. The MUM2B and MP41 cells were purchased from American Type Cell Culture (ATCC, Rockville, MD, USA). MUM2B and 92.1 cells were cultured in RPMI-1640 media with 10% fetal bovine serum, and MP41 cells were cultured with 20% fetal bovine serum. All three cell lines were transduced with pCMV/firefly-luciferase-neomycin lentivirus (Genechem, Shanghai, China).
Oncolytic Virus
HSV-1 modification was described as before.23 Briefly, HSV-GM-CSF was constructed via a deletion of the ICP34.5 and ICP47 from HSV-1 to produce an oncolytic HSV-1 vector, and then human GM-CSF transgene was inserted into the ICP34.5 deletion site. After modification, the structure of HSV-GM-CSF is the same as T-VEC (Imlygic®, Amgen).
qPCR
Total RNA was isolated from frozen cell pellets using TRIzol reagent (Thermo Scientific, Carlsbad, CA, USA). RNA quality and concentration were evaluated with a Nanodrop ND-1000 Spectrophotometer. A total of 1 µg of RNA was reverse-transcribed using a Reverse Transcription System Kit (Promega A3500, Fitchburg, WI, USA) following the manufacturer’s instructions. The following primers were generated: GM-CSF, forward 5ʹ-GCGTCTCCTGAACCTGAGTA-3ʹ, reverse 5ʹ-TGGGTTGCACAGGAAGTTTC-3ʹ; GAPDH, forward 5ʹ-CTGCACCACCAACTGCTTAGC-3ʹ, reverse 5ʹ-CTTCACCACCTTCTTGATGTC-3ʹ. qPCR was performed on triplicate samples using a QuantStudio 6 Flex system (Applied Biosystems, Waltham, MA, USA) with SYBR-Green PCR Master Mix (Applied Biosystems). GAPDH was used as an internal standard. Results are expressed as 2−ΔΔCt.
Western Blot
Cells were incubated with or without virus and lysed using RIPA lysis buffer (50 mM Tris-HCl, pH 7.4, 100 mM 2-mercaptoethanol, 2% w/v SDS, 10% glycerol). Protein concentrations were determined, and then the various proteins were separated by 12% SDS-PAGE. After separation, the proteins were transferred to nitrocellulose membranes and incubated with primary antibodies (Anti-GM-CSF diluted 1:1000 [Proteintech, 17762-1-AP, Rosemont, IL, USA] and Anti-β-actin diluted 1:5000 [Sigma, A5441, Shanghai, China]) followed by secondary antibodies (Thermo Scientific, 31462). Signals were detected with an ECL detection system.
Cell Viability Assay
Cells were seeded in a 96-well plate at a density of 5000 cells per well. Various concentrations of virus suspended in culture medium were used to infect the cells. At 3 days post-infection, the cell viability was determined using a cell counting kit-8 (CCK-8, Dojindo Molecular Technologies, Shanghai, China) in accordance with the manufacturer’s instructions. The absorbance at an optical density of 450 nm (OD) of the samples was measured with a Spectra Microplate Reader. Cell viability was calculated as follows: OD value of the sample/OD value of the control.
Transmission Electron Microscopy (TEM) Imaging
MUM2B cells were treated with HSV-GM-CSF at a MOI of 0.1 in a 10-cm2 dish. At 3 days post-infection, the cells were washed with PBS, then fixed in osmium tetraoxide (OsO4). After being subjected to a serial dehydration in gradient ethanol, the samples were stained with alkaline lead citrate and uranyl acetate. TEM images were taken at 80 kV.
In vivo Tumor Cell Implantation
Animal studies were approved by the Experimental Animal Ethical Committee of the Beijing Neurosurgical Institute (No. 201902015). All experiment operations were in accordance with the Experimental Animal Ethical Committee of the Beijing Neurosurgical Institute’s guidelines of Care and Use of Laboratory Animals.
Intraocular xenografts were established in six-week-old male BALB/c-nude mice (n = 5 for each group, Charles River Laboratories, Beijing, China) by implanting 1×105 MUM2B cells in the right eye of each mouse as previously described.24 For intravitreal virus administration, 5 µL of 1×105 PFU/µL virus was injected at 10 days post-tumor cell implantation.
IVIS Imaging
Bio-luciferase images were obtained via IVIS Spectrum. Images were obtained on 0 day (prior to virus injection) and on 7 days and 14 days post-virus injection. Each mouse was intraperitoneally administered 100 µL of 15 mg/mL D-Luciferin (PerkinElmer, Waltham, Mass, USA) in PBS. Images were captured, and ROIs were assessed.
Histological Analyses
Tumor specimens were obtained from in vivo murine xenografts. Hematoxylin and eosin (H&E) and immunohistochemistry (IHC) staining were performed as previously described.24 Leica Aperio AT2 instruments were used for image acquisition. The following primary antibodies were used: anti-GM-CSF (1:50 dilution; Proteintech, 17762-1-AP) and anti-F4/80 (1:200 dilution; Cell Signaling Technology, 70076, Danvers, MA, USA). The integrated optical density (IOD) was analyzed using ImageJ.
Flow Cytometry Analyses
Tumors were collected 24 days after UM xenograft establishment. Single-cell suspensions were prepared, and then cells were stained with anti-FC receptor antibody (BD Biosciences, Franklin Lakes, NJ, USA), PE-conjugated anti-mouse F4/80 antibody (BioLegend, San Diego, CA, USA), and propidium iodide (PI) (BD Biosciences). Stained cells were washed twice with 1% bovine serum albumin (BSA). A BD C6 flow cytometer was used to acquire 1×105 cells per sample. Results were analyzed using BD FACS Diva software.
Tumor Volumes, Animal Weights, and Survival
Tumor volumes and animal weights were measured every 3 days until 24 days post-treatment. Survival times were also recorded.
Statistical Analyses
All data are expressed as means ± SDs. Statistical analysis was performed using Student’s t-tests. The survival percentage was analyzed using the Kaplan–Meier method and the Log rank test. GraphPad Prism 7.0 was used to prepare all graphs and perform statistical analyses. Differences with p < 0.05 were considered significant. Asterisks are used to denote significance in the figures: *p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.00005; NS, no significance.
Results
Establishment of UM Cell Lines Expressing Luciferase
To observe changes in tumor size within the in vivo xenograft, a plasmid encoding luciferase and neomycin was constructed and used to create a recombinant lentivirus vector. MUM2B, 92.1, and MP41 cells were then transfected with this packaged lentivirus. When G418 was added to the cell cultures, the proportion of infected UM cells increased dramatically. Luciferase expression was examined via the IVIS spectrum imaging system, where all three cell types exhibited strong luciferase expression, indicating that they were suitable for use in our in vitro and in vivo experiments (Figure 1).
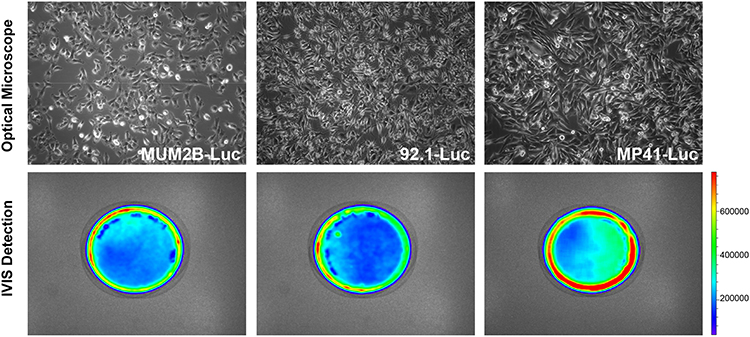 |
Figure 1 Stable UM cell lines expressing luciferase. Images of stable UM cell lines MUM2B, 92.1, and MP41 expressing the luciferase gene acquired with a phase microscope (100×) or IVIS. |
Characterization of HSV-GM-CSF
The recombinant oncolytic HSV-1 virus used in this study was established from the HSV-1 (F) strain. Human GM-CSF transgene was used to replace ICP34.5 in this virus. ICP47 gene was also deleted during the creation of the recombinant HSV-GM-CSF virus (Figure 2A). qPCR was performed to detect GM-CSF gene expression and confirm the insertion of GM-CSF in the recombinant virus (Figure 2B). A Western blot analysis further confirmed the expression of GM-CSF in infected cells (Figure 2C). GM-CSF expression was detected in each HSV-GM-CSF-infected cell line by 48 h post-infection, whereas GM-CSF was not detected in uninfected cells, as expected. TEM was performed to assess cancer cells that had been subjected to HSV-GM-CSF or control treatment. Virus particles were observed clearly in the cell nuclei (red arrow, Figure 2D). These results confirm that the recombinant virus HSV-GM-CSF expresses GM-CSF and can infect UM cells.
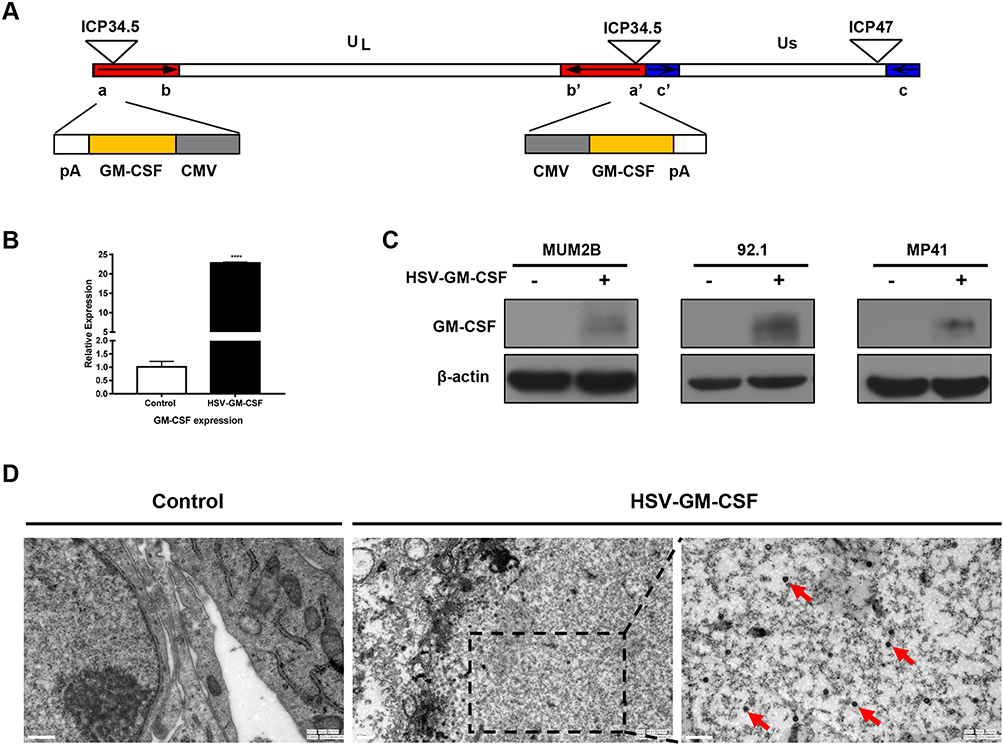 |
Figure 2 Characterization of HSV-GM-CSF. (A) Construction of the recombinant oncolytic virus HSV-GM-CSF based on an HSV-1 strain. (B) qPCR analysis of GM-CSF in samples from the untreated control group and HSV-GM-CSF group. ****p < 0.00005. (C) Western blotting analysis of MUM2B, 92.1, and MP41 cells in the control and HSV-GM-CSF groups. Cells were infected with HSV-GM-CSF at a MOI of 0.1 and collected at 72 h post-infection. (D) Transmission electron microscopy (TEM) images of cells infected at a MOI of 0.1 obtained 72 h post-infection. Red arrows indicate virus in the tumor cells. Scale bar, 0.5 µm. |
Cytotoxic Effect of HSV-GM-CSF in vitro
MUM2B, 92.1 and MP41 cells were infected with HSV-GM-CSF to investigate its oncolytic potency in vitro. Cell viability was assessed at 2 days post-infection (Figure 3A). The IC50 for HSV-GM-CSF-infected MUM2B, 92.1, and MP41 cells was 0.04082, 0.1283, and 0.2191, respectively. IVIS spectra were captured for all three cell lines, and a dramatic decline in radiance was observed after infection in each cell type (Figure 3B). The relative pixel intensity was calculated to quantify the intensity of the IVIS images which showed consistency with IVIS spectra (Figure 3C). These results indicate that the recombinant virus HSV-GM-CSF retains the high oncolytic ability of HSV-1 and is efficient at infecting UM cells.
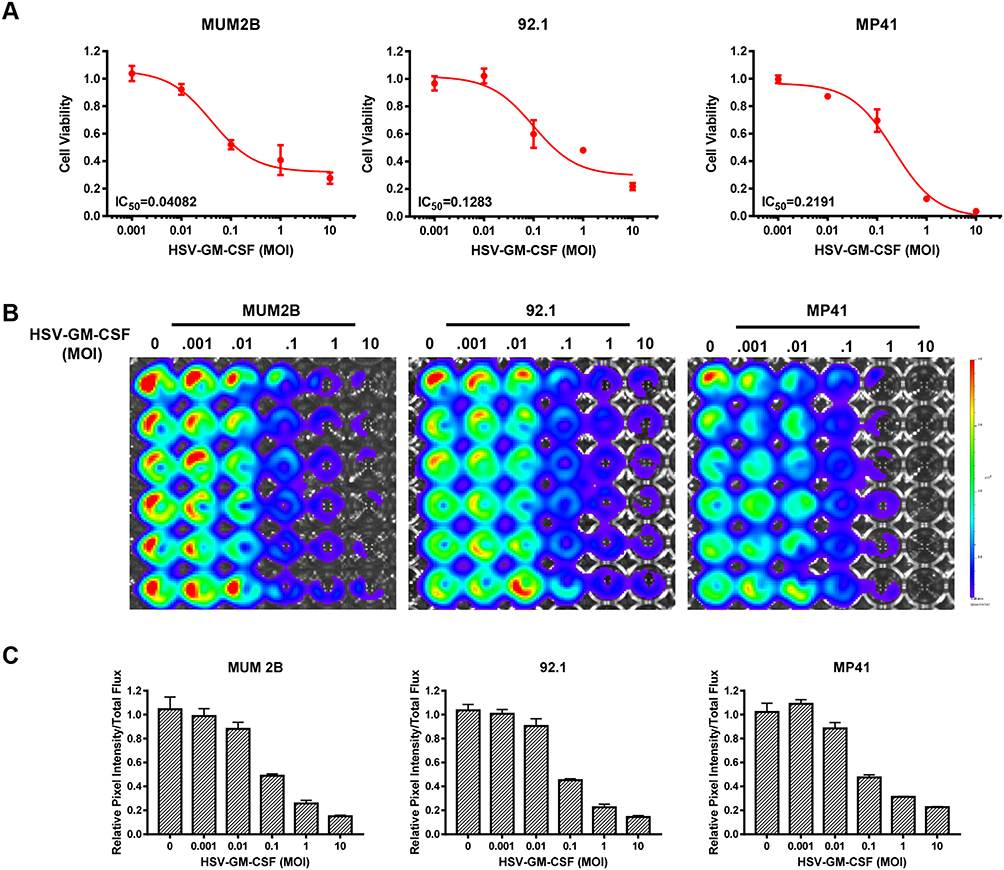 |
Figure 3 In vitro cytotoxic effect of HSV-GM-CSF. (A) Data from cell viability assays performed using MUM2B, 92.1, and MP41 cells infected with HSV-GM-CSF at various MOIs and analyzed 48 h post-infection. (B) IVIS spectra from MUM2B, 92.1, and MP41 cells infected with HSV-GM-CSF at various MOIs. (C) Relative pixel intensity of (B). |
Antitumor Efficacy of HSV-GM-CSF in vivo
We established an in situ tumor xenograft in T-cell-deficient BALB/c-nude mice by implanting MUM2B cells in the posterior chamber of the right eye. Ten days after tumor cell implantation, HSV-1 or HSV-GM-CSF were injected intravitreally (Figure 4A). IVIS imaging was conducted prior to injection, then again at 7 days and 14 days post-injection (Figure 4B). Total flux (p/s) was measured after capturing the IVIS images (Figure 4C). Ocular volumes were recorded 24 days after the virus treatment (Figure 4D). As expected, by 7 days post-injection, the tumor volumes of the virus-treated groups (HSV-1 and HSV-GM-CSF) were significantly smaller compared with the control group. However, at 14 days post-injection, there was an increase in the tumor size of the HSV-1-treated group. In contrast, the tumors in the HSV-GM-CSF group continued to regress. The mean ocular tumor volumes (n = 5/group) calculated at 14 days post-virus injection were 176.3±20.5 mm3, 69.02±6.8 mm3, and 49.02±7.4 mm3 for the control, HSV-1, and HSV-GM-CSF groups, respectively.
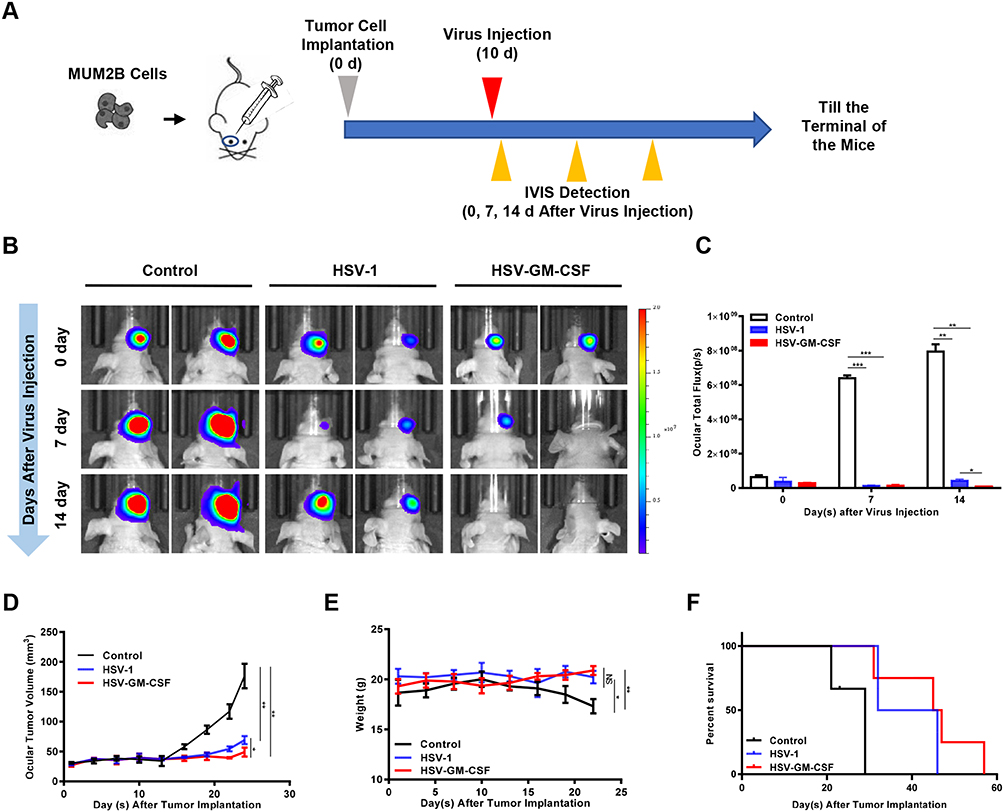 |
Figure 4 Antitumor efficacy of HSV-1 and HSV-GM-CSF in a murine uveal melanoma xenograft. (A) Schematic of the applied in vivo treatment. (B) IVIS images of in situ UM xenografts from the untreated control, HSV-1, and HSV-GM-CSF groups (n = 5 mice per group). (C) Total flux (p/s) of (B). *, p < 0.05; **, p < 0.005; ***, p < 0.0005. (D) Ocular volumes (mm3) of the three groups. *, p < 0.05; **, p < 0.005. (E) Mouse bodyweight (g) of the three groups. Data were analyzed using a Student’s t-test, and values shown are the mean ± SD of each group. *, p < 0.05; **, p < 0.005; NS, no significance. (F) Percentage of survival in the three groups. Data were analyzed using a Log rank test, p < 0.05. |
Mouse bodyweights were tracked over the 24 days following UM cell implantation. At the end of this period, the control group mice weighed significantly less compared with the HSV-1 and HSV-GM-CSF treated group mice (Figure 4E). Follow-up survival was prolonged in the virus-treated groups compared with the control group (Figure 4F). Notably, GM-CSF armed virus (HSV-GM-CSF) prolonged survival compared with the unmodified virus (HSV-1). The median survival times for the control, HSV-1, and HSV-GM-CSF groups were 24, 39, and 46 days, respectively. These data indicate that the treatment of UM xenografts with HSV-1 suppresses tumor growth and that the tumor reduction ability of HSV-1 was enhanced by arming the virus with GM-CSF. These findings are consistent with the tumor size analysis results, suggesting HSV-GM-CSF has an enhanced antitumor efficacy.
Immune Response Induced by HSV-GM-CSF Treatment
To assess the immune response against the virus in infected tumor-bearing mice, tumor specimens were obtained 2 weeks after viral injection. H&E staining and IHC were performed for pathology analysis (Figure 5A). H&E staining revealed that the UM tumor cells displayed typical characteristics, such as nuclear pleomorphism and large nuclei.
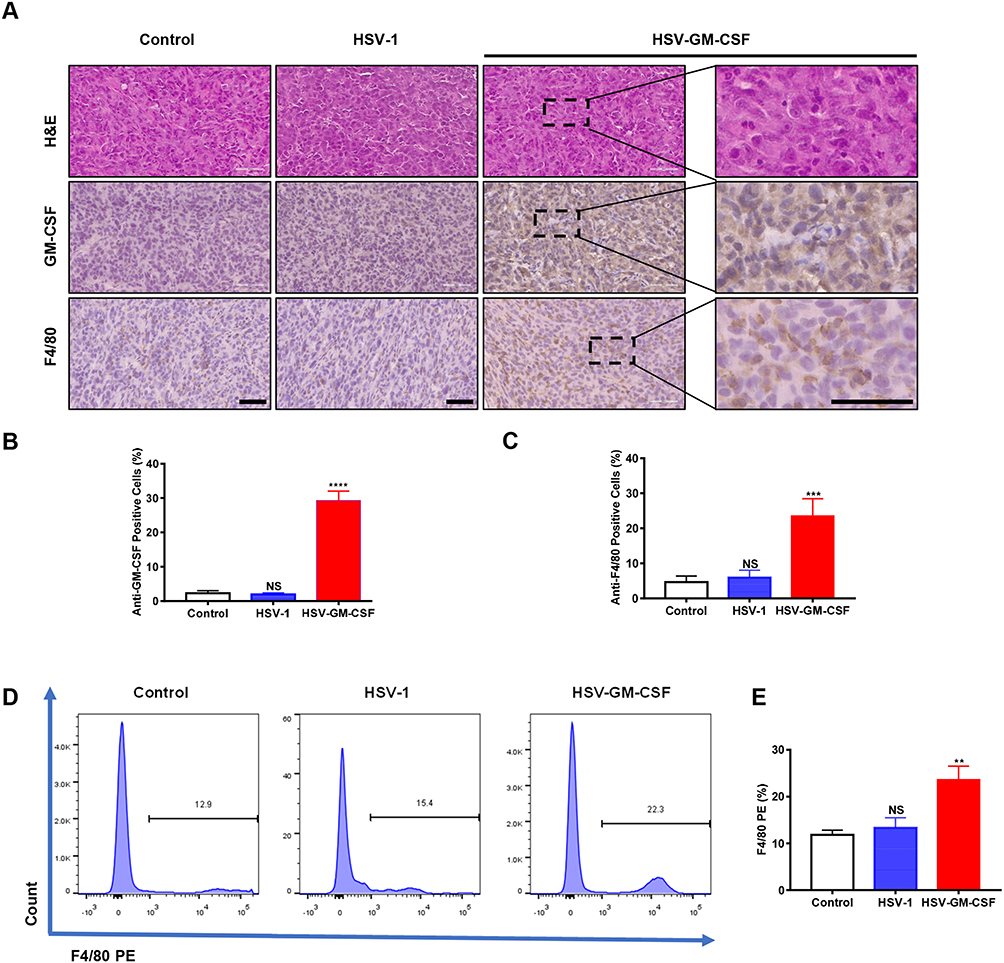 |
Figure 5 Immune response induced by injection with HSV-1 or HSV-GM-CSF. (A) H&E and IHC images of dissected tumors. GM-CSF and F4/80 were detected in the HSV-GM-CSF-treated group. Scale bar, 60 µm. (B) IOD values for anti-GM-CSF-positive cells. ****p < 0.00005; NS, no significance. (C) IOD values for anti-F4/80-positive cells. ***, p < 0.0005; NS, no significance. (D) Data from a FACS analysis performed on in situ tumors from UM xenografts. F4/80 PE was detected for macrophage infiltration. These data were analyzed using a Student’s t-test, and values shown are the mean ± SD. (E) Statistics of (D). Data were analyzed using Student’s t-test and values are shown as the mean ± SD. **, p < 0.005; NS, no significance. |
Anti-GM-CSF staining levels were compared among the control, HSV-1, and HSV-GM-CSF groups. The percentage of GM-CSF positive area was higher in the HSV-GM-CSF group than those in the control and HSV-1 groups (Figure 5B). This indicates that GM-CSF levels are greatly elevated following HSV-GM-CSF treatment. We next evaluated the macrophage infiltration into the xenografts by measuring anti-F4/80 levels. There was no significant difference in macrophage quantity between the HSV-1 and untreated control groups, but the HSV-GM-CSF group had significantly more macrophage infiltrations (Figure 5C). FACS was also performed to verify macrophage infiltration levels inside the tumor masses (Figure 5D). The mean percentages of F4/80 expressing cells in the untreated control, HSV-1, and HSV-GM-CSF groups were 11.83±0.58%, 13.34±1.2%, and 23.64±1.68%, respectively (n=3) (Figure 5E). These results are consistent with our IHC observations. Together, these results indicate that HSV-GM-CSF increases macrophage infiltration into the in situ xenografts.
Discussion
The combination of an oncolytic virus with other molecules has shown as a promising therapeutic in cancer treatment. In the current study, we constructed a recombinant oncolytic HSV-1 virus armed with the human GM-CSF gene. This study is the first to explore the therapeutic effects of treating UM with an oncolytic HSV-GM-CSF virus, and our in vitro and in vivo results demonstrate that this virus has cell-killing effects and tumor reductions after the treatment. Additionally, mice with UM xenografts treated with the HSV-GM-CSF virus survived significantly longer compared with control or the unmodified HSV-1 virus group. Our results indicate that HSV-GM-CSF has superior in vivo antitumor efficacy in UM animal models compared with HSV-1. We also observed the infiltration of innate immune cells following treatment. Significantly more macrophages infiltrated into the tumors of mice treated with HSV-GM-CSF compared with control or HSV-1 treated group. Base on the knowledge that macrophage is correlated with tumor growth in UM, our current study suggests that the treatment of HSV-GM-CSF has the ability to not only direct lysis tumor cells but also attenuating UM tumor growth from an innate immune perspective.
Previous studies have shown that the deletion of non-essential viral ICP34.5 significantly increased virus specificity towards tumor cells.25,26 It was also demonstrated that the deletion of the immediate early protein ICP47 induces transporter for antigen processing (TAP) mediated cytotoxic T-lymphocytes, leading to immune activation to the virus-infected cells.27,28 Therefore, when creating the recombinant HSV-1 virus, we chose to mutate ICP34.5 with the GM-CSF gene and to delete ICP47.
The antitumor efficacy of oncolytic HSV-1 has been demonstrated by its cell-killing effect and its ability to activate the immune system. These properties of HSV-1 can be further enhanced by arming the virus with the gene for GM-CSF, which is a specific and long-lasting immunostimulatory molecule.29 GM-CSF enhances the tumor-suppressing properties of macrophages.30 In the present study, more macrophages were observed in tumor specimens from HSV-GM-CSF-treated mice. It is well known that macrophages can act as antigen-presenting cells (APCs), stimulating reactions that recruit T cells by releasing tumor necrosis factor (TNF).31 Previous reports demonstrated a correlation between UM tumor prognosis and macrophage infiltration.32 Macrophages have also been reported to exert an antitumor effect without the help of T cells.33 Our data support that increased macrophage infiltration is associated with reduced UM xenograft tumor growth.
NK cells were reported to play an important role in the prevention of UM metastases.34 Human leukocyte antigen (HLA) class I antigen expression is essential for tumor-specific antigen presentation and effective antitumor responses. The presence of these molecules can inactivate NK cells.35 Regarding UM, NK cell responses are inversely correlated with HLA class I expression levels on tumor cells, leading to the hypothesis that NK cells remove micro-metastases from circulation that have downregulated HLA class I.36,37 Additionally, UM cells were reported to be sensitive to NK cell lysis.34 Because HSV-1 induces NK cell infiltration, this virus and the GM-CSF-expressing HSV-1 may also have antitumor efficacy against metastases from UM xenografts (data not shown).
Because the in vivo xenograft used in our experiments was built on an immune-incompetent BALB/c-nude mouse, our data have limitations. The antiviral immune response and associated virus clearance, which could limit the application of HSV-GM-CSF in immunocompetent hosts, remain unclear. Establishing a suitable UM cell line that can grow into an immunocompetent xenograft and the application of repeated injections are strategies for addressing these problems in future studies. Other key factors in this process, like cytokines and chemokines, are also important for future exploration.
Conclusion
In summary, we demonstrated here that recombinant oncolytic HSV-1 armed with human GM-CSF (HSV-GM-CSF) induced a potent antitumor response both in vitro and in vivo. HSV-GM-CSF injection induced GM-CSF expression as well as macrophage infiltration. Future studies should focus on the safety of this recombinant virus and its clinical effect in UM patients. Overall, our study introduced a novel therapeutic oncolytic agent that holds promise as a candidate for the treatment of UM.
Abbreviations
UM, Uveal melanoma; HSV-1, Herpes simplex virus type 1; GM-CSF, Granulocyte-macrophage colony-stimulating factor.
Acknowledgments
The authors thank Dr. Vavvas Demetrios and Efstathiou Nikolaos from Massachusetts General Hospital for their help with cell lines. This work was supported by the National Natural Science Foundation of China (No. 81672478, 81972344 and 81772671), Beijing Natural Science Foundation (No. 7202020) and Beijing Laboratory of Biomedical Materials Foundation.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Singh AD, Bergman L, Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18(1):75–84. doi:10.1016/j.ohc.2004.07.002
2. Egan KM, Seddon JM, Glynn RJ, Gragoudas ES, Albert DM. Epidemiologic aspects of uveal melanoma. Surv Ophthalmol. 1988;32(4):239–251. doi:10.1016/0039-6257(88)90173-7
3. Seddon JM, Gragoudas ES, Egan KM, et al. Relative survival rates after alternative therapies for uveal melanoma. Ophthalmology. 1990;97(6):769–777. doi:10.1016/S0161-6420(90)32512-5
4. Kroll S, Char DH, Quivey J, Castro J. A comparison of cause-specific melanoma mortality and all-cause mortality in survival analyses after radiation treatment for uveal melanoma. Ophthalmology. 1998;105(11):2035–2045.
5. Rajpal S, Moore R, Karakousis CP. Survival in metastatic ocular melanoma. Cancer. 1983;52(2):334–336.
6. Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14(9):642–662.
7. Bell J, McFadden G. Viruses for tumor therapy. Cell Host Microbe. 2014;15(3):260–265.
8. Yu W, Fang H. Clinical trials with oncolytic adenovirus in China. Curr Cancer Drug Targets. 2007;7(2):141–148.
9. Li Y, He J, Qiu C, et al. The oncolytic virus H101 combined with GNAQ siRNA-mediated knockdown reduces uveal melanoma cell viability. J Cell Biochem. 2019;120(4):5766–5776.
10. Huang X, Jia R, Zhao X, et al. Recombinant oncolytic adenovirus H101 combined with siBCL2: cytotoxic effect on uveal melanoma cell lines. Br J Ophthalmol. 2012;96(10):1331–1338. doi:10.1136/bjophthalmol-2011-301470
11. Garcia M, Moreno R, Gil-Martin M, et al. A Phase 1 Trial of Oncolytic Adenovirus ICOVIR-5 Administered Intravenously to Cutaneous and Uveal Melanoma Patients. Hum Gene Ther. 2019;30(3):352–364. doi:10.1089/hum.2018.107
12. Kooby DA, Carew JF, Halterman MW, et al. Oncolytic viral therapy for human colorectal cancer and liver metastases using a multi-mutated herpes simplex virus type-1 (G207). FASEB J. 1999;13(11):1325–1334. doi:10.1096/fasebj.13.11.1325
13. Nakano K, Todo T, Chijiiwa K, Tanaka M. Therapeutic efficacy of G207, a conditionally replicating herpes simplex virus type 1 mutant, for gallbladder carcinoma in immunocompetent hamsters. Mol Ther. 2001;3(4):431–437. doi:10.1006/mthe.2001.0303
14. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. doi:10.1126/science.1851332
15. Chambers R, Gillespie GY, Soroceanu L, et al. Comparison of genetically engineered herpes simplex viruses for the treatment of brain tumors in a SCID mouse model of human malignant glioma.. Proc Natl Acad Sci U S A. 1995;92(5):1411–1415. doi:10.1073/pnas.92.5.1411
16. Andtbacka RHI, Kaufman HL, Collichio F, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol. 2015;33(25):2780–2788. doi:10.1200/JCO.2014.58.3377
17. Miyatake S, Martuza RL, Rabkin SD. Defective herpes simplex virus vectors expressing thymidine kinase for the treatment of malignant glioma. Cancer Gene Ther. 1997;4(4):222–228.
18. Liu S, Zhang J, Fang S, et al. Antitumor efficacy of oncolytic HSV-1 expressing cytosine deaminase is synergistically enhanced by DPD down-regulation and EMT inhibition in uveal melanoma xenograft. Cancer Lett. 2020;495:123–134. doi:10.1016/j.canlet.2020.09.013
19. Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17(3):718–730. doi:10.1245/s10434-009-0809-6
20. Shi Y, Liu CH, Roberts AI, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16(2):126–133. doi:10.1038/sj.cr.7310017
21. Becher B, Tugues S, Greter M. GM-CSF: from Growth Factor to Central Mediator of Tissue Inflammation. Immunity. 2016;45(5):963–973. doi:10.1016/j.immuni.2016.10.026
22. Makitie T, Summanen P, Tarkkanen A, Kivela T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Invest Ophthalmol Vis Sci. 2001;42(7):1414–1421.
23. Zhu G, Su W, Jin G, et al. Glioma stem cells targeted by oncolytic virus carrying endostatin-angiostatin fusion gene and the expression of its exogenous gene in vitro. Brain Res. 2011;1390:59–69. doi:10.1016/j.brainres.2011.03.050
24. Liu S, Song W, Liu F, Zhang J, Zhu S. Antitumor efficacy of VP22-CD/5-FC suicide gene system mediated by lentivirus in a murine uveal melanoma model. Exp Eye Res. 2018;172:144–151. doi:10.1016/j.exer.2018.04.009
25. Liu BL, Robinson M, Han Z-Q, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Therapy. 2003;10(4):292–303. doi:10.1038/sj.gt.3301885
26. Kanai R, Zaupa C, Sgubin D, et al. Effect of γ34.5 Deletions on Oncolytic Herpes Simplex Virus Activity in Brain Tumors. J Virol. 2012;86(8):4420–4431. doi:10.1128/JVI.00017-12
27. Tomazin R, van Schoot NEG, Goldsmith K, et al. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J Virol. 1998;72(3):2560–2563. doi:10.1128/JVI.72.3.2560-2563.1998
28. Ahn K, Meyer TH, Uebel S, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47.. EMBO J. 1996;15(13):3247–3255. doi:10.1002/j.1460-2075.1996.tb00689.x
29. Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity.. Proc Natl Acad Sci U S A. 1993;90(8):3539–3543. doi:10.1073/pnas.90.8.3539
30. Grabstein KH, Urdal DL, Tushinski RJ, et al. Induction of macrophage tumoricidal activity by granulocyte-macrophage colony-stimulating factor. Science. 1986;232(4749):506–508. doi:10.1126/science.3083507
31. Villeneuve J, Tremblay P, Vallieres L. Tumor necrosis factor reduces brain tumor growth by enhancing macrophage recruitment and microcyst formation. Cancer Res. 2005;65(9):3928–3936. doi:10.1158/0008-5472.CAN-04-3612
32. Jager MJ, Ly LV, El Filali M, Madigan MC. Macrophages in uveal melanoma and in experimental ocular tumor models: friends or foes? Prog Retin Eye Res. 2011;30(2):129–146. doi:10.1016/j.preteyeres.2010.11.004
33. Galarneau H, Villeneuve J, Gowing G, Julien J-P, Vallieres L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007;67(18):8874–8881. doi:10.1158/0008-5472.CAN-07-0177
34. Maat W, van der Slik AR, Verhoeven DHJ, et al. Evidence for Natural Killer Cell–Mediated Protection from Metastasis Formation in Uveal Melanoma Patients. Invest Ophthalmol Vis Sci. 2009;50(6):2888–2895. doi:10.1167/iovs.08-2733
35. Kaufman DS, Schoon RA, Robertson MJ, Leibson PJ. Inhibition of selective signaling events in natural killer cells recognizing major histocompatibility complex class I.. Proc Natl Acad Sci U S A. 1995;92(14):6484–6488. doi:10.1073/pnas.92.14.6484
36. Blom DJ, Luyten GP, Mooy C, Kerkvliet S, Zwinderman AH, Jager MJ. Human leukocyte antigen class I expression. Marker of poor prognosis in uveal melanoma. Invest Ophthalmol Vis Sci. 1997;38(9):1865–1872.
37. Ma D, Niederkorn JY. Transforming growth factor-beta down-regulates major histocompatibility complex class I antigen expression and increases the susceptibility of uveal melanoma cells to natural killer cell-mediated cytolysis. Immunology. 1995;86(2):263–269.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.