")
Back to Journals » OncoTargets and Therapy » Volume 12
Antitumor effect of kurarinone and underlying mechanism in small cell lung carcinoma cells
Authors Chung TW, Lin CC, Lin SC , Chan HL, Yang CC
Received 13 May 2019
Accepted for publication 16 July 2019
Published 5 August 2019 Volume 2019:12 Pages 6119—6131
DOI https://doi.org/10.2147/OTT.S214964
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Ting-Wen Chung,*,1 Chi-Chien Lin,2,3,* Shih-Chao Lin,4 Hong-Lin Chan,1 Ching-Chieh Yang5,6
1Institute of Bioinformatics and Structural Biology and Department of Medical Sciences, National Tsing Hua University, Hsinchu 300, Taiwan; 2Institute of Biomedical Science, National Chung‑hsing University, Taichung 402, Taiwan; 3Department of Medical Research, China Medical University Hospital, Taichung 404, Taiwan; 4National Center for Biodefense and Infectious Diseases, School of Systems Biology, George Mason University, Manassas, VA, USA; 5Department of Radiation Oncology, Chi-Mei Medical Center, Tainan 710, Taiwan; 6Department of Pharmacy, Chia-Nan University of Pharmacy and Science, Tainan 717, Taiwan
*These authors contributed equally to this work
Background: Kurarinone, a prenylated flavonone isolated from the roots of Sophora flavescens, is known to be cytotoxic against many human cancer cells but not human small cell lung carcinoma (SCLC) yet. Also, the exact molecular mechanism of kurarinone for induction cytotoxicity remains unknown.
Material and methods: We investigated the effects of kurarinone on cell proliferation, apoptosis, and migration in H1688 SCLC cells. Cell viability was determined by the MTT assay. Apoptotic indices such as cell cycle, mitochondrial membrane potential, cytochrome c release, caspase activity, and death receptors were evaluated by flow cytometry. Transwell migration and invasion assays were also included.
Results: Our results indicated that kurarinone significantly decreased H1688 cell viability and induced the accumulation of sub-G1 fractions by activating caspase-3, -9, and PARP cleavage accompanied by the elevated release of cytochrome c and mitochondrial dysfunction in H1688 cells. Additionally, kurarinone promoted Fas and TRAIL receptor-1 and -2 expression via the caspase-8/Bid pathway, suggesting that kurarinone triggered apoptosis via the mitochondria-mediated and receptor-mediated apoptotic pathways. We also observed that kurarinone repressed migration and invasion capabilities of SCLC cells by suppressing the expression of epithelial-mesenchymal transition-related proteins and matrix metalloproteinases.
Conclusion: Our findings provided evidence that kurarinone can induce apoptosis in SCLC cells via multiple mechanisms and delayed the cell migration and invasion of SCLC cells.
Keywords: kurarinone, small cell lung carcinoma, apoptosis, caspase, migration, invasiveness
Introduction
Lung cancer is the second most common cancer in the world and the leading cause of cancer-related deaths.1 Small cell lung cancer (SCLC) is one of the most aggressive cancers, exhibiting rapid tumor growth and early onset of metastases, often at presentation.2 Despite the use of numerous combinations of treatments including chemotherapy, radiation, and targeted therapy, the outcomes of SCLC patients remains poor.3,4 Most SCLC patients experience relapse within 2 years, and the 5-year overall survival is approximately 6.5% for limited-stage and 5% for extensive-stage disease.5 Furthermore, currently-used therapies have been demonstrated to have toxic side effects to the bone marrow, esophagus, normal lung tissue, and heart.6,7 Thus, the identification of novel and promising agents for the treatment of SCLC is greatly needed.
Kurarinone, isolated from Sophora flavescens Ait (Leguminosae), has several known biological activities. This compound has been used in traditional Chinese medicine as an antipyretic, analgesic, anthelmintic, and stomachic drug.8 Several lines of evidence indicate that kurarinone exhibits anti-inflammatory, anti-bacterial, and anti-viral activity.9–11 Kurarinone is also reported to have anti-tumor activity against many types of cancer.12–15 A recent study by Yang et al found that kurarinone exerts potent activity against human non-small cell lung cancer (NSCLC) cell line A549 by activating the mitochondrial apoptosis signaling pathway and repressing the endoplasmic reticulum and AKT pathways.14 In vivo, kurarinone has been observed to inhibit the growth of A549 cells in xenograft mouse models, with no apparent signs of toxicity.
However, to the best of our knowledge, no published study has investigated the effects of kurarinone on SCLC. Hence, this study aimed to evaluate the antitumor activities of kurarinone in human SCLC cells. In addition, the mechanism underlying the antitumor activities of kurarinone was also investigated.
Materials and methods
Chemicals
Kurarinone was purchased from Sigma-Aldrich (St. Louis MO, USA) and directly dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) at the stock concentration of 50 mM. Prior to adding to cells, kurarinone was serially diluted with DMSO to 25, 12.5, 6.25, and 3.125 mM followed by 1:1000 of dilution with complete culture medium. Following the addition of kurarinone, the culture plates were gently rocked to evenly diffuse the kurarinone in wells, and the final concentrations of kurarinone fell between 3.125 and 50 μM throughout the study.
Cell lines
Two human small-cell lung cancer (SCLC) cell lines, H1688 and H146, and an immortalized bronchial epithelial cell line, BEAS-2B, were purchased from the Food Industry Research and Development Institute (Hsinchu, Taiwan). The cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 100 g/mL of penicillin, and 100 μg/mL of streptomycin (all from Gibco Laboratory, Grand Island, NY), at 5% CO2 and 37°C.
MTT cell viability assay
The cells were seeded into 24-well plates at 2×104 cells/well and incubated with different concentrations of kurarinone (3.125–50 μM) or with DMSO (0.1%) as a vehicle control for 24 h. To measure cell viability, 200 μL/well of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-di-phenyltetrazolium bromide solution (MTT) (Sigma-Aldrich) was added to wells and incubated for 4 h at 37°C. The supernatant then was removed, and 600 μL of DMSO was added to each well to dissolve the formazan complex. The amount of colored formazan was determined by its absorbance at 540 nm using a microplate reader (Tecan Sunrise, San Jose, CA, USA). Data are presented as the percent absorbance of kurarinone-treated cells relative to DMSO-treated cells. The 50% inhibitory concentration (IC50) values were calculated using Microsoft Excel software for semi-log curve fitting with regression analysis.
Colony‐forming assays
Colony‐formation assays were carried out to test the effect of kurarinone on the clonogenicity of SCLC cells. Briefly, cells were seeded into 6‐well plates at 500 cells/well and incubated for 24 h. The cells then treated with different concentrations of kurarinone (6.25, 12.5, and 25 μM) for one week to allow colonies to form. Crystal violet (2%) (Sigma-Aldrich) was used to stain colonies, and the number of colonies in each well was counted under an inverted microscope (Olympus, Tokyo, Japan).
Western blot analysis
Cells (2×105/well) were seeded into 6-well plates and treated with the indicated concentrations of kurarinone. After 24 hrs, the cells were lysed in RIPA buffer (Sigma-Aldrich) supplemented with freshly-added 1% protease inhibitor cocktail (Sigma-Aldrich). Lysate protein concentrations were determined using the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) SDS-PAGE and then transferred to Immobilon-P Transfer Membrane (Merck Millipore, Billerica, MA, USA). Membranes were incubated in 5% bovine serum albumin (BSA) (Sigma-Aldrich) blocking buffer for 1 h at room temperature and then overnight with primary antibody at 4°C. Immunoblotting was performed using the following antibodies: anti-cleaved PARP (clone 19F4, 1:2000), anti-cleaved caspase-3 (clone 5A1E; 1:1000), anti-cleaved caspase-8 (clone 11G10; 1:1000), anti-Bcl-2 (50E3; 1:1000), anti-Bcl-xl (clone 54H6; 1:1000), anti-Bax (clone D2E11; 1:1000) (All from Cell Signaling Technology, Danvers, MA, USA), cleaved Bid (cat no. ab10640, 1:1000) (Abcam, Cambridge, MA, USA), anti-N-cadherin (EPR1792Y, 1:50,000) (Epitomics, Burlingame, CA, USA), anti-vimentin (clone 9E7E7, 1:1000), anti-E-cadherin (clone H-108, 1:1000), anti-MMP-3 (clone 1B4, 1:1000) (All from Santa Cruz Biotechnology), anti-MMP-2 (cat no. GTX104577, 1:500),, anti-MMP-9 (cat no. GTX100458; 1:500) (Both from GeneTex, Irvine, CA, USA), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Abcam, clone 9484, 1:1000). Membranes were washed 3 times (10 min each) in Tween buffer before incubating with horseradish peroxidase (HRP)-conjugated goat anti-mouse or rabbit secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) at 4°C overnight. Protein bands were visualized using the enhanced chemiluminescence detection kit reagent (GE Healthcare Life Sciences, Piscataway, NJ, USA) and the Hansor Luminescence Image system (Taichung, Taiwan). All bands in the blots were normalized to GAPDH in each lane. The intensity of the bands was quantified using Image J software version 1.50 (National Institutes of Health, Bethesda, MD, USA).
Analysis of cell apoptosis by flow cytometry
The extent of apoptosis was evaluated by flow cytometry using an Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, Franklin Lakes, NJ, USA). Cells (2×105/well) were treated with kurarinone (0, 6.25, 12.5, or 25 µM) for 24 h and then harvested and washed thrice with phosphate buffered saline (PBS). Cells were incubated with 5 μL of Annexin V-FITC (20 μg/mL) and 5 μL of propidium iodide (PI) (50 μg/mL) at room temperature for 10 min in the dark. Apoptotic cells were detected using an AccuriTM C5 cytometer (BD Biosciences) and analyzed using BD Accuri C6 Software version 1.0.264.21.
Determination of DNA content by flow cytometry
Cells were seeded into 6-well plates (2×105 cells/well) and treated with kurarinone (6.25, 12.5, or 25 μM) for 24 h. The cells were harvested using trypsin, washed twice with PBS, and fixed in 70% ethanol overnight at −20°C. The fixed cells were stained in propidium iodine solution containing 1 mL of PBS, 50 μg/mL of propidium iodide (Sigma-Aldrich), 100 μg/mL of RNase A, and 0.1% Triton X-100 (Sigma-Aldrich) in constant darkness at room temperature for 20 min. Apoptotic cells in the sub-G1 population were detected using an AccuriTM C5 cytometer.
Measurement of δψm
Changes in mitochondrial membrane potential were assessed using the JC-1 Mitochondrial Potential Assay Kit (Invitrogen Life Technologies, Carlsbad, CA, USA). H1688 cells were seeded into 6-well plates (2×105 cells/well) and treated with the indicated concentrations of kurarinone for 24 h. The cells were harvested and then incubated with 1 mL medium containing JC-1 fluorescent dye for 20 min in the dark at 37°C. Cells then were washed twice in staining buffer followed by flow cytometry analysis for detecting ΔΨm. JC-1 monomers and J-aggregates were detected in the FL1 and FL2 channels, respectively, and variations in the red/green fluorescence intensity ratio reflect changes in the mitochondrial membrane potential.
In vitro assay for cytochrome c release from mitochondria by flow cytometry
Measurement of cytochrome c release via FITC-anti-cytochrome c antibody (Santa Cruz Biotechnology) was described in a previous study.16 Briefly, cells were seeded in 6-well plates (2×105 cells/well) and then treated with the indicated concentrations of kurarinone for 24 h. Afterwards, cells were resuspended thoroughly in 100 µL digitonin lysis buffer (50 µg/mL digitonin and 100 mM KCl in 1× PBS) for 5 min on ice to permeabilize the plasma membranes and then fixed with 4% paraformaldehyde (Sigma-Aldrich) in PBS at room temperature for 30 min. Cells were washed 3 times with PBS and incubated in 0.5 mL labeling buffer (2% BSA in 0.05% Triton X-100 PBS) containing 10 μL of FITC-anti-cytochrome c antibody at 4°C for 1 h followed by detecting cytochrome c-expressed cells with flow cytometry.
Caspase activity assay by flow cytometry
Cells were seeded in 6-well plates (2×105 cells/well) and then treated with the indicated concentrations of kurarinone for 24 h. Cells were harvested and tested for caspase-3, −8, and −9 activities using the appropriate CaspGLOW fluorescein active caspase staining kits (Biovision, Milpitas, CA, USA) according to the manufacturer’s protocol. The caspase activity was detected using an AccuriTM C5 cytometer.
Analysis of death receptors and ligands by flow cytometry
Cells were seeded in a 6-well plate at 2×105 cells per well and then treated with indicated concentrations of kurarinone for 24 h followed by harvesting and staining with the following antibodies from eBioscience (San Diego, CA, USA): anti-DR4-PE (clone DJR1, 1:1000), anti-DR5-PE (clone DJR2-4, 1:1000,), anti-Fas-PE (clone DX2, 1:1000), anti-TRAIL-PE (clone N2B2, 1:1000), or anti-FasL-PE (clone MFL3, 1:1000) at 4°C for 1 h. The expression level of each protein was determined by flow cytometry.
Scratch wound healing assay
Cells were seeded into 6-well plates at 2×105 cells/well and then treated with the indicated concentrations of kurarinone for 24 h. After treatment, the cells were collected and seeded (1.2×104 cells/mL) into Culture-Insert (ibidi GmbH, Planegg, Germany) in 70-μl cell suspensions. After cell attachment, the Culture-Insert was gently removed to form a cell-free gap. Images were taken before and 24 h after treatment under an optical microscope (Carl Zeiss, Jena, Germany) at 100 × magnification. The wound areas were quantified and analyzed using AxioVision Rel. 4.8 software (Carl Zeiss).
Transwell migration and invasion assays
For cell migration assays, 1×105 of H1688 cells were planted into the top chamber of a 24-well Transwell plate(Millipore), and 600 μL of medium containing 10% FBS was added to the lower chamber. Kurarinone of varying concentrations was added to both chambers. Cells in the upper chamber were then allowed to migrate for 24 h prior to removal with a cotton swab. The migrated cells were fixed in 4% paraformaldehyde, stained with 0.5% crystal violet (Sigma‐Aldrich), and visualized under an inverted microscope (Olympus) at 100× magnification. The crystal violet in the filter membrane containing stained cells was dissolved using ethanol/acetic acid (49.9%, 0.1%, v/v) solution, and the OD of the solution was measured at 600 nm. For cell invasion assay, the upper chamber was coated with Matrigel (Sigma-Aldrich). Subsequent operations were similar to the cell migration assays. Data are expressed as the migration or invasion rate as compared to the DMSO-treated control group.
Statistical analysis
Data are reported as the mean ± SD throughout the study. Data acquisition and analysis of variance (ANOVA) by Tukey’s posttest was performed for comparisons between groups using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). p<0.05 was considered statistically significant. Similar results were obtained from 3 independent experiments.
Results
Kurarinone decreased cell viability and induced apoptosis in small-cell lung cancer cell lines
As shown in Figure 1A and B, 24-h treatment with kurarinone (0−50 μM) decreased the proliferation of H1688 and H1466 cells in a dose-dependent manner. Kurarinone exhibited a moderate antiproliferative effect on human bronchial epithelial cells BEAS-2B (Figure 1A). MTT assay results show that the IC50 values for kurarinone were 12.5±4.7, 30.4±5.1 and 55.8±4.9 μM for H1688, H146, and BEAS-2B cells, respectively (Table 1). Similarly, colony formation assays showed dose-dependent inhibition of H1688 cells (Figure 1B) after 1 week of treatment with kurarinone, confirming the growth inhibition effect of kurarinone. Our data indicates that H1688 cells were more sensitive to kurarinone than H146 and BEAS-2B cells and as such, H1688 cells could be an adequate in vitro model to investigate the anti-cancer mechanism of kurarinone.
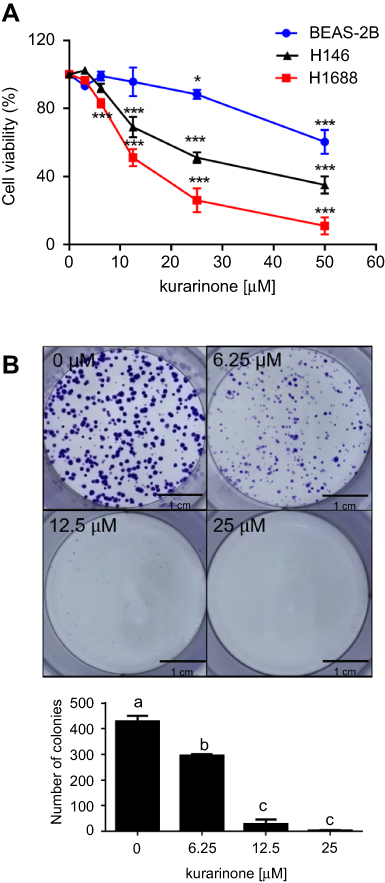 |
Figure 1 (A) Cell viability of kurarinone-treated H1688, H146, and BEAS-2B cells. Cells were treated with 0.1% DMSO (0 μM of kurarinone), or kurarinone at 3.125, 6.25, 12.5, 25, or 50 μM for 24 h. Cell viability was examined by MTT assay. *P<0.05, ***P<0.001 versus 0.1% DMSO group. (B) Colony formation assay of H1688 cells following treatment with kurarinone for one week. Data are presented as the mean ± SD in 3 replicates for each treatment. Different letters (a,b,c) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
 |
Table 1 IC50 of kurarinone for cell viability of human SCLC cell lines H1688 and H146 and human bronchial epithelial cells BEAS-2B |
To determine whether apoptosis was involved in the cytotoxic effects induced by kurarinone, we aimed to investigate the apoptotic indicators, including Annexin V, poly(ADP-ribose) polymerase (PARP), caspase cascade, and mitochondria membrane permeability. PARP is the caspase-3 substrate which could be subsequently cleaved by active caspase-3 during apoptosis.17 Thus, the expression of poly(ADP-ribose) polymerase (PARP) can be used as a marker for detecting apoptotic cells.18,19 Western blot analysis of the expression of cleaved PARP showed that kurarinone dose-dependently increased the cleaving of PARP in H1688 cells at 24 h (Figure 2A). Moreover, the apoptosis and cell-cycle distribution in H1688 cells were determined by using Annexin V-FITC and propidium iodide staining and flow cytometry analysis. As shown in Figure 2B, 24-h kurarinone treatment of H1688 cells resulted in dose-dependent increases in both the early (Annexin V+/PI−) and late (Annexin V+/PI+) apoptotic cell populations (Figure 2B). The total apoptotic cell populations (early and late apoptosis) were 2.4%±1.8, 10.3%±0.9, 33.3%±2.9 and 73.1%±3.0 at 0, 6.25, 12.5 and 25 μM of kurarinone, respectively. Additionally, the fractions of sub-G1 phase cells (apoptotic cells) increased markedly from 1.4% of DMSO-treated cells to 59.3% along with kurarinone treatments for 24 h (Figure 2C). These results suggest that kurarinone treatment induced considerable apoptosis in H1688 cells.
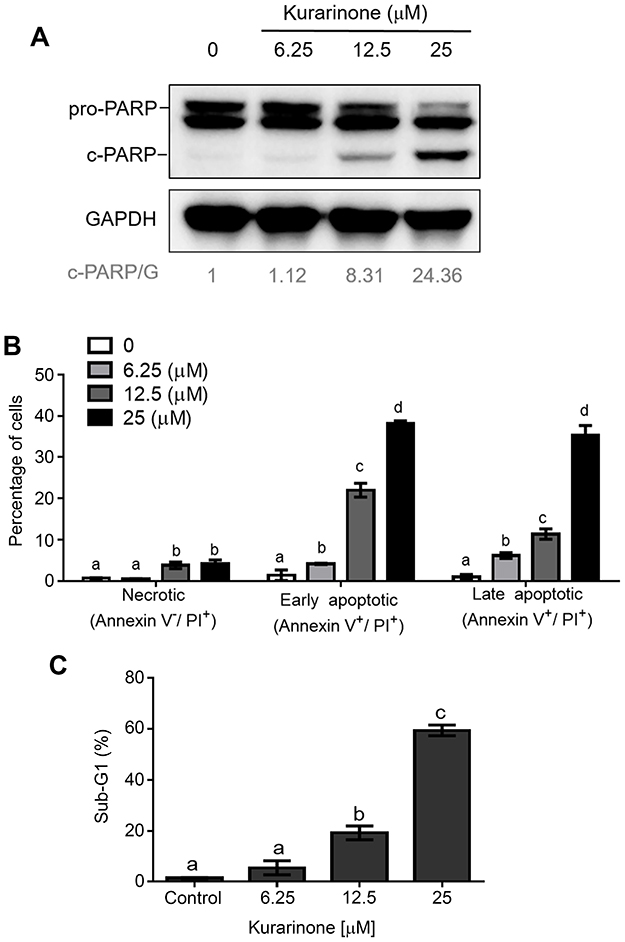 |
Figure 2 Apoptosis induced by kurarinone on H1688 cells. Cells were treated with or without kurarinone at indicated concentrations for 24 h. (A) Expression levels of cleaved poly (ADP-ribose) polymerase (PARP) were investigated by Western blotting using GAPDH as a loading control. (B) Cell apoptosis was determined via flow cytometry using the Annexin V-FITC apoptosis detection kit and PI. The percentage of necrotic (Annexin V−/PI+), early apoptotic (Annexin V+/PI−), and late apoptotic (Annexin V+/PI+) (mean ± SD) from triplicate samples of each treatment were plotted in the bar graphs. (C) The cell cycle was investigated using flow cytometry analysis via PI staining. The percentages of sub-G1 cells (mean ± SD) were plotted based on triplicated samples. Different letters (a,b,c,d) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
Kurarinone treatment altered mitochondrial membrane potentials and induced the intrinsic mitochondrial apoptotic pathway in H1688 cell
Mitochondria play a key role in activating apoptotic signaling via intrinsic and extrinsic apoptotic pathways, which subsequently converge to induce permeabilization of the mitochondrial membranes in mammalian cells. As such, the change of mitochondrial membrane potential and release of cytochrome c are regarded as mitochondrial apoptotic indications.20,21 To explore whether the mitochondria-mediated apoptotic pathway is involved in kurarinone-induced apoptosis, we first analyzed disruption or loss of the mitochondrial membrane permeability (Δψm) using JC-1 fluorescent cationic dye. JC-1 forms aggregates (red fluorescence) in the mitochondria of live cells, but these aggregates leak out from the damaged mitochondria into the cytosol as monomers (green fluorescence) in apoptotic cells. Our results in Figure 3A show that 24-h kurarinone treatments increased the ratio of green to red fluorescence, with a 21.1-fold increase at 12.5 μM and 31.0-fold increase at 25 μM. Also, we detected elevated cytochrome c in the cytosol of cells in a dose-dependent style upon kurarinone treatments (Figure 3B), indicating the disruption of the mitochondrial membrane within H1688 cells
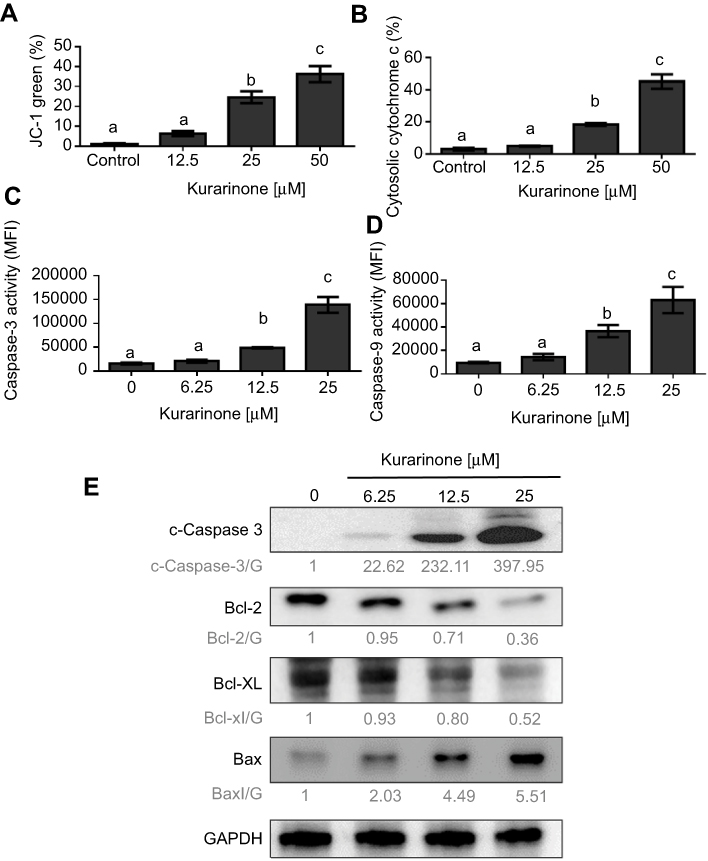 |
Figure 3 Changes on the mitochondrial membrane permeability (Δψm) and intrinsic mitochondrial apoptotic pathway in H1688 cells after kurarinone treatments. (A) Cells stained with the JC-1 probe for analyzing Δψm by flow cytometry. (B) Cytochrome c release was quantified with FITC-conjugated antibody via flow cytometry analysis. The activities of (C) caspase-3 and (D) caspase-9 were measured via flow cytometry. (E) Expression levels of cleaved caspase-3, Bcl-2, Bcl-XL, and Bax were determined by Western blot analysis along with loading control GAPDH Data were presented as mean ± SD from triplicate samples for each treatment and plotted as the bar graphs. Different letters (a,b,c) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
Mitochondrial dysfunction activates pro-caspase-9, which activates downstream caspase-3, leading to the cleavage of substrates such as PARP. Anti-apoptotic proteins, Bcl-2 and Bcl-xl, as well as pro‐apoptotic molecule, Bax, are also frequently involved in mitochondrial dysfunction.22 To explore the effect of kurarinone on the expression of these apoptotic proteins, H1688 cells were exposed to kurarinone for 24 h and the cells or cell lysates were subjected to flow cytometry or Western blot analysis, respectively. Kurarinone treatment significantly increased caspase-3 (Figure 3C and E) and caspase-9 activity (Figure 3D) in H1688 cells. Kurarinone treatment also resulted in the downregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xl and the upregulation of the apoptotic protein Bax (Figure 3E). Taken together, these results suggest that kurarinone induced apoptosis in SCLC cells via the intrinsic mitochondrial apoptotic pathway.
Kurarinone induced activation of the caspase-8/tBid pathway in H1688 cells
Once active, caspase-8 directly cleaves caspase-3 and indirectly cleaves Bid (23 kDa) into tBid, initiating the mitochondria-dependent apoptosis pathway.23,24 We investigated whether caspase-8 would be activated upon kurarinone treatment by treating H1688 cells with different doses of kurarinone for 24 h. Our results show that kurarinone significantly increased caspase-8 activity (Figure 4A) and increased the levels of cleaved caspase-8 and Bid (Figure 4B).
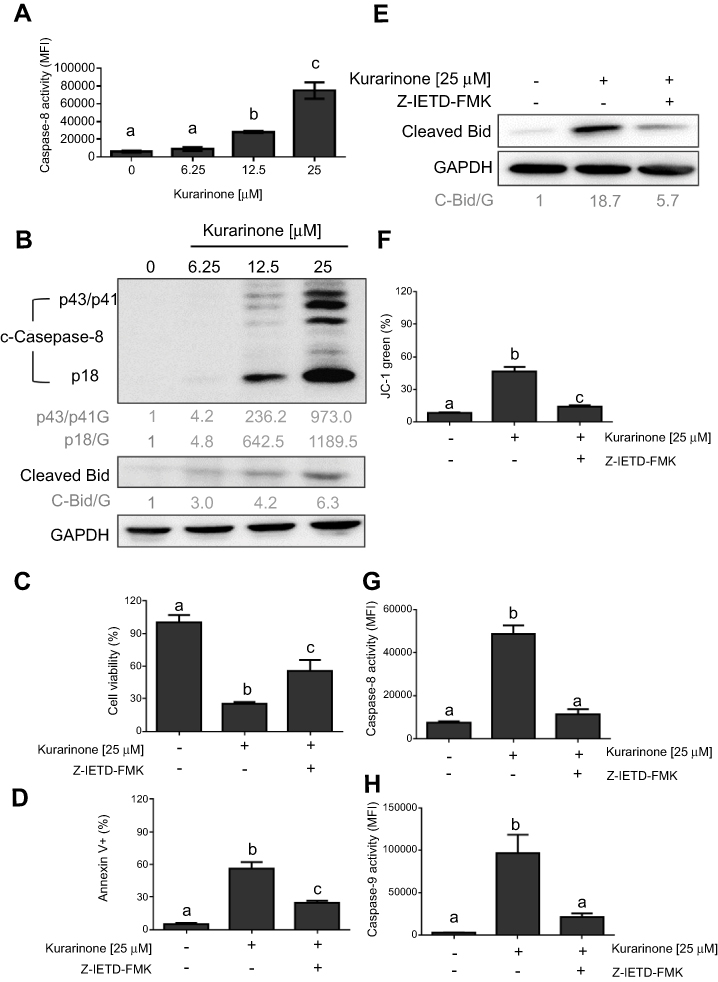 |
Figure 4 Activation of caspase-8/tBid pathway by kurarinone in H1688 cells. (A) caspase-8 activity measured by flow cytometry in the presence or absence of kurarinone. (B) Protein expression levels of cleaved caspase-8 (43/41 kDa and 18 kDa) and truncated Bid 50 μM of caspase-8 inhibitor Z-IETD-FMK was applied 1 h before 25 μM of kurarinone treatment for next 24 h and followed by measuring (C) cell viability of H1688 cells, (D) cell apoptosis, (E) protein expression levels of cleaved Bid, (F) mitochondrial membrane potential, and activities of (G) caspase-8, and (H) caspase-9. Bar graphs present the mean ± SD of 3 replicates for each treatment. Different letters (a,b,c) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
To confirm the role of caspase-8 in kurarinone-induced intrinsic mitochondrial apoptosis, we applied a selective caspase-8 inhibitor, Z-IETD-FMK, to H1688 cells in the presence or absence of kurarinone. Our results show that pretreatment with Z-IETD-FMK markedly increases cell viability (Figure 4C), inhibits cell apoptosis (Figure 4D), and prevents tBid formation (Figure 4E), Δψm loss (Figure 4F) and caspase-8 (Figure 4G), and caspase-9 activation (Figure 4H). Taken together, these data indicate that caspase-8 plays a critical role in kurarinone -induced apoptosis via the indirect activation of the mitochondrial pathway via cleaved Bid.
Since caspase-8 cleavage is induced through the stimulation of death receptors such as Fas (APO-1/CD95) and TRAIL receptor-1 and −2 (TRAIL-R1/DR4, TRAIL-R2/DR5) via their respective ligands,25 we examined the surface expression levels of these proteins using flow cytometry after a 24 h treatment with kurarinone. We observed an increase in the expression of DR4, DR5, and TRIAL (Figure 5A), as well as Fas and FasL (Figure 5B) after treatment with 12.5 and 25 μM kurarinone. These results suggest that kurarinone induces apoptosis by activating the death-receptor pathway.
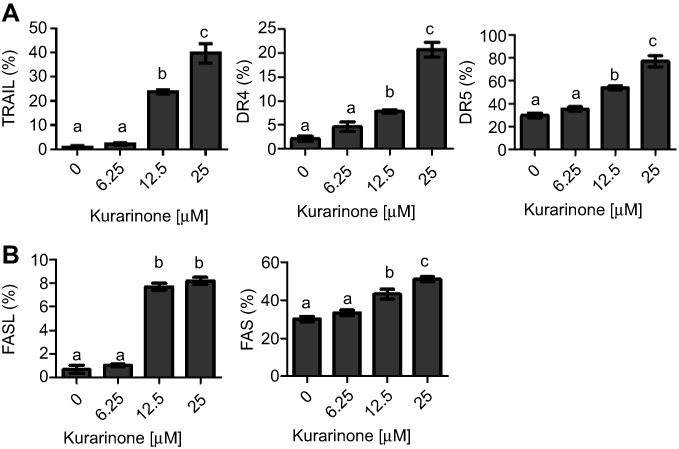 |
Figure 5 Death receptors were up-regulated in H1688 cells after kurarinone treatment. Flow cytometric analysis of (A) TRAIL, DR4, and DR5, (B) FAS and FASL expression after 24-h exposure to kurarinone. Bar graphs present the mean ± SD of 3 replicates for each treatment. Different letters (a,b,c) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
Kurarinone inhibited the migration and invasion of H1688 cells
Cell-matrix interactions and cell motility are important for cancer cell metastasis.26 Therefore, we evaluated the effects of kurarinone on H1688 cell migration and invasion using wound healing analysis and transwell assays. The results from the wound-healing assay indicated that kurarinone treatment for 24 h significantly inhibited the migration of H1688 cells in a dose-dependent manner (Figure 6A). Furthermore, the transwell migration (Figure 6B) and invasion (Figure 6C) assays showed that kurarinone treatment significantly and dose-dependently decreased the cell numbers on the transmembrane filters, proving that cell migration and invasion of H1688 cells were suppressed upon kurarinone treatment.
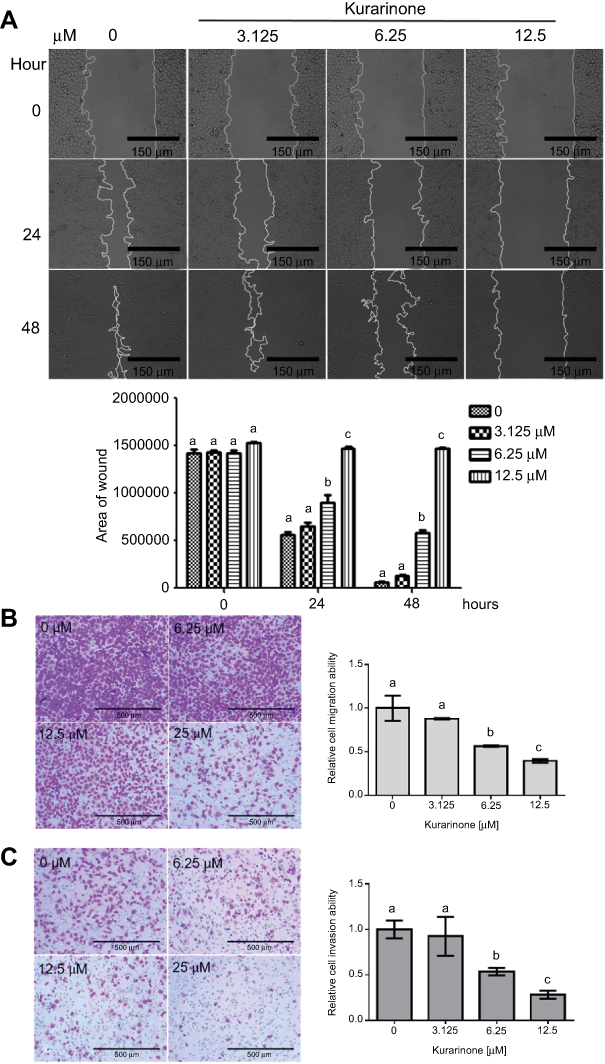 |
Figure 6 Cell migration and invasion of H1688 cells after 24-h treatment with kurarinone (3.125, 6.25, or 12.5 μM). Cell migratory capacity was detected by (A) wound scratch assays as described in Materials and Methods, presenting representative images at 100× magnification and (B) transwell assays. (C) Cell invasiveness of H1688 cells was also evaluated by transwell assays. The wound area (mean ± SEM) is shown from 3 independent experiments and bar graphs represents the mean ± SD of 3 replicates for each treatment. Different letters (a,b,c) indicate statistically significant differences between groups (one way ANOVA followed by Tukey test, p<0.05). |
Given the epithelial-mesenchymal-transition (EMT)-related proteins and matrix metalloproteinases (MMPs) were reported to link with the metastasis, irregular tumor growth, and tissue invasion of SCLC,27 we measured the expressions of the mesenchymal markers N-cadherin and vimentin, epithelial marker E-cadherin, and three MMPs in H1688 cells via Western blotting analysis. We found that N-cadherin and vimentin decreased whereas E-cadherin increased dose-dependently following kurarinone treatment (Figure 7A). In addition, kurarinone treatment resulted in a dose-dependent decrease in the expression of MMP-2, MMP-3, and MMP-9 (Figure 7B). These data suggest that kurarinone treatment repressed the metastatic properties of H1688 cells via reducing the migration and invasiveness.
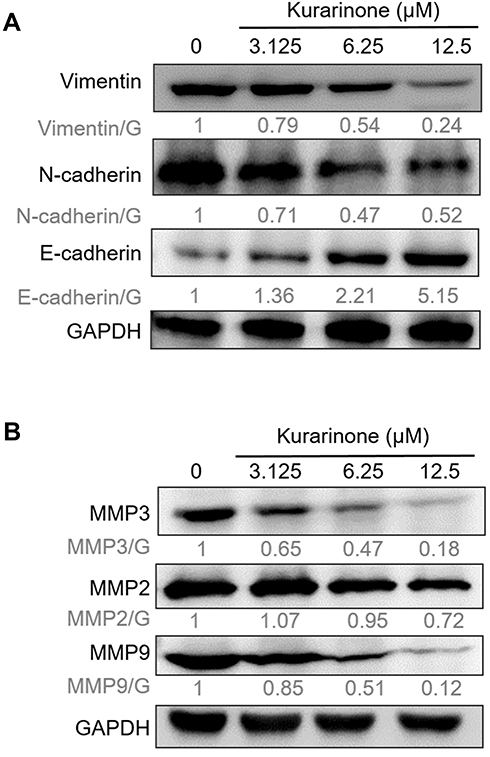 |
Figure 7 EMT-related proteins and MMPs in H1688 cells were modulated by kurarinone. Cells were treated with 0.1% DMSO or kurarinone (3.125, 6.25, or 12.5 μM) for 24 h followed by examining expression levels of (A) vimentin, N-cadherin, E-cadherin, and (B) MMP2, MMP-3, and MMP-9 via Western blotting along with GAPDH as a loading control. |
Discussion
Although the anticancer effects of kurarinone to other cancer types have been studied extensively, its effects and mechanisms of action are not clearly understood for SCLC. This study investigated the effects of kurarinone on human SCLC cell line, H1688, in terms of inhibition of tumor growth and explored its potential mechanisms. Consistent with previous research, kurarinone treatment inhibited the growth of H1688 cancer cells, inducing apoptosis via receptor and mitochondrial pathways, including induction of cleaved caspase-8, −3, Bid, PARP and reduction of Bax and Bcl-xl. Moreover, H1688 cancer cell migration and invasion were inhibited by kurarinone via suppressing the expressions of EMT-related proteins and MMPs. Thus, kurarinone may be an effective anti-cancer and anti-metastatic therapeutic strategy for human SCLC.
Currently, the concurrent chemoradiation (a two-drug combination of etoposide and cisplatin) remains the standard treatment for limited-stage SCLC.3 Other treatment options include cyclophosphamide, doxorubicin, and vincristine (the CAV regimen).28 For extensive-stage disease, the recommended treatment is chemotherapy with radiation for symptomatic relief.29 However, the treatment results for SCLC have remained stagnant over the decades and the side effects of chemotherapy, even at usual therapeutic doses, aggravate the patient conditions. Therefore, the kurarinone might serve as a less-harmful natural derivative in alternative combinational treatment of SCLC. The prenylated flavonone kurarinone has been investigated as a potential anticancer drug because of its anti-inflammatory, and antitumor activities.10,12,13 Although the mechanism of action of kurarinone against cancer cells is still unclear, it is thought that the apoptotic pathway exerted by flavonone is the major factor accounting for the tumor cell death, similar to many chemotherapy regimens.30 One previous study reported by Yang et al indicated that kurarinone has proapoptotic activities where kurarinone was shown to induce apoptosis in NSCLC A549 cells via the mitochondria-dependent pathway.14 Congruent with the previous study, we found that kurarinone increased the expressions of cleaved caspase-3, caspase-9 and cytochrome c (Figure 3B–D), as well as the caspase-3-mediated PARP cleavage. The mitochondrial membrane pro-apoptotic protein Bak was increased and anti-apoptotic proteins, Bcl-xL and BCl-2, were decreased following kurarinone treatments (Figure 3E), arguing the apoptosis of human SCLC cells induced by kurarinone is at least a caspase-dependent and mitochondrial-mediated intrinsic apoptotic pathway.
However, our findings differ from the NSCLC A549 study in reference to the receptor-mediated apoptosis pathway, which they did not investigate. Death receptors such as Fas ligand (FASL) and TNF-related apoptosis-inducing ligand (TRAIL) are known as apoptosis-inducing ligands that stimulate death receptors. We observed in the current study that kurarinone significantly increased caspase-8 and cleaved Bid activity or expression, (Figure 4) in addition to the up-regulation of FAS, FASL, TRAIL, DR4, and DR5 (Figure 5). All of which points to that kurarinone could not only activate the death receptor-mediated apoptosis (the extrinsic pathway) but also induce mitochondrial-dependent apoptotic machinery (the intrinsic pathway), contributing to the inhibition of tumorigenicity of H1688 cells as summarized in Figure 8. Of note, Bid, a member of the Bcl-2 family, is considered as an adaptor that connects the death receptor pathway and the mitochondrial pathway as activated Bid will be cleaved by caspase-8 or −10 and turn into its truncated form (tBid), which translocates into mitochondria, facilitating the subsequent mitochondrial pathway.25,31,32 With multiple pathways of apoptosis implemented by kurarinone in SCLC cells, we believe that kurarinone might be more effective in treating SCLC than treating NSCLC.
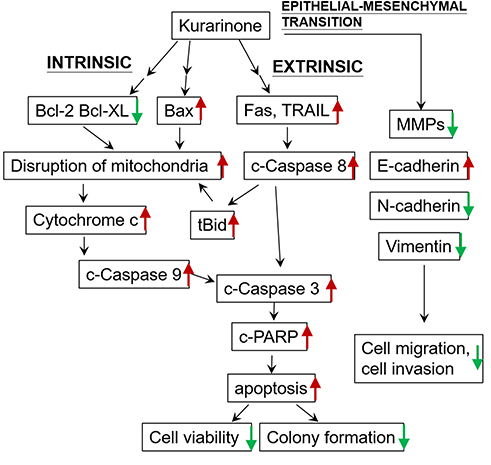 |
Figure 8 Schematic overview of cancer inhibitory pathways initiated by kurarinone. The cleaved form of a protein is labeled with a lower case c ahead of the protein name and a t indicates a truncated form. Red arrows indicate up-regulation or increase whereas green arrows signify down-regulation or decrease. |
Metastasis remains the major cause leading to the failure of SCLC treatment. Previous studies report that the EMT, which alters cell to cell adhesion, plays an important role in cancer metastasis.33,34 As a result, the EMT might also be associated with molecular mechanisms in SCLC spread and metastasis, possibly contributing to its poor prognosis. To this end, chemicals that possess anti-metastasis activities, such as targeting the EMT, are attractive candidates for treating metastatic SCLC. Our results elucidate that kurarinone modulated the expressions of cell adhesion proteins and reduced the migratory capacity. On the other hand, the invasiveness is also an essential characteristic of metastatic cancer cells. Invasive metastatic cells manifest the secretion of MMPs to traverse biological barriers.35 It has also been demonstrated in tumor cell models that the metastatic potential and tumor cell growth depends on the levels of MMP.36 We demonstrated that kurarinone significantly decreased the expressions of cancer-associated MMPs (MMP-2, MMP-3, and MMP-9), functioning in the tumor invasion process of SCLC cells (Figure 7). Therefore, it is strongly suggested that kurarinone may serve as a promising anti-metastasis agent for SCLC therapy.
Given the 5-year survival rate of SCLC patients in 2015 was only 6.5%, according to the data from National Cancer Institute, compared to 24.6% for NSCLC, and nearly 60% of SCLC patients had distant metastasis when diagnosed,37 it is urgent to have multiple therapeutic strategies available for SCLC patients, especially those whose distal tissues have been invaded by SCLC. As a result, our study is aimed at not only to demonstrate the feasibility of kurarinone to induce intrinsic and extrinsic apoptosis on SCLC cells but also provide a perspective in utilizing flavanone for metastatic SCLC cells. Future studies will certainly be needed for evaluation of the efficacy of kurarinone in SCLC patients and we believe that our findings have just gradually furthered the potential development toward the clinical use.
Acknowledgments
We greatly appreciate Nicole Bracci at George Mason University for reviewing and providing suggestions to improve this manuscript. This study was financially supported by the iEGG and Animal Biotechnology Center from The Feature Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE-107-S-0023-E) in Taiwan.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi:10.3322/caac.21442
2. Caprario LC, Kent DM, Strauss GM. Effects of chemotherapy on survival of elderly patients with small-cell lung cancer: analysis of the SEER-medicare database. J Thorac Oncol. 2013;8(10):1272–1281. doi:10.1097/JTO.0b013e3182a007ba
3. Watkins JM, Wahlquist AE, Zauls AJ, et al. Involved-field radiotherapy with concurrent chemotherapy for limited-stage small-cell lung cancer: disease control, patterns of failure and survival. J Med Imaging Radiat Oncol. 2010;54(5):483–489. doi:10.1111/j.1754-9485.2010.02201.x
4. Weckler BC, Baldes N, Schirren J. Survival following multimodality treatment including surgery for stage IA–IIIB small-cell lung cancer. Thorac Cardiovasc Surg. 2019;67(4):291–298. doi:10.1055/s-0038-1667145
5. Zhu H, Guo H, Li M, et al. Increased serum carcinoembryonic antigen level can predict poor survival of patients with small cell lung cancer. Transl Res. 2015;166(4):355–365. doi:10.1016/j.trsl.2015.04.005
6. Hatfield LA, Huskamp HA, Lamont EB. Survival and toxicity after cisplatin plus etoposide versus carboplatin plus etoposide for extensive-stage small-cell lung cancer in elderly patients. J Oncol Pract. 2016;12(7):666–673. doi:10.1200/JOP.2016.012492
7. Singh S, Parulekar W, Murray N, et al. Influence of sex on toxicity and treatment outcome in small-cell lung cancer. J Clin Oncol. 2005;23(4):850–856. doi:10.1200/JCO.2005.03.171
8. De Naeyer A, Vanden Berghe W, Pocock V, Milligan S, Haegeman G, De Keukeleire D. Estrogenic and anticarcinogenic properties of kurarinone, a lavandulyl flavanone from the roots of Sophora flavescens. J Nat Prod. 2004;67(11):1829–1832. doi:10.1021/np040069a
9. Chen L, Cheng X, Shi W, et al. Inhibition of growth of Streptococcus mutans, methicillin-resistant Staphylococcus aureus, and vancomycin-resistant enterococci by kurarinone, a bioactive flavonoid isolated from Sophora flavescens. J Clin Microbiol. 2005;43(7):3574–3575. doi:10.1128/JCM.43.7.3574-3575.2005
10. Kim BH, Na KM, Oh I, et al. Kurarinone regulates immune responses through regulation of the JAK/STAT and TCR-mediated signaling pathways. Biochem Pharmacol. 2013;85(8):1134–1144. doi:10.1016/j.bcp.2013.01.005
11. Pan ZS, Yu QH, Yan H, Zhang Y. [Clinical study on treatment of chronic hepatitis B by kurarinone combined with interferon alpha-1b]. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2005;25(8):700–703.
12. Berghe WV, De Naeyer A, Dijsselbloem N, David JP, De Keukeleire D, Haegeman G. Attenuation of ERK/RSK2-driven NFkappaB gene expression and cancer cell proliferation by kurarinone, a lavandulyl flavanone isolated from Sophora flavescens ait. roots. Endocr Metab Immune Disord Drug Targets. 2011;11(3):247–261.
13. Zhou W, Cao A, Wang L, Wu D. Kurarinone synergizes TRAIL-induced apoptosis in gastric cancer cells. Cell Biochem Biophys. 2015;72(1):241–249. doi:10.1007/s12013-014-0444-0
14. Yang J, Chen H, Wang Q, et al. Inhibitory effect of kurarinone on growth of human non-small cell lung cancer: an experimental study both in vitro and in vivo studies. Front Pharmacol. 2018;9:252. doi:10.3389/fphar.2018.00252
15. Xie L, Gong W, Chen J, et al. The flavonoid kurarinone inhibits clinical progression of EAE through inhibiting Th1 and Th17 cell differentiation and proliferation. Int Immunopharmacol. 2018;62:227–236.
16. Campos CB, Paim BA, Cosso RG, Castilho RF, Rottenberg H, Vercesi AE. Method for monitoring of mitochondrial cytochrome c release during cell death: immunodetection of cytochrome c by flow cytometry after selective permeabilization of the plasma membrane. Cytometry A. 2006;69(6):515–523. doi:10.1002/cyto.a.20273
17. Feng Y, Xu X, Zhang Y, et al. HPIP is upregulated in colorectal cancer and regulates colorectal cancer cell proliferation, apoptosis and invasion. Sci Rep. 2015;5:9429. doi:10.1038/srep09429
18. Schreiber V, Hunting D, Trucco C, et al. A dominant-negative mutant of human poly(ADP-ribose) polymerase affects cell recovery, apoptosis, and sister chromatid exchange following DNA damage. Proc Natl Acad Sci U S A. 1995;92(11):4753–4757. doi:10.1073/pnas.92.11.4753
19. Zhang Y, Li D, Jiang Q, et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018;9(7):743. doi:10.1038/s41419-018-1111-y
20. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116(2):205–219. doi:10.1016/s0092-8674(04)00046-7
21. Wang L, Ma G, Zhang Y, et al. Effect of mitochondrial cytochrome c release and its redox state on the mitochondrial-dependent apoptotic cascade reaction and tenderization of yak meat during postmortem aging. Food Res Int. 2018;111:488–497. doi:10.1016/j.foodres.2018.05.049
22. Um HD. Bcl-2 family proteins as regulators of cancer cell invasion and metastasis: a review focusing on mitochondrial respiration and reactive oxygen species. Oncotarget. 2016;7(5):5193–5203. doi:10.18632/oncotarget.6405
23. Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. doi:10.1016/s0092-8674(00)81590-1
24. Nam YJ, Kim A, Lee MS, Shin YK, Sohn DS, Lee CS. Lamotrigine attenuates proteasome inhibition-induced apoptosis by suppressing the activation of the mitochondrial pathway and the caspase-8- and bid-dependent pathways. Neurochem Res. 2016;41(10):2503–2516. doi:10.1007/s11064-016-1962-5
25. Kim WS, Lee KS, Kim JH, et al. The caspase-8/Bid/cytochrome c axis links signals from death receptors to mitochondrial reactive oxygen species production. Free Radic Biol Med. 2017;112:567–577. doi:10.1016/j.freeradbiomed.2017.09.001
26. Gkretsi V, Stylianopoulos T. Cell adhesion and matrix stiffness: coordinating cancer cell invasion and metastasis. Front Oncol. 2018;8:145. doi:10.3389/fonc.2018.00145
27. Ito T, Kudoh S, Ichimura T, Fujino K, Hassan WA, Udaka N. Small cell lung cancer, an epithelial to mesenchymal transition (EMT)-like cancer: significance of inactive Notch signaling and expression of achaete-scute complex homologue 1. Hum Cell. 2017;30(1):1–10. doi:10.1007/s13577-016-0149-3
28. Johnson BE, Bridges JD, Sobczeck M, et al. Patients with limited-stage small-cell lung cancer treated with concurrent twice-daily chest radiotherapy and etoposide/cisplatin followed by cyclophosphamide, doxorubicin, and vincristine. J Clin Oncol. 1996;14(3):806–813. doi:10.1200/JCO.1996.14.3.806
29. Foster NR, Renfro LA, Schild SE, et al. Multitrial evaluation of progression-free survival as a surrogate end point for overall survival in first-line extensive-stage small-cell lung cancer. J Thorac Oncol. 2015;10(7):1099–1106. doi:10.1097/JTO.0000000000000548
30. Aleo E, Henderson CJ, Fontanini A, Solazzo B, Brancolini C. Identification of new compounds that trigger apoptosome-independent caspase activation and apoptosis. Cancer Res. 2006;66(18):9235–9244. doi:10.1158/0008-5472.CAN-06-0702
31. Kohler B, Anguissola S, Concannon CG, Rehm M, Kogel D, Prehn JH. Bid participates in genotoxic drug-induced apoptosis of HeLa cells and is essential for death receptor ligands’ apoptotic and synergistic effects. PLoS One. 2008;3(7):e2844. doi:10.1371/journal.pone.0002844
32. Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10(3):161–167. doi:10.1038/sj.cr.7290045
33. Vincent CT, Fuxe J. EMT, inflammation and metastasis. Semin Cancer Biol. 2017;47:168–169. doi:10.1016/j.semcancer.2017.09.003
34. Joseph JP, Harishankar MK, Pillai AA, Devi A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018;80:23–32. doi:10.1016/j.oraloncology.2018.03.004
35. Welch DR. Tumor cell invasion-not all barriers are created equal. Cancer Res. 2016;76(7):1675–1676. doi:10.1158/0008-5472.CAN-16-0550
36. Deryugina EI, Quigley JP. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix Biol. 2015;44–46:94–112. doi:10.1016/j.matbio.2015.04.004
37. Nakazawa K, Kurishima K, Tamura T, et al. Specific organ metastases and survival in small cell lung cancer. Oncol Lett. 2012;4(4):617–620. doi:10.3892/ol.2012.792
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.