")
Back to Journals » OncoTargets and Therapy » Volume 7
Antitumor activity of sorafenib and imatinib in a patient with thymic carcinoma harboring c-KIT exon 13 missense mutation K642E
Authors Catania C , Conforti F, Spitaleri G, Barberis M, Preda L, Noberasco C, Lazzari C, Toffalorio F, de Marinis F , Manzotti M, De Pas T
Received 9 December 2013
Accepted for publication 14 February 2014
Published 8 May 2014 Volume 2014:7 Pages 697—702
DOI https://doi.org/10.2147/OTT.S58902
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Chiara Catania,1 Fabio Conforti,1 Gianluca Spitaleri,1 Massimo Barberis,2 Lorenzo Preda,3 Cristina Noberasco,1 Chiara Lazzari,1 Francesca Toffalorio,1 Filippo de Marinis,1 Michela Manzotti,2 Tommaso Martino De Pas1
1Division of Thoracic Oncology, 2Division of Pathology, 3Division of Radiology, European Institute of Oncology, Milan, Italy
Abstract: We report the case of a man with an advanced nonkeratinizing squamous cell thymic carcinoma harboring c-KIT exon 13 missense mutation K642E. This aberration is rare and has never been described previously in patients with thymic cancers. It has been found in a small number of cases of gastrointestinal stromal tumor and also in several cases of acral and mucosal melanomas. Some of the patients with gastrointestinal stromal tumor or melanoma harboring this rare mutation have had a tumor response when treated with imatinib. In contrast, in our case, the mutation was associated with primary resistance to full doses of imatinib but, at the same time, it was not a cause of resistance to sorafenib.
Keywords: sorafenib, imatinib, thymic carcinoma
Case report
A 58-year-old man without relevant comorbidities was diagnosed in July 2009 with a thymic carcinoma and multiple synchronous hepatic metastases by total body computed tomography (CT) and total body fluorodeoxyglucose positron emission tomography. The histological diagnosis was made from a liver biopsy, which showed proliferation of epithelial spindle cells arranged in bundles. The nuclei were cigar-shaped with granular chromatin. There was no necrosis and the mitotic rate was two mitoses per ten high-power fields. The cells showed immunostaining for cytokeratins AE1–AE3 and cytokeratins 5/6, transformation-related protein 63, and tyrosine-protein kinase Kit, but not for thyroid transcription factor, Wilms tumor 1, cluster of differentiation 1a, terminal deoxynucleotidyltransferase-positive cells, CD5, chromogranin A, or synaptophysin. A diagnosis was made of poorly differentiated nonkeratinizing squamous cell carcinoma with a prevalent low-grade spindle cell pattern consistent with metastatic thymic carcinoma (Figure 1A and B).
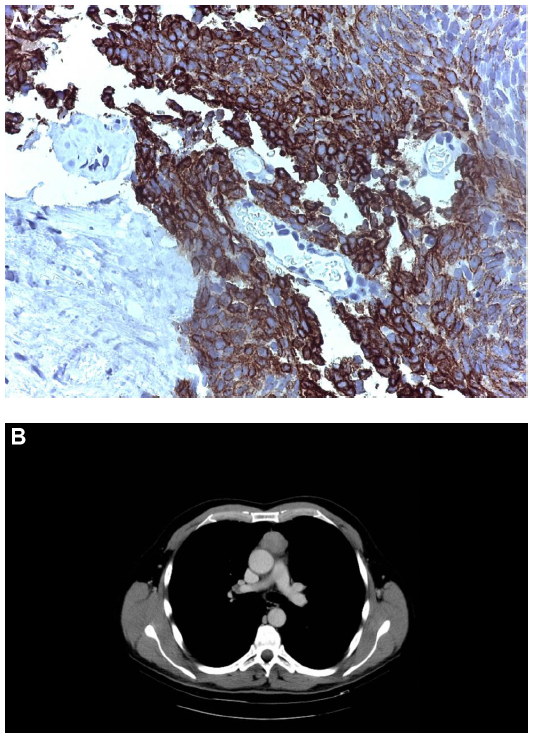 | Figure 1 Histological and radiological diagnosis. |
In accordance with the disease stage, the patient was treated with three lines of chemotherapy from July 2009 to July 2010. The first-line chemotherapy, to which the patient had a partial response, was ifosfamide 5 g/m2 on day 1, carboplatin 300 mg/m2 on day 1, and etoposide 120 mg/m2 on days 1–3 of a 3-week schedule. The second-line chemotherapy, which also produced a partial response, was paclitaxel 100 mg/m2 on days 1 and 8 and gemcitabine 1,500 mg/m2 on days 1 and 8 every 3 weeks. The third-line chemotherapy consisted of epirubicin 30 mg/m2 and ifosfamide 3 g/m2 on days 1–3 of a 3-week schedule, and was followed by liver and lung progression.
In July 2010, targeted therapy with sorafenib was started at a daily dose of 800 mg/day. A partial response was obtained, consisting, according to Response Evaluation Criteria In Solid Tumors (RECIST) criteria, of a 30% reduction of the sum of the maximal diameters of lesions in all sites of disease.1 In detail, the two pulmonary lesions showed a complete response, while in the other sites of disease, along with the dimensional reduction, a significant reduction of CT scan attenuation (Hounsfield) units was seen.2 This was probably related to intralesional devascularization with associated necrosis in response to the targeted therapy (Figure 2A–D). Disease progression occurred after 6 months in February 2011 and was limited to the liver.
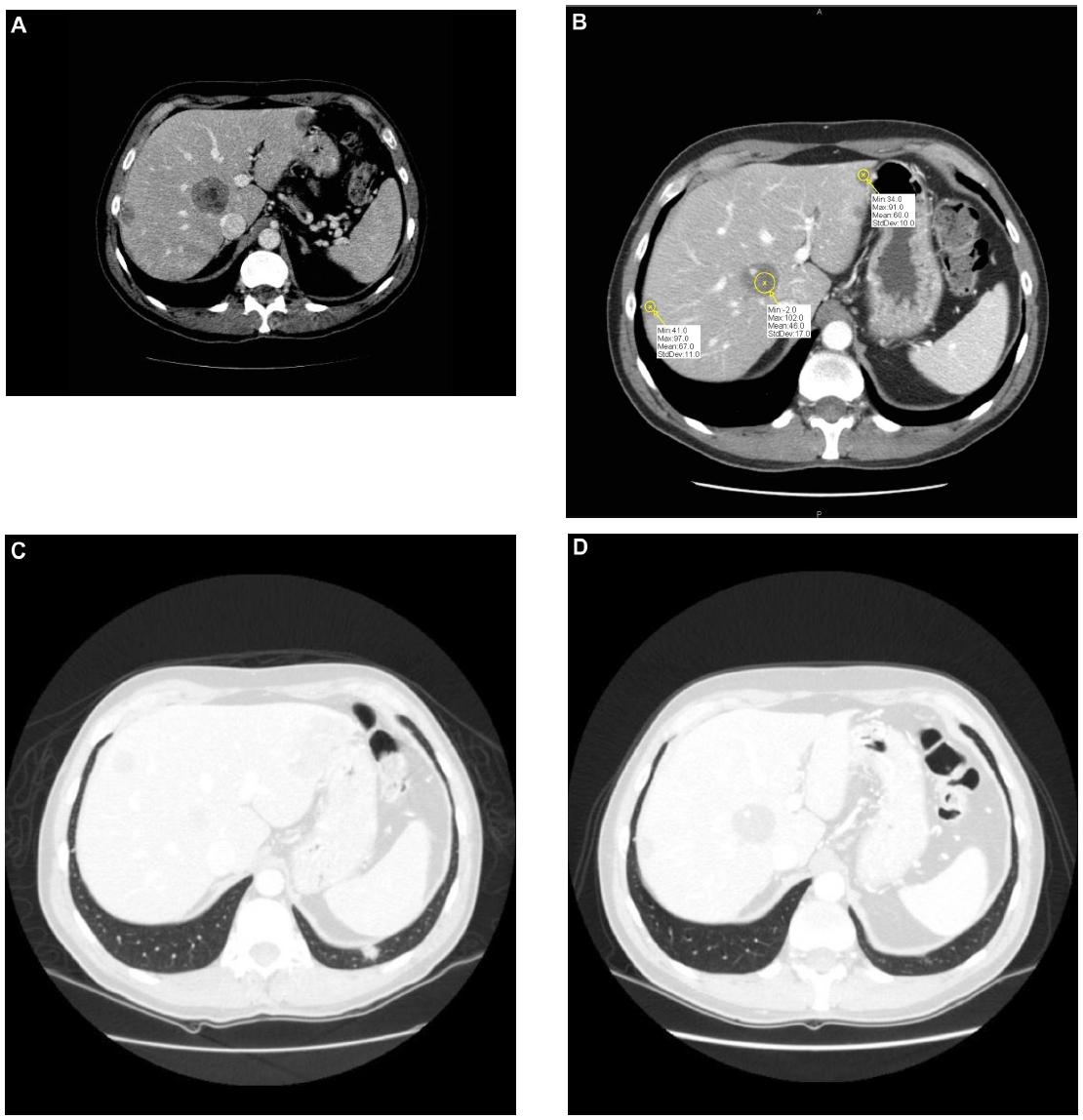 | Figure 2 Liver and lung respose to sorafenib. |
In March 2011, the patient participated in a Phase I clinical trial and received experimental treatment with a polo-like kinase inhibitor given in combination with an antiangiogenic agent. Polo-like kinases are a family of serine/threonine kinases with a highly conserved N-terminal Ser/Thr kinase catalytic domain and a C-terminal region that play crucial roles in cell cycle progression. Most polo-like kinase inhibitors competitively bind to the adenosine triphosphate binding site, which has unique features. The patient received this treatment because he had requested an treatment and because of the possible activity of an antiangiogenic drug in thymic carcinoma.
After the failure of this treatment, manifested by progressive liver disease, he was treated in June 2011 by three cycles of intra-arterial polychemotherapy with mitomycin 1 mg/m2 and 5-fluoruracil 1,000 mg/m2 by continuous infusion over 24 hours and cisplatin 10 mg/m2 on days 1–3 of a 6-week schedule, with a major but short-lasting tumor response followed by development of new hepatic lesions. A tumor specimen taken by liver biopsy at the time of diagnosis was then subjected to molecular analysis with polymerase chain reaction-based direct sequencing, and a missense mutation in v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (c-KIT) exon 13 (K642E) was found (Figure 3).
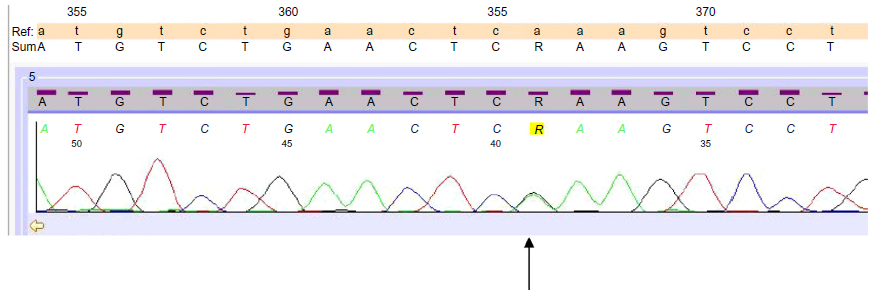 | Figure 3 Mutation p.K642E (cod A/Gaa). |
On the basis of this finding, in November 2011, the patient started treatment with imatinib 800 mg/day. A CT scan performed after 6 weeks of treatment showed many tumor metastases of increased size but with areas of reduced contrast enhancement. Because these radiological findings had also been present in the baseline CT scan, making the whole radiological evaluation difficult to interpret, administration of imatinib was continued for another 6 weeks. During this period the patient’s clinical condition deteriorated, he had abdominal pain and weight loss, and the subsequent CT scan showed clear progression at all sites of disease.
There was a significant increase in both number and size of the secondary lesions, particularly within the liver, with increasingly evident solid portions showing uptake of contrast. Administration of imatinib was interrupted in January 2012. The patient died in July 2012.
Discussion
Thymic carcinomas are rare tumors, representing about 1% of all thymic malignancies.3 They are highly aggressive diseases, for which limited therapeutic options are available.4 The poor prognosis of these tumors highlights the importance of finding new therapeutic strategies able to target molecules involved in the pathogenesis of thymic carcinoma. One of the most interesting targets is c-KIT, a tyrosine kinase transmembrane receptor that transduces signals promoting cell growth, proliferation, and survival, and is overexpressed in a high percentage of thymic carcinomas (46%–86%), according to different studies.5–7
C-KIT acts through the Rat sarcoma/mitogen-activated protein kinase, phosphatidylinositol-3 kinases/AKT, and Janus kinase/signal transducers and activators of transcription pathways.7 This tyrosine kinase is targeted by many drugs, including imatinib, sunitinib, and sorafenib, but these agents also have many other mechanisms of action, such as inhibition of vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), Raf, Fms-like tyrosine kinase-3 receptor, the receptor encoded by the ret proto-oncogene and Abelson kinase (c-ABL).8–10
However, despite the high prevalence of c-KIT overexpression, three different trials testing the activity of imatinib in unselected patients with thymoma or thymic carcinoma failed to demonstrate a possible therapeutic role for this drug. The failure of these trials is a demonstration that, as in gastrointestinal stromal tumor (GIST), the mere overexpression of c-KIT in patients with thymic cancers is not predictive of a response to imatinib. Moreover, it leads to the hypothesis that the absence of antitumor activity demonstrated in these studies could have been due to poor selection of patients. Indeed, the most important factors predicting a response to imatinib seem to be c-KIT and PDGFR mutations, and none of the patients enrolled in these trials had mutations in the c-KIT gene.11–14
Mutations of c-KIT have been extensively studied in GIST, in which it has been clearly proven that the mutations are highly heterogeneous. Some of these mutations appear to be associated with response to imatinib, while others are associated with resistance.15–17 Consistent with these findings, a few case reports on the effect of imatinib in patients with c-KIT-mutated thymic cancers have described favorable results: stable disease lasting 6 months was described in one patient with thymic carcinoma and a V560 deletion in exon 11 and a partial response lasting for 8 months in another with a missense mutation Y553N in exon 11.18,19
Unfortunately, c-KIT mutations are present in only a small proportion of thymic carcinomas. Among 109 cases examined in six different studies, nine had a mutation (8%), comprising six in exon 11, one in exon 9, one in exon 14, and one in exon 17.5,6,20–23 In all of these cases, tumors were poorly differentiated squamous cell carcinoma without keratinization.23
Unlike the situation for imatinib, correlations between specific c-KIT mutations and response to sorafenib and sunitinib are still poorly known. These drugs inhibit c-KIT, and interestingly, they have also been demonstrated to be active in GIST harboring mutations predictive of resistance to imatinib, such as mutations in exons 13 and 14.24 Two cases of c-KIT-mutated thymic cancers responsive to sorafenib have been reported: in one case, a patient with the missense mutation D820E in exon 17 had a partial response lasting more than 15 months, while in the other case, stable disease was achieved in the presence of exon 11 deletion.25,26
Besides inhibiting c-KIT, sunitinib and sorafenib also inhibit VEGFR and PDGFR, which are involved in angiogenesis and might play important roles in the pathogenesis of thymic cancers. In fact, it has been shown that VEGF-A, VEGFR-1, and VEGFR-2 are overexpressed in thymic cancers and that the levels of VEGF expression correlate with tumor invasion and clinical stage.27–29
Their multiple activities make these drugs an interesting therapeutic option also in the subgroup of thymic cancers without mutations of c-KIT, which are actually the majority of cases. Consistent with this, Ströbel et al reported disease response in all four cases of wild-type c-KIT thymic cancer treated with sunitinib.30
In this paper, we have described the case of a patient affected by thymic carcinoma harboring the missense mutation K642E in exon 13 of the c-KIT gene who was treated with both sorafenib and imatinib. This mutation causes substitution of a positively charged lysine side chain with a negatively charged glutamic acid chain in the autoinhibitory c-KIT domain, leading to constitutive and ligand-independent activation of the molecule.31
This aberration is rare and has never been described previously in patients with thymic cancers. It has been found in a small number of cases of GIST, including two germinal mutations determining a familial GIST syndrome, and also in several cases of acral and mucosal melanomas. Some of the patients with GIST or melanoma harboring this rare mutation had a tumor response when treated with imatinib.14–16,32–36
In contrast, in our case, the mutation was associated with primary resistance to full doses of imatinib but, at the same time, was not a cause of resistance to sorafenib. As a consequence, although the treatment of patients according to druggable target mutations can usually be translated over different types of tumors, our experience suggests that data collected in specific tumor types might not always be applicable to others.
A similar conclusion could be drawn from other experiences: for example, small molecule BRAF inhibitors showed promising results in BRAF-mutant metastatic melanoma, while they have been less effective in BRAF-mutant bowel adenocarcinoma.37,38 It should be noted that only a limited region of our patient’s tumor was resequenced, so different alterations could have been present in areas not investigated.
Molecular screening for c-KIT mutations in thymic carcinoma may be a useful tool to identify patients who could be successfully treated with KIT inhibitors. In conclusion, this report suggests that patients with thymic carcinoma harboring the missense mutation K624E in exon 13 of the c-KIT gene can be resistant to imatinib and may obtain a tumor response when treated with sorafenib, possibly related to the antiangiogenic activity of imatinib.
Author contributions
This study was conceived and executed by all the authors listed.
Disclosure
None of the authors nor the European Institute of Oncology received any payment or support in kind for any aspect of the submitted work.
References
Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. J Natl Cancer Inst. 2000;92:205–216. | |
Choi H, Charnsangavej C, Faria SC, et al. Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol. 2007;25:1753–1759. | |
Kuhn E, Wistuba II. Molecular pathology of thymic epithelial neoplasm. Hematol Oncol Clin North Am. 2008;22:443–445. | |
Loehrer PJ Sr, Kim K, Aisner SC, et al. Cisplatin plus doxorubicin plus cyclophosphamide in metastatic or recurrent thymoma: final result of an Intergroup trial – the Eastern Cooperative Oncology Group, Southwest Oncology Group, and Southeastern Cancer Study Group. J Clin Oncol.1994;12:1164–1168. | |
Petrini I, Zucali PA, Lee HS, et al. Expression and mutational status of c-kit in thymic epithelial tumors. J Thorac Oncol. 2010;5:1447–1453. | |
Pan CC, Chen PC, Chiang H. KIT (CD117) is frequently overexpressed in thymic carcinomas but is absent in thymomas. J Pathol. 2004;202:375–381. | |
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–878. | |
Chow LQ, Eckhardt SG. Sunitinib: from rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. | |
Rini BI. Sorafenib. Expert Opin Pharmacother. 2006;7:453–461. | |
Radford IR. Imatinib. Novartis. Curr Opin Investig Drugs. 2002;3:492–499. | |
Salter JT, Lewis D, Yiannoutsos C, Loehrer PJ Sr, Risley L, Chiorean EG. Imatinib for the treatment of thymic carcinomas. J Clin Oncol. 2008; 26 Suppl:Abstr 8116. | |
Giaccone G, Rajan A, Ruijter R, et al. Imatinib mesylate in patients with WHO B3 thymomas and thymic carcinomas. J Thorac Oncol. 2009;4:1270–1273. | |
Palmieri G, Marino M, Buonerba C, et al. Imatinib mesylate in thymic epithelial malignancies. Cancer Chemother Pharmacol. 2012;69:309–315. | |
Heinrich MC, Corless CL, Demetri GD, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–4349. | |
Heinrich MC, Corless CL, Blanke CD, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–4774. | |
Debiec-Rychter M, Sciot R, Le Cesne A, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–1103. | |
Gao J, Tian Y, Li J, et al. Secondary mutations of c-KIT contribute to acquired resistance to imatinib and decrease efficacy of sunitinib in Chinese patients with gastrointestinal stromal tumors. Med Oncol. 2013;30:522. | |
Ströbel P, Hartmann M, Jakob A, et al. TC with overexpression of mutated KIT and the response to imatinib. N Engl J Med. 2004;350:2625–2626. | |
Buti S, Donini M, Sergio P, et al. Impressive response with imatinib in a heavily pretreated patient with metastatic c-KIT mutated TC. J Clin Oncol. 2011;29:803–805. | |
Tsuchida M, Umezu H, Hashimoto T, et al. Absence of gene mutations in KIT-positive thymic epithelial tumors. Lung Cancer. 2008;62:321–325. | |
Yoh K, Nishiwaki Y, Ishii G, et al. Mutational status of EGFR and KIT in thymoma and thymic carcinoma. Lung Cancer. 2008;62:316–320. | |
Girard N, Shen R, Guo T, et al. Comprehensive genomic analysis reveals clinically relevant molecular distinctions between thymic carcinomas and thymomas. Clin Cancer Res. 2009;15:6790–6799. | |
Schirosi L, Nannini N, Nicoli D, et al. Activating c-KIT mutations in a subset of thymic carcinoma and response to different c-KIT inhibitors. Ann Oncol. 2012;23:2409–2414. | |
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–5359. | |
Bisagni G, Rossi G, Cavazza A, et al. Long lasting response to the multikinase inhibitor bay 43-9006 (sorafenib) in a heavily pretreated metastatic thymic carcinoma. J Thorac Oncol. 2009;4:773–775. | |
Dişel U, Oztuzcu S, Beşen AA, et al. Promising efficacy of sorafenib in a relapsed thymic carcinoma with C-KIT exon 11 deletion mutation. Lung Cancer. 2011;71:109–112. | |
Cimpean AM, Raica M, Encica S, et al. Immunohistochemical expression of vascular endothelial growth factor A (VEGF), and its receptors (VEGFR1, 2) in normal and pathologic conditions of the human thymus. Ann Anat. 2008;190:238–245. | |
Sasaki H, Yukiue H, Kobayashi Y, et al. Elevated serum vascular endothelial growth factor and basic fibroblast growth factor levels in patients with thymic epithelial neoplasms. Surg Today. 2001;31:1038–1040. | |
Tomita M, Matsuzaki Y, Edagawa M, et al. Correlation between tumor angiogenesis and invasiveness in thymic epithelial tumors. J Thorac Cardiovasc Surg. 2002;124:493–498. | |
Ströbel P, Bargou R, Wolff A, et al. Sunitinib in metastatic thymic carcinomas: laboratory findings and initial clinical experience. Br J Cancer. 2010;103:196–200. | |
Tarn C, Merkel E, Canutescu AA, et al. Analysis of KIT mutations in sporadic and familial gastrointestinal stromal tumors: therapeutic implications through protein modeling. Clin Cancer Res. 2005;11:3668–3677. | |
Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–4346. | |
Rivera RS, Nagatsuka H, Gunduz M, et al. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Arch. 2008;452:27–32. | |
Isozaki K, Terris B, Belghiti J, et al. Germline-activating mutation in the kinase domain of KIT gene in familial gastrointestinal stromal tumors. Am J Pathol. 2000;157:1581–1585. | |
Graham J, Debiec-Rychter M, Corless CL, et al. Imatinib in the management of multiple gastrointestinal stromal tumors associated with a germline KIT K642E mutation. Arch Pathol Lab Med. 2007;131:1393–1396. | |
Lutzky J, Bauer J, Bastian BC. Dose-dependent, complete response to imatinib of a metastatic mucosal melanoma with a K642E KIT mutation. Pigment Cell Melanoma Res. 2008;21:492–493. | |
Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365. | |
Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy. Pharmacol Ther. 2014;142:176–182. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.