")
Back to Journals » Drug Design, Development and Therapy » Volume 9
Antitumor activity of SA12, a novel peptide, on SKBr-3 breast cancer cells via the mitochondrial apoptosis pathway
Authors Yang L, Cui Y, Shen J, Lin F, Wang X, Long M, Wei J, Zhang H
Received 14 October 2014
Accepted for publication 2 December 2014
Published 2 March 2015 Volume 2015:9 Pages 1319—1330
DOI https://doi.org/10.2147/DDDT.S75780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Shu-Feng Zhou
Longfei Yang,* Ying Cui,* Jianjun Shen, Fang Lin, Xi Wang, Min Long, Junxia Wei, Huizhong Zhang
Department of Medical Laboratory and Research Center, Tangdu Hospital, Fourth Military Medical University, Xi’an, People’s Republic of China
*These authors contributed equally to this work
Abstract: Breast cancer is considered to be the most common malignancy in women. Treatment of breast cancer has been focused on molecular targeted therapy, and anticancer peptides are considered to be some of the most promising candidate drugs. In the current study, we used mRNA-peptide display technology to screen antibreast cancer peptides and identified a novel peptide, SA12, which showed significant activity in the inhibition of proliferation and induction of apoptosis in SKBr-3 breast cancer cells. The mechanism by which SA12 peptide triggers apoptosis was further investigated using a pull-down assay, reverse transcription-polymerase chain reaction, and Western blotting analysis. The results demonstrated that this peptide could interact with tumor-associated proteins MECP2 and CDC20B, which further induced apoptosis of tumor cells via the mitochondrial pathway involving the Bcl-2 family and related caspases. We propose that the novel SA12 peptide has the potential to provide a new strategy for the development of targeted therapy in breast cancer.
Keywords: targeted therapy, mRNA display, MECP2, Bcl-2, caspase
Introduction
Breast cancer is considered to be the most common malignancy in women. Although significant progress has been made in the last two decades, breast cancer remains one of the leading causes of death in females worldwide.1–5 Breast cancer is a complicated process involving multiple factors which are critical in cell signal pathways, so designing drugs that can target factors related to survival of cancer cells is an important strategy in the treatment of breast cancer.6 Considerable progress has been made in targeted therapy for breast cancer, especially the use of monoclonal antibodies, thereby increasing the cure rate and prolonging survival in patients with the disease.4,5 However, the cure rate for breast cancer remains unsatisfactory, and development of new and more effective targeted therapy regimens remains need to be continue undoubtedly.
Since peptide ligands are able to bind to their receptors with high affinity, specificity, and saturation characteristics, peptide drugs have attracted more and more attention in targeted therapy for cancer.7,8 Further, anticancer peptides which specifically target to the oncoprotein are considered to be broad spectrum, high efficiency, low side effects, and other advantages in targeted therapy.9 For discovery of novel anticancer peptides, in vitro peptide selection using mRNA displays enables the screening and directed evolution of new peptide drugs from constructed libraries.10,11 In this technology, mRNAs are covalently attached to their encoded peptides via a puromycin molecule, an analog of the 3′ end of aminoacyl-tRNA, which can enter the ribosomal A site to bind covalently to the C-terminal end of the encoded peptide in the ribosomal P site, resulting in stable conjugation of a genotype and the corresponding phenotype.12–14 The covalently conjugated phenotype-genotype model in the mRNA display is highly stable, so specific selection under diverse conditions is becoming possible, avoiding attenuation of the library and increasing the success rate of peptides selection.15 This technology was developed for construction of a peptide library for the discovery of new peptide ligands and novel anticancer peptide drugs for testing in preclinical experiments.
In this study, we constructed an mRNA-peptide display library and used total protein from SKBr-3 breast cancer cells as bait to screen for specific antibreast cancer peptides. One of the captured peptides (SA12) demonstrated a significant role in the inhibition of proliferation and induction of apoptosis in SKBr-3 cells, which was closely related to changes in the bioactivity of their target proteins, MECP2 and CDC20B. Further, the signal pathway of apoptosis induced by SA12 was revealed to be the mitochondrial pathway, involving the Bcl-2 family and related caspases. The novel SA12 peptide could potentially be a new candidate or strategy for development of targeted therapy in breast cancer.
Materials and methods
Construction of mRNA-peptide display library
A complementary DNA (cDNA) library encoding dodecapeptides was constructed as previously described16 but with several improvements. The cDNA library consisted of short cassettes encoding 12 amino acids arranged in a random manner. The T7 promoter gene for activating the transcription process and a Flag tag gene encoding eight amino acids as the invariant region for the purpose of purification were introduced into the sequence upstream of the expression cassette. The cDNA library was transcribed by T7 RNA polymerase (Promega, Madison, WI, USA), after which a puromycin oligonucleotide linker was added to the 3′ end by using T4 RNA ligase (New England Biolabs UK Ltd, Hitchin, UK). The puromycin-terminated mRNA was then translated in vitro, yielding a library of dodecapeptides linked via the puromycin moiety to their encoding mRNAs. These mRNA-display peptides were then purified on oligo-(dT)30 cellulose, and the mRNA portion was reverse transcribed into cDNA, resulting in a library consisting of different displayed dodecapeptides. The library was then confirmed by polymerase chain reaction (PCR) and agarose gel electrophoresis.
Selection of breast cancer-associated protein binding peptides
Total protein from an SKBr-3 cell line was prepared using radio-immunoprecipitation assay lysis buffer (1% NP-40, 0.1% sodium dodecyl sulfate, 0.5% sodium deoxycholate, 150 mM NaCl, and 10 mM Tris-HCl) containing a 1/100 phenylmethanesulfonyl fluoride solution for selection of breast cancer-associated protein binding peptides from the library, and then immobilized onto the polystyrene surface. The mRNA-peptide display library was added and incubated with immobilized SKBr-3 total protein for 20 minutes, washed thoroughly, and then enzymolyzed the combined mRNA-peptide fusion with RNase H. The cDNA released in the supernatant were then reverse transcribed and amplified by PCR to complete the first round of selection, and then six subsequent rounds were carried out for selection of higher affinity peptides. The supernatant from the last round of selection was amplified by PCR, and the PCR products were purified and cloned into a PMD18-T vector and sequenced.
Peptide sequencing and synthesis
Amino acid motifs on peptides that bond to the breast cancer protein were determined via their encoding cDNA. The final peptide, SA12, and an SA12 peptide labeled with fluorescein isothiocyanate (FITC-Ahx-SA12) were then synthesized and further characterized by GL Biochem Ltd (Shanghai, People’s Republic of China) using electrospray ionization-mass spectrometry detection to confirm the molecular weight before subsequent bioactivity assays. The peptides were thoroughly dissolved in dimethyl sulfoxide under ultrasound and vortex for 5 times alternately, and then diluted in Dulbecco’s Modified Eagle’s Medium to the required concentrations, with a consequent dimethyl sulfoxide content of less than 0.1%.
Cell lines and cell culture
The SKBr-3 cell line was initially purchased from the American Tissue Culture Collection (Rockville, MD, USA) and preserved by our laboratory. The cells were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (Gibco, Waltham, MA, USA), supplemented with 10% fetal calf serum (HyClone, Logan, UT, USA) and 1% glutamine (Invitrogen, Waltham, MA, USA), and incubated in a humidified atmosphere with 5% CO2 at 37°C.
Fluorescence-labeled SA12 peptide internalization assay
Internalization of SA12 peptide was detected by the fluorescence-tagged peptide FITC-Ahx-SA12. SKBr-3 cells were seeded into six-well plates at a density of 5×105 cells/well and cultivated for 24 hours. The FITC-Ahx-SA12 peptide solution was then added into the wells at a final concentration of 80 μM with fresh Dulbecco’s Modified Eagle’s Medium and maintained for 30 minutes or one hour. After incubation, the culture medium was discarded and the cells were washed twice with phosphate-buffered saline. Photographs were taken using a fluorescence microscope (Olympus, Tokyo, Japan) in a dark room.
Cell viability assay
Cell viability was determined by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay. SKBr-3 cells were seeded into 96-well plates at a density of 5×103 cells/well and maintained in the incubator for 24 hours. The medium was then replaced with the novel SA12 peptide in Dulbecco’s Modified Eagle’s Medium at concentrations of 80 nM, 800 nM, 8 μM, or 80 μM. The LL12 peptide (Leu-Phe-Val-Ser-Leu-Leu-Arg-Ile-Phe-Pro-Leu-Leu), which was confirmed to be inactive in previous experiments at a concentration of 80 μM was used as the negative control and solvent only was set as the blank control. The cells were subjected to MTT assay after treatment for 24, 48, 72, and 96 hours. The medium was replaced with 100 μL of 0.05% MTT, and the plate was incubated for a further 4 hours at 37°C. After incubation, the reaction was stopped by addition of 150 μL of dimethyl sulfoxide per well for 10 minutes. Finally, absorbance was determined on a microreader (Bio-Rad, Hercules, CA, USA) at 490 nm. The percentage cell of viability was calculated as (Atreated/Acontrol) ×100%. The cell proliferation curve was plotted using the percentage of cell viability at each time point. All experiments were performed three times and the mean value was used.
Annexin V-FITC/PI apoptosis detection
SKBr-3 cells treated with SA12 peptide at a concentration of 80 μM for 6, 18, 24, 36, and 48 hours, respectively, were collected and subjected to Annexin V-FITC/propidium iodide (PI) staining using an apoptosis detection kit (Keygen, Nanjing, People’s Republic of China) following the manufacturer’s instructions. SA12 was replaced with solvent only for the control group. The apoptosis rate was then analyzed with WinMDI software (Scripps Institute, La Jolla, CA, USA) on a FACSCalibur™ system (BD Biosciences, San Jose, CA, USA).
TUNEL assay
Coverslips placed in six-well plates were seeded with SKBr-3 cells at a density of 5×105 cells/well. After 24 hours of incubation, the medium was replaced with SA12 peptide in Dulbecco’s Modified Eagle’s Medium at a concentration of 80 μM for 24, 36, and 48 hours, respectively. A TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) assay was performed using an in situ cell apoptosis detection kit (Keygen) as described by the manufacturer. TUNEL-positive cells were observed and photographed using a phase-contrast microscope (Olympus). TUNEL-positive cells were counted on the illustrations, and at least 500 total cells were examined in each experimental group.
In vitro pull-down and mass spectrometry analysis
Total protein was extracted from the SKBr-3 cells using RIPA lysis buffer as described above. The purified total protein was then added into a tube containing SA12 peptide that was preconjugated onto superparamagnetic beads with a Dynabeads M-270 carboxylic acid kit as described by the manufacturer (Invitrogen). The mixture was incubated with tilting and rotation for 30 minutes to capture the target protein, after which the tube was then placed on the magnetic particle concentrator for 4 minutes. The supernatant was then pipetted off, and magnetic beads on the tube wall was collected and washed three times using 1 mL of phosphate-buffered saline containing 0.1% Tween-20. Loading buffer for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was added into the washed beads and the mixture was boiled for 5 minutes to obtain the captured proteins.
The captured proteins were separated by 12% SDS-PAGE and stained with Coomassie blue. Visible bands were subjected to in-gel digestion and analyzed in a quadrupole time-of-flight mass spectrometer equipped with a nanoelectrospray source (Beijing Proteome Research Center, Beijing, People’s Republic of China). The acquired data were searched against human proteins in the UniProt database. Subsequently, Western blotting analysis was performed for further evidence of interanction between SA12 and the targeted protein using antibody of determined proteins.
RT-PCR analysis
Total RNA was extracted from SKBr-3 cells (treated or not treated with 80 μM SA12) using TRIzol reagent (TaKaRa, Dalian, People’s Republic of China), and 1 μg of RNA from each sample was used for cDNA synthesis using a Primerscript RT kit (TaKaRa) according to the manufacturer’s instructions. PCR amplification was then performed using the primer pairs listed in Table 1. The reverse transcription-polymerase chain reaction products were then electrophoresed through 1.5% agarose gel and signals were quantified by densitometric analysis using a MultiImage™ light cabinet (Alpha, San Leandro, CA, USA). The β-actin mRNA expression level was used for the inner control.
 | Table 1 Primer sequences used in reverse transcription-polymerase chain reaction analysis |
Western blotting analysis
Cells treated or not treated with 80 μM SA12 for 48 hours were washed twice with phosphate-buffered saline and lysed on culture dishes using RIPA lysis buffer. Total protein was separated by 12% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The membrane was blocked by incubation with TBST (Tris-buffered saline, 0.1% Tween-20) containing 5% (w/v) non-fat dried milk for one hour at room temperature, and then immunoblotted overnight at 4°C. After washing three times with TBST, the membrane was incubated with horseradish peroxidase-conjugated secondary antibody for 2 hours. Signals were visualized by electrochemiluminescence. The β-actin protein expression level was used for the inner control. The blotting bands were semiquantified using Gel-Pro Analyzer 4 software. The primary antibodies used in this experiment were goat anti-human MECP2, rabbit anti-human CDC20B, mouse anti-human P53, rabbit anti-human phosphatase and tensin homolog (PTEN), rabbit anti-human cytochrome-c, mouse anti-human Bax, rabbit anti-human Bak, rabbit anti-human caspase-3, rabbit anti-human caspase-8, rabbit anti-human caspase-9, mouse anti-human Bcl-2, mouse anti-human β-actin, and mouse anti-human Mcl-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse immunoglobulin G and goat anti-rabbit immunoglobulin G (Zhongshanjinqiao, Beijing, People’s Republic of China).
Statistical analysis
The data are expressed as the mean ± standard deviation. Statistical evaluation of the data was performed by one-way analysis of variance. Date comparisons were conducted by the one-sample or paired-samples Student’s t-test. A P-value less than 0.05 was considered to be statistically significant.
Results
Generation of mRNA-peptide display library
The cDNA library encoding dodecapeptides was constructed as described in the Materials and methods section. The library consisted of short expression cassettes encoding 12 amino acids arranged randomly with the T7 promoter upstream of the expression cassette, and a Flag tag gene encoding eight amino acids followed in the downstream of T7 promoter for the purposes of purification. The final fusion cDNA library was confirmed by PCR, and the agarose gel electrophoresis result showed that the size of the fusion cDNA was consistent with the predicted value of 110 base pairs (Figure 1).
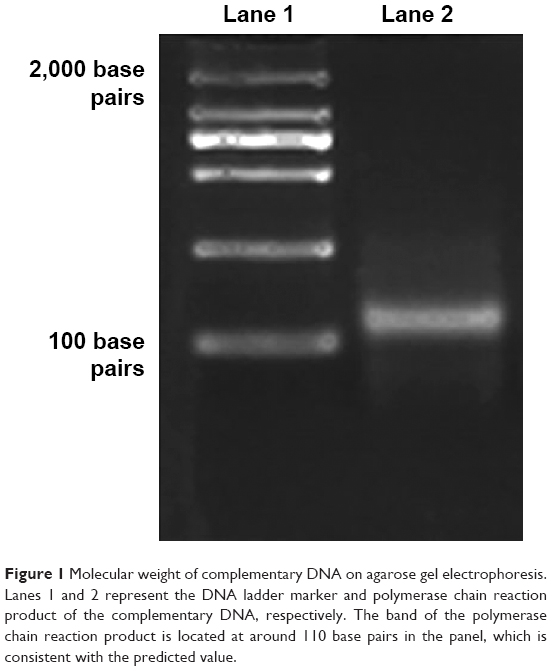 | Figure 1 Molecular weight of complementary DNA on agarose gel electrophoresis. Lanes 1 and 2 represent the DNA ladder marker and polymerase chain reaction product of the complementary DNA, respectively. The band of the polymerase chain reaction product is located at around 110 base pairs in the panel, which is consistent with the predicted value. |
Selection of breast cancer cell-associated protein binding peptides
We used total protein from SKBr-3 cells as the target for selection of breast cancer cell-associated protein binding peptides. The mRNA-peptide display library was screened with immobilized SKBr-3 total protein followed by reverse transcription and PCR to amplify the sequences for six rounds of biopanning. After subsequent selection and sequencing, several binding peptides were finally obtained via triplet codes. The antitumor activity of these peptides was identified in a preliminary experiment, and a dodecapeptide designated as SA12 (Ser-Val-Pro-Leu-Phe-Asn-Phe-Ser-Val-Tyr-Leu-Ala; patent ZL201310060261.8, People’s Republic of China) was further characterized as having excellent bioactivity in tumor suppression. As shown in the electrospray ionization-mass spectrometry report for SA12, the molecular weight was found to be 1,357.27 ([M+H]+), which was conjugated with a H+, and 1,379.94 ([M+Na]+), which was conjugated with an Na+, both of which were identical to the predicted molecular weight for SA12 of 1,356.60 (Figure 2).
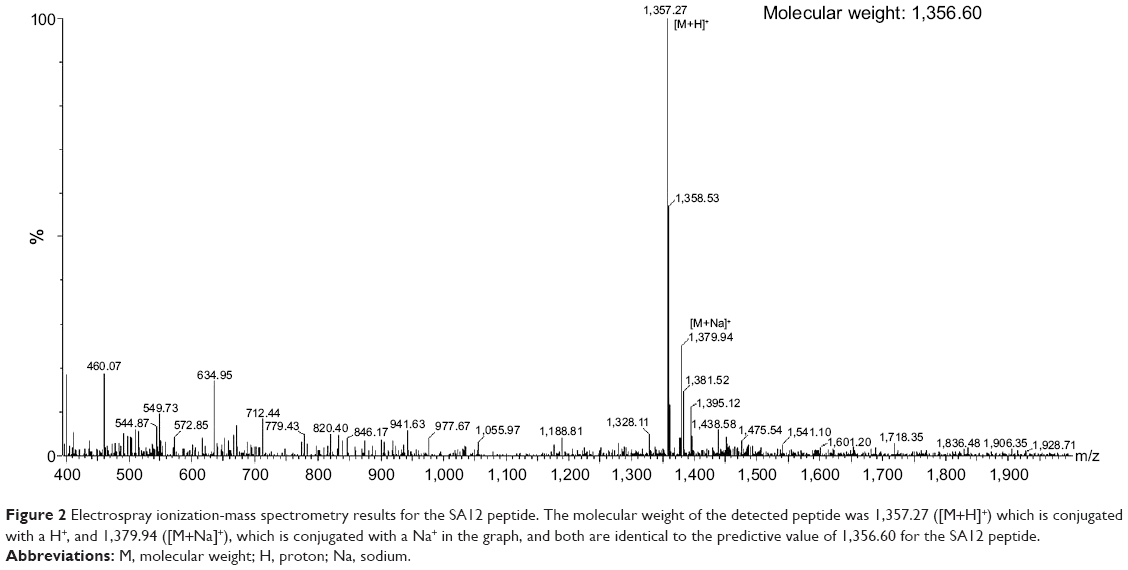 | Figure 2 Electrospray ionization-mass spectrometry results for the SA12 peptide. The molecular weight of the detected peptide was 1,357.27 ([M+H]+) which is conjugated with a H+, and 1,379.94 ([M+Na]+), which is conjugated with a Na+ in the graph, and both are identical to the predictive value of 1,356.60 for the SA12 peptide. |
SA12 peptide internalized and localized at the nucleus in SKBr-3 cells
To analyze the distribution of the SA12 peptide when it interacted with SKBr-3 cells, we synthesized a fluorescence-labeled SA12 peptide, FITC-Ahx-SA12. The SKBr-3 cells were treated with FITC-Ahx-SA12 at a concentration of 80 μM, and fluorescence was observed under a fluorescence microscope (Olympus). The results showed that slight fluorescence (Figure 3C) appeared in the field of vision as early as 30 minutes after treatment with FITC-Ahx-SA12; clear localization was present in the cell nucleus region within 60 minutes after treatment, but there was only minor localization in the cytoplasm (Figure 3D). These results indicate that the SA12 peptide could be rapidly internalized, and its intracellular localization in SKBr-3 cells was in the nucleus.
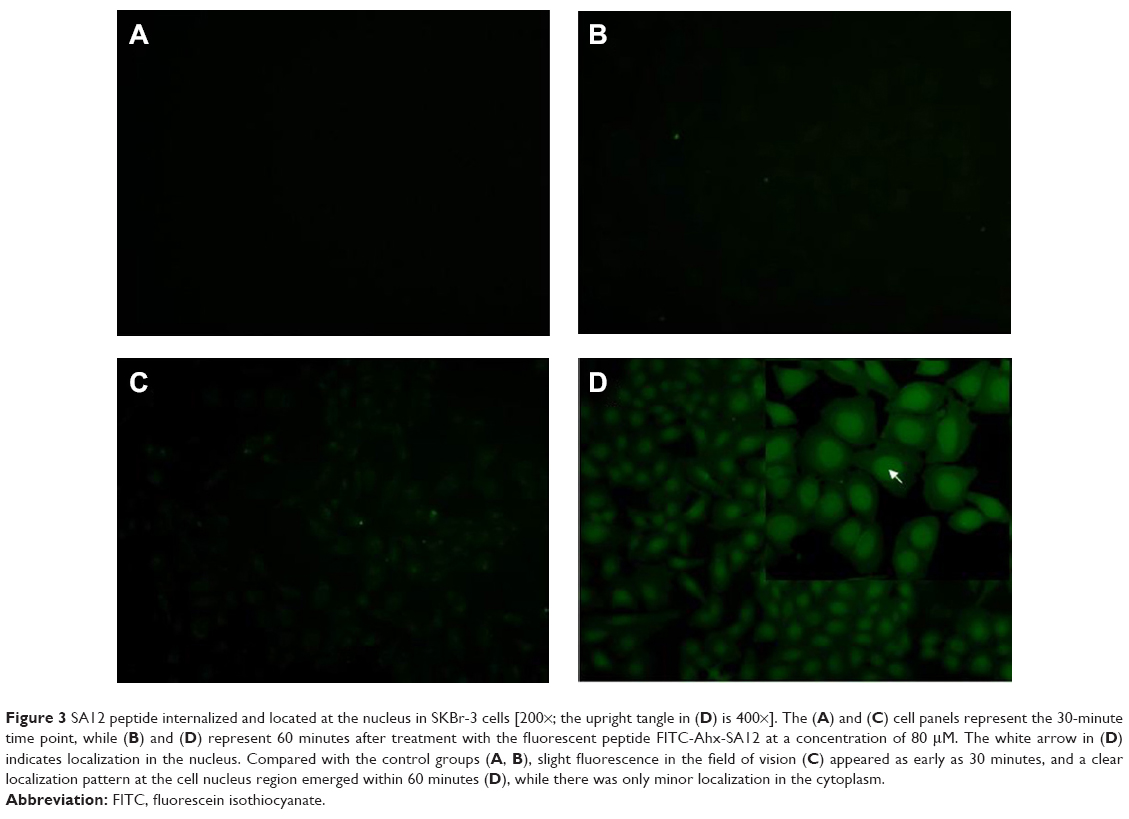 | Figure 3 SA12 peptide internalized and located at the nucleus in SKBr-3 cells [200×; the upright tangle in (D) is 400×]. The (A) and (C) cell panels represent the 30-minute time point, while (B) and (D) represent 60 minutes after treatment with the fluorescent peptide FITC-Ahx-SA12 at a concentration of 80 μM. The white arrow in (D) indicates localization in the nucleus. Compared with the control groups (A, B), slight fluorescence in the field of vision (C) appeared as early as 30 minutes, and a clear localization pattern at the cell nucleus region emerged within 60 minutes (D), while there was only minor localization in the cytoplasm. |
SA12 inhibited proliferation of SKBr-3 cells in a time-dependent manner
In the current study, we tested the effect of SA12 on the proliferation of SKBr-3 cells using MTT assays. SKBr-3 cells were incubated with SA12 peptide at different concentrations for 24, 48, 72, and 96 hours, and the ability of SA12 to inhibit cell proliferation increased with increasing concentrations of SA12, with the strongest inhibition observed at 80 μM (P<0.05). Compared with the control group, incubation with SA12 at a concentration of 80 μM resulted in 27.61%, 40.15%, 43.19%, and 46.8% inhibition of cell proliferation at 24, 48, 72, and 96 hours, respectively, whereas the negative control peptide, LL12, did not show any effect on cell viability at the same concentrations or at any time point (Figure 4). These results show that the SA12 peptide could suppress proliferation of SKBr-3 cells in a time-dependent manner.
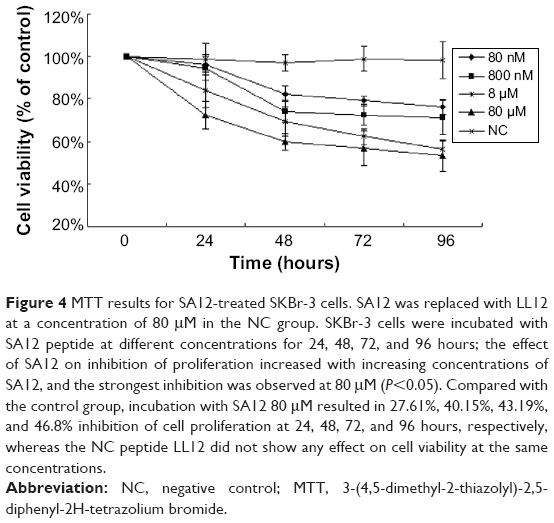 | Figure 4 MTT results for SA12-treated SKBr-3 cells. SA12 was replaced with LL12 at a concentration of 80 μM in the NC group. SKBr-3 cells were incubated with SA12 peptide at different concentrations for 24, 48, 72, and 96 hours; the effect of SA12 on inhibition of proliferation increased with increasing concentrations of SA12, and the strongest inhibition was observed at 80 μM (P<0.05). Compared with the control group, incubation with SA12 80 μM resulted in 27.61%, 40.15%, 43.19%, and 46.8% inhibition of cell proliferation at 24, 48, 72, and 96 hours, respectively, whereas the NC peptide LL12 did not show any effect on cell viability at the same concentrations. |
SA12 induced apoptosis of SKBr-3 cells
To reveal the mechanism of inhibition of proliferation, we investigated the role of SA12 in inducing apoptosis in SKBr-3 cells. SKBr-3 cells were treated with SA12 peptide and then subjected to Annexin V-FITC/PI staining apoptosis assay by flow cytometry. As shown in Figure 5, the apoptosis rate in SKBr-3 cells increased slightly from 6 hours to 18 hours when the cells were treated with SA12 at a concentration of 80 μM, while apoptosis was significantly induced at the 24-hour time point using the 80 μM concentration. Compared with the control groups, the apoptosis rate in SKBr-3 cells treated with SA12 80 μM for 24, 36, and 48 hours increased up to 12.73%, 14.91%, and 18.01%, respectively (Figure 5). The apoptotic cells presented mainly as both Annexin V-FITC-positive and PI-positive, indicating that the inducing and promoting effect of the SA12 peptide in SKBr-3 cells was mainly in the late phase of apoptosis.
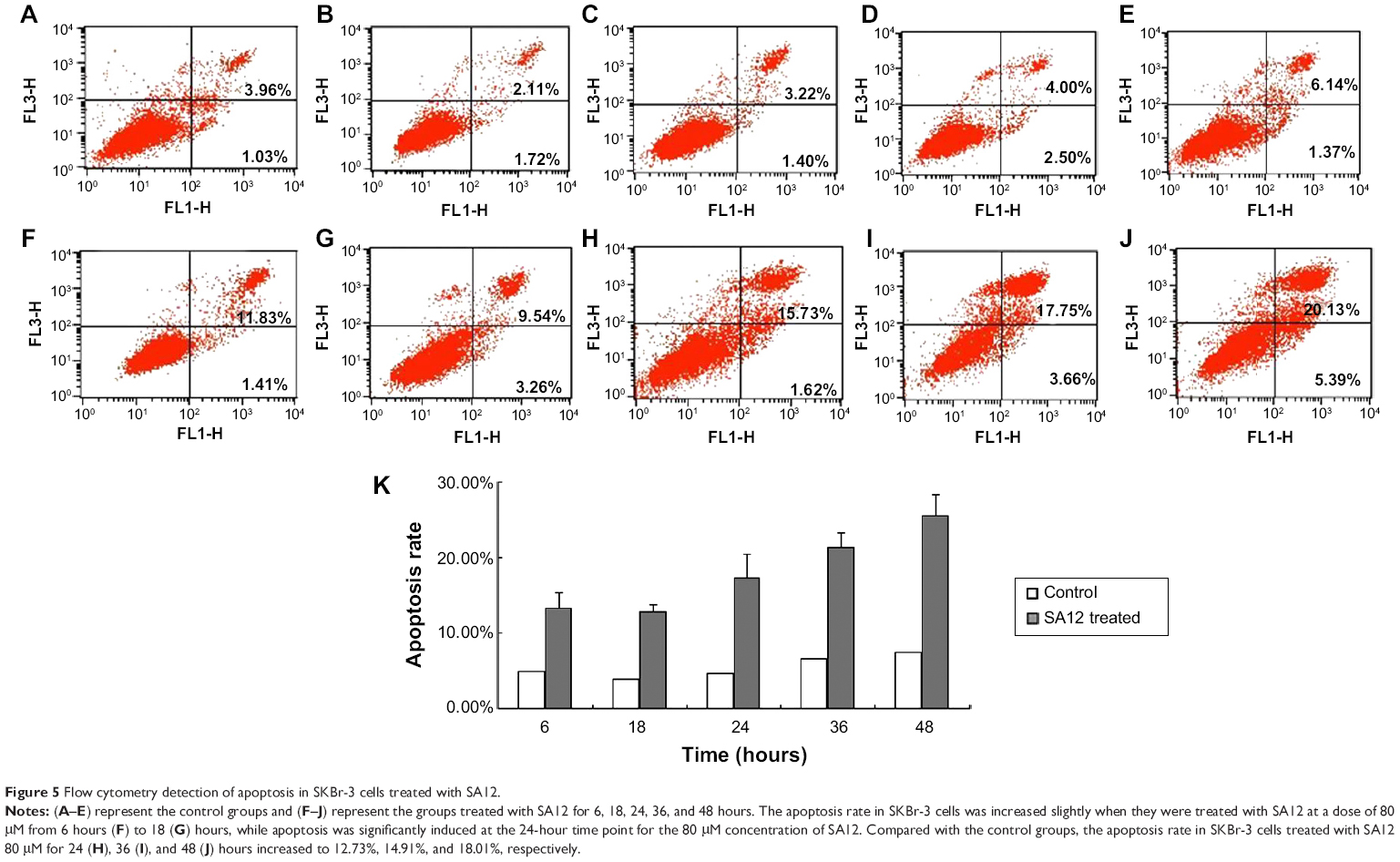 | Figure 5 Flow cytometry detection of apoptosis in SKBr-3 cells treated with SA12. |
We also used the TUNEL assay to confirm induction of apoptosis by SA12 in the SKBr-3 cell line. SKBr-3 cells seeded on coverslips were treated with SA12 at a concentration of 80 μM for 24, 36, and 48 hours, and then subjected to TUNEL assay as described in the Materials and methods section. Photographs of TUNEL assay were obtained from the microscope. Compared with the controls, the percentage of TUNEL-positive cells increased by 16.55% when treated with SA12 80 μM for 24 hours, by 18.90% when treated for 36 hours, and by 29.81% when treated for 48 hours (Figure 6). These results are consistent with the results of the Annexin V-FITC/PI staining apoptosis assay, confirming that treatment with SA12 at a dose of 80 μM for over 24 hours significantly induced apoptosis in SKBr-3 cells. This is also consistent with the tendency seen in the MTT assay, ie, repression of cell viability by SA12 in a time-dependent manner.
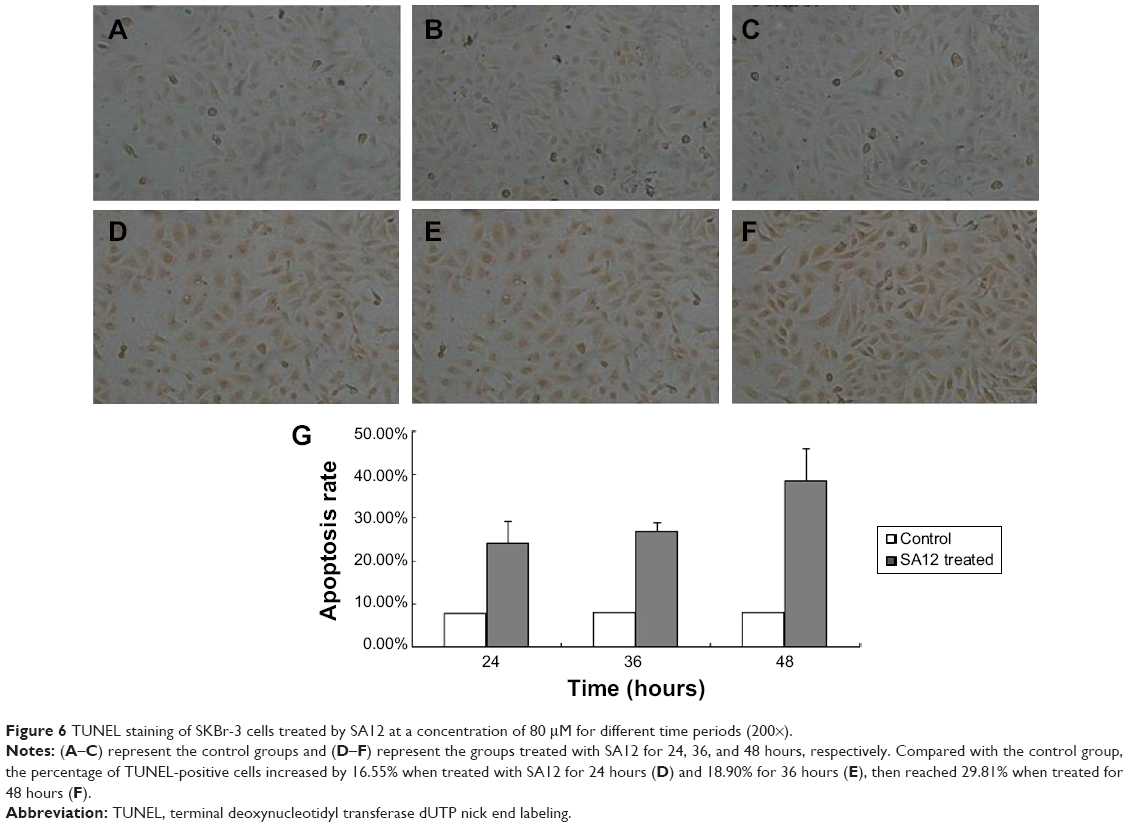 | Figure 6 TUNEL staining of SKBr-3 cells treated by SA12 at a concentration of 80 μM for different time periods (200×). |
SA12 interacted with MECP2 and CDC20B directly in vitro
To identify the intracellular targets of the novel anti-tumor peptide SA12, we carried out pull-down assays on SKBr-3 cells. Total protein from SKBr-3 cells was added to SA12 peptide conjugated on superparamagnetic beads to perform the in vitro pull-down experiments, and the resulting magnetic beads were boiled in protein loading buffer and separated by SDS-PAGE (Figure 7A). Major protein bands from pull-down fractions separated by SDS-PAGE were subjected to mass spectrometry analysis; obvious interacting proteins were obtained and identified as MECP2 and CDC20B, and both of these two proteins were found to be tumor-associated proteins. To confirm the above results, we carried out Western blotting experiments using the pull-down fractions and polyclonal antibodies against these two proteins. A protein band around 52.4 kDa was clearly visible in the anti-MECP2 immunoblot, and a major protein band around 52.8 kDa was observed in the anti-CDC20B immunoblot, but no visible band was present in the LL12 captured group (Figure 7B). These results provide further evidence that SA12 interacted with the target proteins, MECP2 and CDC20B, directly in vitro.
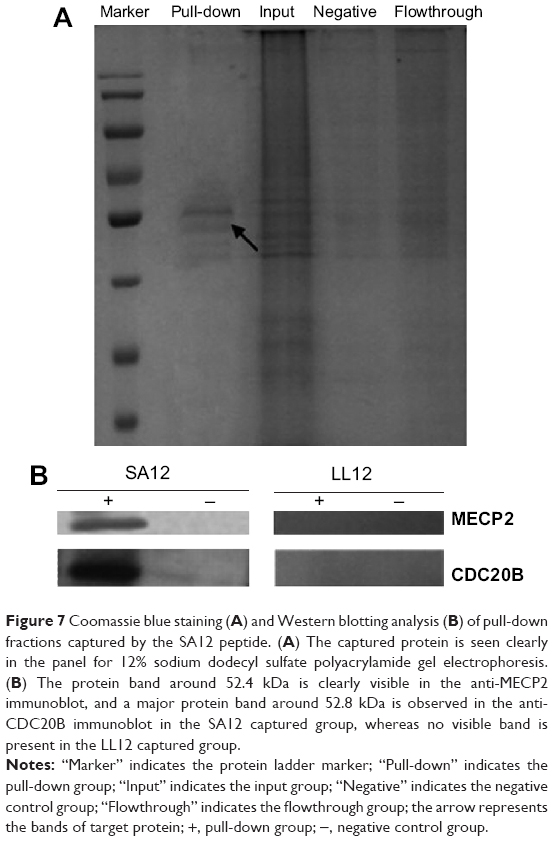 | Figure 7 Coomassie blue staining (A) and Western blotting analysis (B) of pull-down fractions captured by the SA12 peptide. (A) The captured protein is seen clearly in the panel for 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis.(B) The protein band around 52.4 kDa is clearly visible in the anti-MECP2 immunoblot, and a major protein band around 52.8 kDa is observed in the anti-CDC20B immunoblot in the SA12 captured group, whereas no visible band is present in the LL12 captured group. |
SA12 upregulated expression of P53 and PTEN tumor suppression proteins
Given that apoptosis is usually correlated with the tumor suppression proteins P53 and PTEN, we investigated whether the apoptosis induced by SA12 was involved in the changes in P53 and PTEN expression. As shown in Figure 8, an obvious accumulation of P53 and PTEN was found in the RT-PCR and Western blotting assays. Compared with the control group, gene expression levels of P53 and PTEN were increased by 95.34% and 42.53%, respectively, in the SA12-treated SKBr-3 cells at a concentration of 80 μM for 48 hours. Moreover, protein expression levels of P53 and PTEN were increased by 183.49% and 153.92%, respectively (P<0.05), which suggest that the apoptosis-inducing effect of SA12 was exactly associated with P53 and PTEN protein expression, although other tumor suppressor genes that might be involved in this process should be further explored.
 | Figure 8 Reverse transcription-polymerase chain reaction (A, C) and Western blotting (B, D) analysis of SA12-treated SKBr-3 cells. Data are shown as the relative value of gene/β-actin and protein/β-actin. (A, C) Compared with the control groups, the gene expression of P53, PTEN, Bax, and Bak was upregulated by treatment with SA12 at a concentration of 80 μM for 48 hours, while NF-κB, PI3K, Akt, Bcl-2, and Mcl-1 were downregulated (P<0.05). (B, D) Compared with the control groups, protein expression of P53, PTEN, Bax, and Bak was upregulated; moreover, the release of cytochrome-c into the cytosol and activation of caspase-9 and caspase-3 were increased by treatment with SA12 at a concentration of 80 μM for 48 hours, while Bcl-2 and Mcl-1 were downregulated (P<0.05), which is consistent with the reverse transcription-polymerase chain reaction results. |
To investigate whether the phosphatidylinositol-4 5-bisphosphate 3-kinase (PI3K)/PTEN/Akt pathway was involved in the apoptosis process induced by SA12, we analyzed gene expression levels of nuclear factor kappa B, PI3K, Akt, and PTEN by RT-PCR, and found that expression of mRNA for nuclear factor kappa B, PI3K, and Akt was downregulated, while PTEN was upregulated in SKBr-3 cells treated with SA12 at a concentration of 80 μM for 48 hours compared with the control group (Figure 8). These results suggest that apoptosis induced by SA12 was also associated with these members of the PI3K/PTEN/Akt signal pathway.
SA12 induced apoptosis via the mitochondrial pathway
To identify the mechanism of apoptosis induced by the SA12 peptide, we carried out RT-PCR assays with SA12-treated SKBr-3 cells. Results of RT-PCR showed that the proapoptotic genes Bax and Bak were upregulated by 258.6% and 143.3%, respectively, when treated with SA12 at a concentration of 80 μM for 48 hours compared with the control group, while the antiapoptotic genes Bcl-2 and Mcl-1 were downregulated by 43.4% and 70.28%, respectively (P<0.05, Figure 8A and C, respectively).
To obtain further evidence as to whether the extrinsic pathway or mitochondrial pathway was involved in the apoptosis process induced by SA12, we performed Western blotting using the polyclonal antibodies of the anticaspase family and anti-Bcl-2 family proteins related to cell apoptosis. As shown in Figure 8B and D, Bax and Bak expression levels were upregulated by 90.29% and 64.11%, respectively, in treatment with SA12 at a concentration of 80 μM for 48 hours. Moreover, release of cytochrome-c into the cytosol and activation of caspase-9 and caspase-3 were increased by 120.38%, 145.19%, and 79.63%, respectively, while Bcl-2 and Mcl-1 were downregulated by 37.73% and 44.09% (P<0.05). In contrast, there was no visible change in caspase-8. These results indicate that the mitochondrial pathway was the primary apoptosis pattern caused by the novel peptide SA12.
Discussion
Peptides offer certain advantages when used as drugs, including high biological activity, high specificity, and low toxicity.17 Thus, peptides have been developed as promising therapeutic agents in the treatment of cancers.8 In the current study, we developed a novel peptide designated as SA12, which exhibited significant potency in the inhibition of proliferation and induction of apoptosis in SKBr-3 breast cancer cells, using mRNA-peptide display technology. Further, we confirmed that the SA12 peptide interacted with MECP2 and CDC20B proteins in SKBr-3 cells.
Cell proliferation and apoptosis are important in tumor progression, which is also related to complicated signaling pathways. Although CDC20B has not been widely investigated, both MECP2 and CDC20B are considered to be crucial in tumor progression.18–20 MECP2 was the first identified member of the methylated DNA binding proteins and is an important regulatory factor in gene expression, and has been classically described as a global transcription repressor.21 As a transcriptional repressor, MECP2 contains two functional domains, ie, a methylated DNA binding domain and a transcriptional repression domain, both of which are involved in the regulation of transcription activity, especially for tumor suppressor genes, thus plays an important role in the development of cancer.19,21 Hypermethylation of tumor suppression gene promoter in CpG islands region is critical in the progression of carcinogenesis.22 MECP2, through binding to the methylated CpG of the gene 5′ flanking sequence, prevents formation of a gene transcriptional complex, and silences the tumor suppressor genes. On the other hand, when MECP2 binds to the methylated CpG, the histone is deacetylated by histone deacetylase which is bound on MECP2 structurally, leading to conformational changes in chromatin and transcriptional repression, and inhibition of expression on tumor suppressor genes.23 Progression of breast cancer involves multiple genetic events which can activate dominant-acting oncogenes and disrupt the function of specific tumor suppressor genes.24 When SKBr-3 breast cancer cells were treated with the SA12 peptide, repression of tumor cell proliferation was observed. The mechanism was explained as that when SA12 interacted with MECP2, the binding ability of MECP2 to the methylated CpG of the tumor suppressor gene was blocked, resulting in termination of transcription repression and re-expression of tumor suppressor genes. Although more research is needed to reveal the complicated effect of SA12 on MECP2, SA12 is considered to play an important role in the re-activation of tumor suppressor genes prevented by MECP2 protein.
The expression levels of tumor suppressor genes like P53 and PTEN are crucial in the fate of tumor cells. Mutation or deletion of P53 and PTEN are found to be involved in the development of a variety of tumors, including breast cancer.24 P53 is described as a transcription factor regulating downstream genes important in cell cycle arrest, DNA repair, and apoptosis,25 so mutation or loss of P53 in many cancers leads to genomic instability, impaired cell cycle regulation, and inhibition of apoptosis. PTEN was identified as one of tumor suppressor genes frequently deleted or mutated in various primary cancers, including brain, prostate, and breast cancer.26 In this study, expression levels of P53 and PTEN were elevated in SKBr-3 cells after treatment with SA12 peptide, the mechanism was explained as the inhibition of MECP2 on the transcription of P53 and PTEN were prevented by SA12, leading to re-expression of P53 and PTEN, and finally corrected unbalance between tumor suppressor genes and oncogenes in tumor cells. Additionally, the PI3K/PTEN/Akt signal cascade has been shown to play a critical role in the initiation and progession of carcinogenesis,27,28 we observed that treatment with SA12 peptide also influences the expression of PI3K and Akt, so the PI3K/PTEN/Akt pathway also contributes to the inhibition of breast cancer cells by SA12.
Apoptosis occurs through two main pathways leading to activation of the death signal, ie, the extrinsic or cytoplasmic pathway and the intrinsic or mitochondrial pathway.29,30 The apoptosis pathway evoked by SA12 in SKBr-3 cells was shown to be the mitochondrial pathway, and both Bcl-2 family and related caspases are proven to be involved in this process. The Bcl-2 family is one of the most important regulators of the mitochondrial apoptosis pathway, and includes proapoptotic members such as Bax, Bak, Bad, Bcl-Xs, Bid, Bik, Bim, and Hrk, and antiapoptotic members such as Bcl-2, Bcl-XL, Bcl-W, Bfl-1, and Mcl-1.31,32 Proapoptotic members of the Bcl-2 family act as promoters of apoptosis, whereas antiapoptotic members act as repressors by blocking the release of cytochrome-c.33 The subsequent effects are mainly dependent on the balance of Bcl-2 and Bax,29 and the final step leading to execution of the death signal is the activation of a series of proteases known as caspases.34,35 When treated with SA12, proapoptotic members (Bax and Bak) were increased, while antiapoptotic members (Bcl-2 and Mcl-1) were decreased, all of which might be caused by SA12-activated expression of P53 and PTEN genes. The imbalance of Bax/Bcl-2 promoted additional release of cytochrome-c, which contributes to the activation of caspase-9 and the subsequent activation of caspase-3. Thus, the mitochondrial pathway is considered to be responsible for the apoptosis induced by the novel peptide SA12.
Conclusion
In summary, we have successfully developed a novel peptide of SA12 that can inhibit proliferation and induce apoptosis in SKBr-3 cells through interaction with the MECP2 protein. We propose that SA12 has potential for development as a drug candidate for the targeted treatment of breast cancer.
Acknowledgment
We thank all the members of the Department of Medical Laboratory and Research Center for their technical assistance.
Disclosure
The authors report no conflicts of interest in this work.
References
Coveler AL, Bates NE, Disis ML. Progress in the development of a therapeutic vaccine for breast cancer. Breast Cancer. 2010;2:25–36. | ||
Kozioł M, Püsküllüoglu M, Zygulska A. [PARP inhibitors and their role in the therapy of triple-negative metastatic breast cancer]. Przegl Lek. 2012;69(6):265–270. Polish. | ||
Ladjemi MZ, Jacot W, Chardès T, Pèlegrin A, Navarro-Teulon I. Anti-HER2 vaccines: new prospects for breast cancer therapy. Cancer Immunol Immunother. 2010;59(9):1295–1312. | ||
Murphy CG, Morris PG. Recent advances in novel targeted therapies for HER2-positive breast cancer. Anticancer Drugs. 2012;23(8): 765–776. | ||
Wicki A, Rochlitz C. Targeted therapies in breast cancer. Swiss Med Wkly. 2012;142:w13550. | ||
Molnar-Stanciu D, Guimas V, Bensalem A, Thiery-Vuillemin A. [Targeted therapy and breast cancer: state of the art]. Pathol Biol (Paris). 2012;60(4):254–263. French. | ||
Boohaker RJ, Lee MW, Vishnubhotla P, Perez JM, Khaled AR. The use of therapeutic peptides to target and to kill cancer cells. Curr Med Chem. 2012;19(22):3794–3804. | ||
Thundimadathil J. Cancer treatment using peptides: current therapies and future prospects. J Amino Acids. 2012;2012:96734. | ||
Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81(1):136–147. | ||
Takahashi TT, Austin RJ, Roberts RW. mRNA display: ligand discovery, interaction analysis and beyond. Trends Biochem Sci. 2003;28(3):159–165. | ||
Wilson DS, Keefe AD, Szostak JW. The use of mRNA display to select high-affinity protein-binding peptides. Proc Natl Acad Sci U S A. 2001;98(7):3750–3755. | ||
Ikeda S, Saito I, Sugiyama H. Facile synthesis of puromycin-tethered oligonucleotides at the 3′-end. Tetrahedron Lett. 1998;39:5975–5978. | ||
Miyamoto-Sato E, Yanagawa H. Puromycin technology for in vitro evolution and proteome exploration. Viva Origino. 2006;3(34):148–154. | ||
Miyamoto-Sato E, Takashima H, Fuse S, et al. Highly stable and efficient mRNA templates for mRNA-protein fusions and C-terminally labeled proteins. Nucleic Acids Res. 2003;31(15):e78. | ||
Wang H, Liu R. Advantages of mRNA display selections over other selection techniques for investigation of protein-protein interactions. Expert Rev Proteomics. 2011;8(3):335–346. | ||
Shiheido H, Takashima H, Doi N, Yanagawa H. mRNA display selection of an optimized MDM2-binding peptide that potently inhibits MDM2-p53 interaction. PLoS One. 2011;6(3):e17898. | ||
Albericio F, Kruger HG. Therapeutic peptides. Future Med Chem. 2012;4(12):1527–1531. | ||
Wade PA. Methyl CpG-binding proteins and transcriptional repression. Bio Essays. 2001;23(12):1131–1137. | ||
Sansom OJ, Maddison K, Clarke AR. Mechanisms of disease: methyl-binding domain proteins as potential therapeutic targets in cancer. Nat Clin Pract Oncol. 2007;4(5):305–315. | ||
Wang Z, Wan L, Zhong J, et al. CDC20: a potential novel therapeutic target for cancer treatment. Curr Pharm Des. 2013;19(18):3210–3214. | ||
Adkins NL, Georgel PT. MECP2: structure and function. Biochem Cell Biol. 2011;89(1):1–11. | ||
Ghavifekr Fakhr M, Farshdousti Hagh M, Shanehbandi D, Baradaran B. DNA methylation pattern as important epigenetic criterion in cancer. Genet Res Int. 2013;2013:317569. | ||
Nan X, Ng HH, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389. | ||
Lee EY, Muller WJ. Oncogenes and tumor suppressor genes. Cold Spring Harb Perspect Biol. 2010;2(10):a003236. | ||
Lacroix M, Toillon R-A, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13(2):293–325. | ||
Petrocelli T, Slingerland JM. PTEN deficiency: a role in mammary carcinogenesis. Breast Cancer Res. 2001;3(6):356–360. | ||
McCubrey JA, Steelman LS, Abrams SL, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006;46(2006):249–279. | ||
Blanco-Aparicio C, Renner O, Leal JF, Carnero A. PTEN, more than the AKT pathway. Carcinogenesis. 2007;28(7):1379–1386. | ||
Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55(3):178–194. | ||
Cosentino K, García-Sáez AJ. Mitochondrial alterations in apoptosis. Chem Phys Lipids. 2014;181:62–75. | ||
Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(4):a008714. | ||
García-Sáez AJ. The secrets of the Bcl-2 family. Cell Death Differ. 2012;19(11):1733–1740. | ||
Scorrano L, Korsmeyer SJ. Mechanisms of cytochrome c release by proapoptotic Bcl-2 family members. Biochem Biophys Res Commun. 2003;304(3):437–444. | ||
McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2013;5(4):a008656. | ||
Fiandalo MV, Kyprianou N. Caspase control: protagonists of cancer cell apoptosis. Exp Oncol. 2012;34(3):165–175. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.