")
Back to Journals » International Journal of Nanomedicine » Volume 16
Antitumor Activity of α-Linolenic Acid-Paclitaxel Conjugate Nanoparticles: In vitro and in vivo
Authors Xu MQ, Hao YL, Wang JR, Li ZY, Li H, Feng ZH, Wang H, Wang JW, Zhang X
Received 30 July 2021
Accepted for publication 8 October 2021
Published 27 October 2021 Volume 2021:16 Pages 7269—7281
DOI https://doi.org/10.2147/IJN.S331578
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Yan Shen
Mei-Qi Xu,1,2 Yan-Li Hao,1,2 Jing-Ru Wang,1,2 Zhuo-Yue Li,1,2 Hui Li,1 Zhen-Han Feng,1,2 Hui Wang,1,2 Jing-Wen Wang,1,2 Xuan Zhang1,2
1Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, School of Pharmaceutical Sciences, Peking University, Beijing, 100191, People’s Republic of China; 2Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Beijing, 100191, People’s Republic of China
Correspondence: Xuan Zhang
Department of Pharmaceutics, School of Pharmaceutical Sciences, Peking University, Xueyuan Road 38, Beijing, 100191, People’s Republic of China
Tel +86-10-82805765
Fax +86-10-8280576
Email [email protected]
Purpose: Small molecule modified antitumor drug conjugate nanoparticles have the advantages of high drug loading, simple synthesis and preparation, and better biocompatibility. Due to the large demand for exogenous α-linolenic acid (ALA) by tumor cells, we synthesized α-linolenic acid-paclitaxel conjugate (ALA-PTX) and prepared α-linolenic acid-paclitaxel conjugate nanoparticles (ALA-PTX NPs), in order to obtain better tumor cellular uptake and antitumor activity in vitro and in vivo.
Methods: We synthesized and characterized ALA-PTX, and then prepared and characterized ALA-PTX NPs. The cellular uptake, uptake pathways, intracellular behavior, in vitro and in vivo antitumor activity of ALA-PTX NPs were evaluated.
Results: The size of ALA-PTX NPs was approximately 110.7± 1.7 nm. The drug loading was approximately 90% (w/w) with CrEL-free and organic solvent-free characteristics. The cellular uptake of ALA-PTX NPs was significantly higher than that of PTX injection by MCF-7, MCF-7/ADR and HepG2 cells. In these three cell lines, the cellular uptake of ALA-PTX NPs at 6h was approximately 1.5– 2.6 times higher than that of PTX injection. ALA-PTX NPs were ingested through clathrin-mediated endocytosis, then transferred to lysosomes, and could dissolve in cells to play an antitumor activity. The in vitro and in vivo antitumor activity of ALA-PTX NPs was confirmed in MCF-7/ADR and HepG2 cell models and tumor-bearing nude mouse models.
Conclusion: ALA-PTX NPs developed in our study could provide a new method for the preparation of nano-delivery systems suitable for antitumor therapy that could increase tumor cellular uptake and enhance antitumor activity.
Keywords: α-linolenic acid, α-linolenic acid-paclitaxel conjugate, α-linolenic acid-paclitaxel conjugate nanoparticles, cellular uptake, antitumor activity
Introduction
The application of nanomedicine in cancer treatment has attracted considerable attention as a way to enhance tumor targets and reduce adverse effects.1,2 Many nanomaterials, such as organic, inorganic, lipid, protein or polymer, are used as carrier materials for constructing antitumor nanomedicine.3,4 However, in the process of research and application, some problems have also been found to limit its clinical conversion: burst release, low drug loading (usually less than 10%), and systemic toxicity caused by a large number of carrier materials.5
The carrier-free NPs has opened up a new way for the construction of stable NPs and the development of antitumor drugs platforms.6–8 The carrier-free NPs have the higher delivery efficiency and the better safety.9–12 Many studies have constructed macromolecule-drug conjugate NPs, but there are still some problems including uneven drug loading and low occupancy of antitumor drugs. In order to overcome these shortcomings, small molecule modified antitumor drug conjugates with higher drug occupancy rates have been synthesized and these conjugates can form NPs in water. These NPs have a significantly high drug loading, simple synthesis and preparation, and better biocompatibility.
Polyunsaturated fatty acids (PUFAs) are essential components of cell membranes. In addition to its important physiological functions such as anti-inflammatory, maintaining cardiovascular health, and promoting brain development, it has also been found to have significant antitumor activity, which can increase the sensitivity of chemotherapeutic drugs, and shows great tumor treatment potential.13,14 Therefore, these PUFAs can be considered ideal functional materials for modifying chemotherapy drugs.9–11 In our previous research, we used conjugated linoleic acid (CLA) as a functional material to modify the chemotherapy drug paclitaxel (PTX) and synthesized conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) which could cross the blood-brain barrier and target brain tumor.15 CLA-PTX can form stable nanoparticles in water. The cellular uptake of CLA-PTX NPs by tumor cells was significantly enhanced.16 CLA-PTX NPs have good stability, long-term circulation, and good antitumor activity.16
Omega-3 PUFA have been shown to regulate cell growth and differentiation, anti-inflammatory, regulate immunity, and inhibit the growth and proliferation of cancer cells, invasion and angiogenesis.17–19 The latest research claimed that omega-3 PUFA may be the key to opening the blood-brain barrier.20 Omega-3 PUFA could be recognized by specialized transporters MFSD2A and promoted their crossing the blood-brain barrier.20 This ability to easily transmembrane may enable omega-3 PUFA modified drugs have higher uptake, which is helpful for the design of new drugs.
α-Linolenic acid (ALA) is a kind of omega-3 PUFAs, that cannot be synthesized endogenously. Due to increased proliferation and metabolism, tumor cells are highly dependent on the uptake of exogenous ALA.21 Studies have shown that ALA can induce breast cancer cell apoptosis by inhibiting the HER2 signal transduction pathway.22–24 Wang et al modified the 10-hydroxyl group of camptothecin derivatives (SN38) with ALA to obtain ALA-SN38, which can form stable NPs in water with good antitumor activity.9 Therefore, ALA is an ideal small molecule functional material for constructing small molecule modified antitumor drug conjugate NPs.
In the present research, we synthesized α-linolenic acid-paclitaxel conjugate (ALA-PTX). ALA-PTX was characterized by NMR and mass spectrometry analysis. α-Linolenic acid-paclitaxel conjugate nanoparticles (ALA-PTX NPs) were then prepared and characterized. We investigated the cellular uptake and uptake pathways in MCF-7, MCF-7/ADR and HepG2 cell lines. The intracellular behavior of ALA-PTX NPs in the aspects of intracellular dissolution, co-localization with endocytic proteins, intracellular transport and efflux pathways were also investigated. The in vitro and in vivo antitumor activity of ALA-PTX NPs were evaluated.
Materials and Methods
Materials
Paclitaxel (PTX) was obtained from Ouhe Co., Ltd. (Beijing, China). α-linolenic acid (ALA) was supplied from Sigma-Aldrich (St. Louis, MO, USA). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine [methoxy(polyethylene glycol)2000] (DSPE-PEG) was purchased from NOF Co. (Tokyo, Japan). Paclitaxel injection was commercially available from local hospital of Beijing (Bristol Myers Squibb Co., Princeton, NJ, U.S.A.). Hypertonic sucrose, filipin, ethylisopropylamiloride (EIPA) and chlorpromazine (CPZ) were supplied from Sigma-Aldrich (St. Louis, MO, USA). Anti-Clathrin heavy chain antibody, anti-caveolin-1-N-terminal (Alexa Fluor 488) antibody and goat anti-Rabbit IgG H&L (Alexa Fluor 647) were obtained from Abcam (Cambridge, MA, USA). Anti-Rab3A, anti-Rab8A, the dye DiI, DiO and Hoechst 33342 were purchased from Beyotime Biotechnology Co., Ltd. (Shanghai, China). LysoBriteTM NIR were obtained from AAT Bioquest (Sunnyvale, CA). All other chemicals were of analytical or HPLC grade.
Cell culture medium, RPMI 1640 and DMEM medium were obtained from Macgene Biotech Co. Ltd. (Beijing China). Fetal bovine serum (FBS) was supplied from GIBCO (Invitrogen Co. (Carlsbad, USA)).
Cell Lines
Human breast carcinoma MCF-7 cells were obtained from the Cell Resource Center, Peking Union Medical College (Beijing, China). Drug-resistant human breast carcinoma MCF-7/ADR cells and human hepatocellular carcinoma HepG2 were supplied from the Chinese Academy of Sciences Cell Bank (Shanghai, People’s Republic of China). Cells were cultured according to the recommended conditions in ATCC.
Animals
Female BALB/C nude mice (4–6 weeks) were obtained from the Experimental Animal Center of Peking University Health Science Center. All care and handling of the animals were performed with the approval of the Experimental Animal Center of Peking University Health Science Center. Additionally, this study was performed following the National Institutes of Health guidelines for the use of experimental animals.
Synthesis of ALA-PTX
ALA-PTX was synthesized according to our previous PTX conjugate method.16 Briefly, ALA-PTX was synthesized through a direct esterification reaction on the C2′-hydroxyl of PTX catalyzed by DCC/DMAP (Sigma-Aldrich, St. Louis, MO, USA). Then, the reaction mixture was filtered to remove N,N’-dicyclohexylurea (DCU), and the filtrate was dried by rotary evaporation. The crude product was purified by silica-gel column chromatography and eluted with petroleum ether/acetone (gradually decreasing from 20:1 to 3:1, v/v). The resulting eluent was dried by rotary evaporation to obtain ALA-PTX.
Characterization of ALA-PTX
1H NMR Spectra
ALA-PTX was dissolved in deuterated chloroform and determined by nuclear magnetic resonance spectroscopy (400 MHz 1H NMR, Bruker AVANCEZ 400).
Mass Spectrometry
ALA-PTX was characterized by mass spectrometry (MALDI-TOF-MS, Waters).
HPLC Analysis
The purity of ALA-PTX was determined by high-performance liquid chromatography (HPLC) according to our previous HPLC method using an LC-20AT liquid chromatograph (SHIMADZU, Japan) and SPD-M20A diode array detector (SHIMADZU, Japan). The details of the HPLC method are shown in our previous report,16,25 as shown in Table S1.
Preparation of ALA-PTX NPs
ALA-PTX NPs were prepared using a simple precipitation method.16,25,26 In brief, ALA-PTX and DSPE-PEG (ALA-PTX: DSPE-PEG = 1: 0.1, w/w) were dissolved in DMSO. A volume of 1 mL of the above solution was added dropwise into distilled water (3 mL) with stirring (300–500 rpm) at room temperature. Then, ALA-PTX NPs formed spontaneously. The resulting NP dispersion was dialyzed against distilled water for 24 h (MWCO = 3500 Da), and the obtained NP dispersion was subjected to ultrafiltration at 4000 rpm for 5 min.
For preparation of DiI-loaded ALA-PTX NPs, ALA-PTX, DSPE-PEG and the fluorescent dye DiI (100:10:5, w/w) were dissolved in DMSO. Then, a procedure similar to that used for the preparation of ALA-PTX NPs was performed.
DiO&DiI- loaded ALA-PTX NPs were prepared by slowly adding the stock solution with the drug mass ratio: ALA-PTX:DSPE-PEG:DiO:DiI = 100:10:2.5:2.5 to 10 times the volume of distilled water under constant stirring under constant stirring.
Characterization of ALA-PTX NPs
Particle Size and Zeta Potential
The particle size and zeta potential of ALA-PTX NPs were measured by a dynamic light scattering detector (Malvern, Worcestershire, UK).
Transmission Electron Microscopy (TEM)
The morphology of ALA-PTX NPs was characterized by TEM (JEM-1400Plus, JEOL, Japan).
Fourier Transform Infrared Spectroscopy (FTIR)
The lyophilized powder of ALA-PTX NPs was characterized by FT-IR measurements recorded using a Fourier transform infrared spectrometer (Nicolet-6700, Thermo Fisher, USA).
In vitro Cellular Uptake
MCF-7, MCF-7/ADR or HepG2 cells were seeded in a 12-well plate at a density of 2×105 cells/well in 1 mL of growth medium and incubated at 37 °C for 24 h. Then, the cells were treated with PTX injection, ALA-PTX solution or ALA-PTX NPs (PTX 10 μM) for 2, 4 or 6 h at 37 °C. After incubation, the cells were washed three times with cold PBS (pH 7.4) and lysed with 0.1% SDS (0.1 mL). A volume of 5 μL of the lysate was assessed to determine the protein concentration using a PierceTM BCA protein assay kit (Thermo Scientific, Waltham, USA). Then, the lysate was extracted with 0.3 mL acetonitrile and centrifuged at 10,000 rpm for 10 min. Then, the resulting supernatant (20 μL) was used to measure the content of PTX or ALA-PTX through HPLC analysis. The cellular uptake of PTX injection, ALA-PTX solution and ALA-PTX NPs was calculated using the following formula:

The Cellular Uptake Pathway of the ALA-PTX NPs
MCF-7, MCF-7/ADR or HepG2 cells were plated in 12-well plates at 2×105 cells per well for 24 h of proliferation. Then, the cell culture medium was replaced with serum-free medium containing inhibitors (listed in Table S2) and ALA-PTX NPs (10 μM) and incubated at 37 °C for 4 h. The other steps were consistent with the experimental steps used for in vitro cellular uptake.
The Intracellular Colocalization
DiI-loaded ALA-PTX NPs (ALA-PTX 20 μM) were exposed to MCF-7/ADR cells and incubated for 4 h at 37 °C. Then, the cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 for 5 min and blocked with BSA solution.
Clathrin
The cells were then incubated with the anti-clathrin heavy chain primary antibody overnight at 4 °C. Then, Alexa Fluor 647-labeled secondary antibodies were used to detect the primary antibodies. Five minutes before the end of the incubation, the nuclear dye Hoechst 33342 (20 μg/mL) was added to the medium. Finally, cells were observed with CLSM (λex= 549 nm and λem= 565 nm for DiI; λex= 350 nm and λem= 460 nm for Hoechst 33342; λex = 652 nm and λem = 668 nm for Alexa Fluor 647). The cells were then incubated with the anti-clathrin heavy chain antibody overnight at +4°C. Finally, Hoechst 33342 and Alexa Fluor 647-labeled secondary antibodies were used to detect the primary antibodies.
Caveolin-1
The cells were incubated with the anti-caveolin-1-N-terminal (Alexa Fluor 488) antibody (Abcam, Cambridge, MA, USA) overnight at 4 °C. Hoechst 33342 (Beyotime Biotechnology Co., Ltd., Shanghai, China) was used to stain the cell nuclei at a concentration of 20 µg/mL for 10 min at room temperature. Finally, the cells were observed with CLSM (λex= 549 nm and λem= 565 nm for DiI; λex= 350 nm and λem= 460 nm for Hoechst 33342; λex = 495 nm and λem = 519 nm for Alexa Fluor 488).
Exocytosis Vesicles
The cells were incubated with primary antibodies (anti-Rab3A or anti-Rab8A). Finally, Alexa Fluor 647-labeled secondary antibodies were used to detect the primary antibodies. Five minutes before the end of the incubation, the nuclear dye Hoechst 33342 was added to the medium. Finally, cells were observed with CLSM (λex= 549 nm and λem= 565 nm for DiI; λex= 350 nm and λem= 460 nm for Hoechst 33342; λex = 652 nm and λem = 668 nm for Alexa Fluor 647).
Lysosomes
The lysosome tracker in the near infrared light area (was added to the medium 30 min before the end of the incubation period. Finally, cells were observed with CLSM (λex = 636 nm and λem = 650 nm for LysoBrite).
Intracellular Dissolution of ALA-PTX NPs
MCF-7/ADR cells were seeded at a density of 15×106 cells/well in confocal dishes (cover-glass-bottom dish) and incubated at 37 °C for 24 h. Then, cells were treated with 20 µM DiO&DiI-ALA-PTX NPs in serum-free medium dilutions and incubated at 37°C and 5% CO2 for 2 hours, 6 hours, 12 hours, and 24 hours. Use the FRET SE mode of the laser scanning confocal microscope to detect, delineate the target region (Region of interest, ROI) in the FRET sample image (n=15), and calculate the FRET efficiency of the ROI. (DiO: 488 nm excitation, 500–550 nm emission; DiI: 561 nm excitation, 580–650 nm emission; FRET: 488 nm excitation, 580–650 nm emission) The FRET efficiency was calculated according to the following formula:
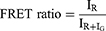
IR is the fluorescence intensity of DiI at 565 nm, and IG is the fluorescence intensity of DiO at 501 nm.
In vitro Antitumor Activity
The in vitro cytotoxicity of ALA-PTX NPs was evaluated in MCF-7/ADR using the sulfonyl rhodamine B (SRB) assay (Sigma-Aldrich, St. Louis, MO, USA).15,16,27 MCF-7/ADR cells (6×103 cells/well) were seeded in 96-well plates, incubated for 24 h, and then treated with ALA-PTX NPs at 37°C for 48 h. The cell viability was determined using SRB, which allowed quantification of the living cells by measuring absorbance at 540 nm by a 96-well plate reader (model 680; Bio-Rad Laboratories Inc., Hercules, CA, USA). The half-maximal inhibitory concentration (IC50) was calculated according to the dose-effect curves using GraphPad Prism 8 software.
In vivo Antitumor Activity
HepG2 and MCF-7/ADR cell lines were used to construct two xenograft tumor models. HepG2 cells or MCF-7/ADR cells (5×106 cells in 100 μL PBS) were subcutaneously injected into the right armpit of the forelimb of 6-week-old female BALB/c nude mice. When the tumors reached 100–300 mm3, the mice were randomly assigned to three groups (n=5) and treated with 5% glucose injection, PTX injection (10 mg/kg of PTX, iv, q3d × 4) or ALA-PTX NPs (40 mg/kg of ALA-PTX, iv, q3d × 4) through tail intravenous injection. Tumor size was measured every two days by a caliper, and the tumor volume was calculated by V = 0.5 × (width)2 × (length).
Statistical Analysis
All experimental data are expressed as the mean ± SD. The significance among groups was determined using one-way analysis of variance (ANOVA) with Bonferroni post hoc correction for comparisons between individual groups. Statistical significance was shown at p<0.01.
Results
Synthesis and Characterization of ALA-PTX
ALA-PTX was synthesized by ALA and PTX in a one-step method, coupling ALA and PTX at the 2ʹ-hydroxyl position (Figure 1A). As shown in Figure 1B and C, the characteristic peaks of ALA and PTX could all be found in ALA-PTX. As shown in Figure 1D, the anion mass spectra of ALA-PTX exhibited a single and sharp peak at m/z 1112.55, which corresponds to the [M-H]−. Therefore, ALA-PTX was successfully synthesized, and the yield of ALA-PTX was 68.9%. As shown in Figure 1E, the HPLC chromatogram showed that the peak time of ALA-PTX was 21.4 min. In addition, there were no significant impurity peaks on the chromatogram, and the main peak area accounted for 98.4% of all peak areas, so the purity of ALA-PTX was about 98.4%.
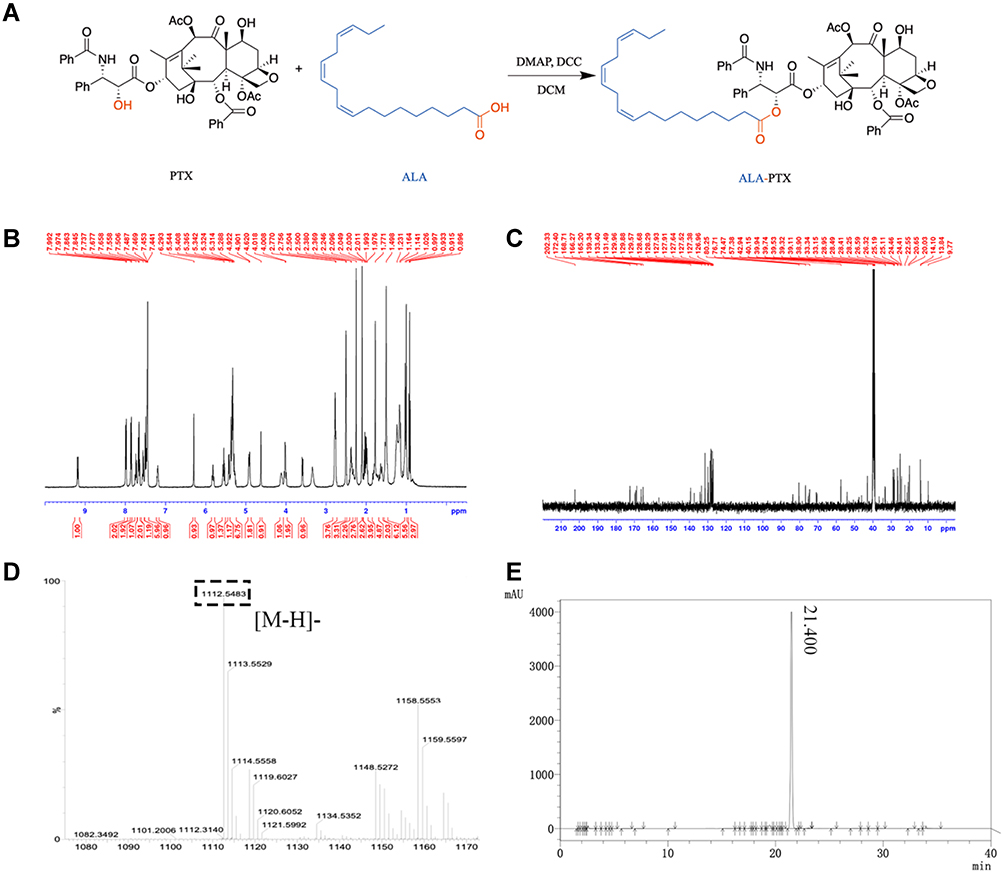 |
Figure 1 Synthesis and characterization of ALA-PTX conjugate. (A) Synthetic route of ALA-PTX. (B) 1H NMR spectra of ALA-PTX. (C) 13C NMR spectra of ALA-PTX. (D) Anion mass spectra of ALA-PTX. (E) HPLC chromatogram of ALA-PTX. |
Preparation and Characterization of ALA-PTX NPs
ALA-PTX NPs was prepared by using a simple precipitation method (Figure 2A). The drug loading was approximately 90% (w/w) with CrEL-free and organic solvent-free characteristics. The size of ALA-PTX NPs was approximately 110.7±1.7 nm. The polydispersity was 0.173. The zeta potential of the ALA-PTX NPs was −20.6±1.5 mV. The typical particle size and zeta potential of ALA-PTX NPs were shown in Figure 2B and C. ALA-PTX NPs exhibited a homogeneous distribution and good dispersibility in aqueous solution, confirmed by TEM images (Figure 2D). FTIR was used to characterize the groups on the surface of ALA-PTX NPs. As shown in Figure 2E, both ALA and PTX feature groups existed on the surface of ALA-PTX NPs.
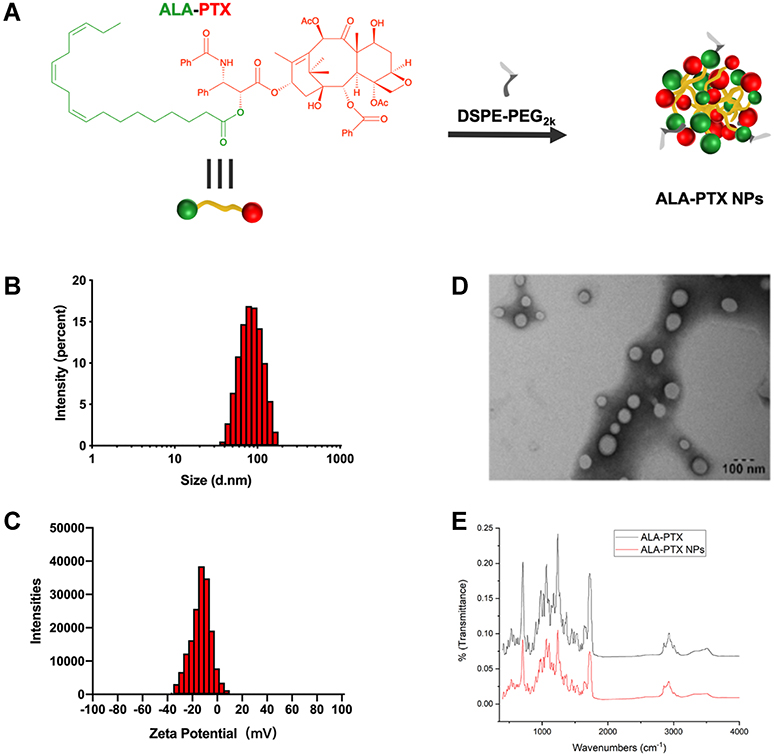 |
Figure 2 Preparation and characterization of ALA-PTX NPs. (A) Preparation of ALA-PTX NPs. (B) Particle size of ALA-PTX NPs. (C) Zeta potential of ALA-PTX NPs. (D) TEM images of ALA-PTX NPs. (E) FTIR spectra of ALA-PTX and ALA-PTX NPs. |
In vitro Cellular Uptake of the ALA-PTX NPs
The in vitro cellular uptake of ALA-PTX NPs was investigated with MCF-7, MCF-7/ADR and HepG2 cell lines. As shown in Figure 3A, compared to PTX injection, ALA-PTX in solution exhibited enhanced uptake by all three cell lines after incubation for 2, 4, or 6 h (p<0.05 or p<0.01), indicating that the functional modification of PTX with ALA increased its cellular uptake. The reason might be that tumor cells are highly dependent on the uptake of exogenous ALA.21 In addition, the uptake of ALA-PTX NPs was significantly higher than that of PTX injection and ALA-PTX in solution by all three cell lines (p<0.05 or p<0.01), demonstrating that ALA-PTX NPs were taken up by tumor cells at higher levels.
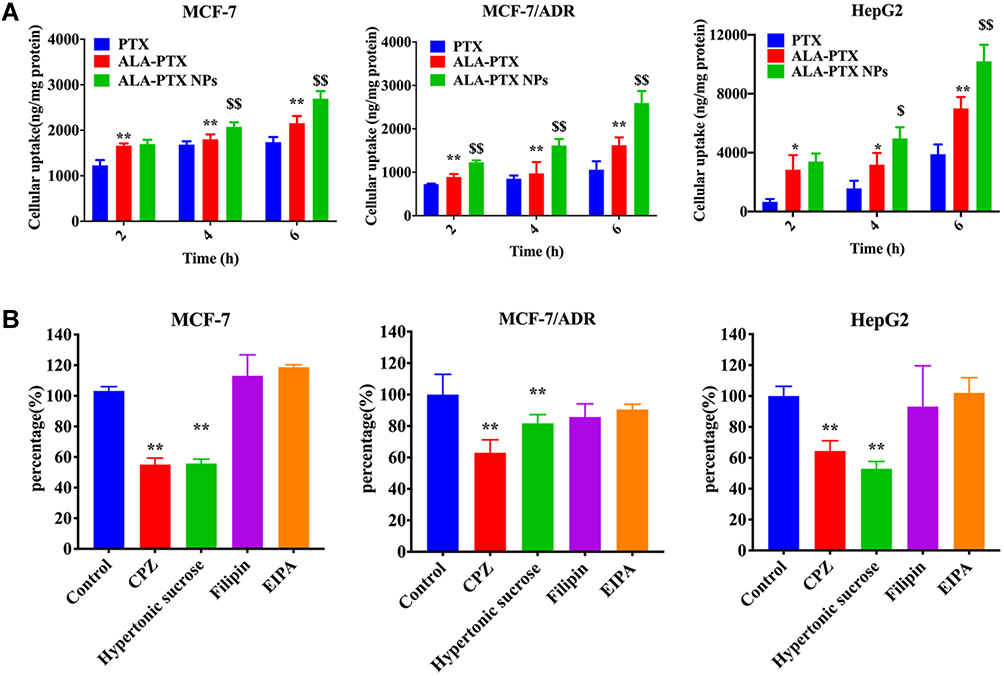 |
Figure 3 In vitro cellular uptake and antitumor efficacy of ALA-PTX NPs. (A) Uptake of ALA-PTX NPs by MCF-7, MCF-7/ADR and HepG2 cells. *p<0.05, **p<0.01, compared with PTX injection; $p<0.05, $$p<0.01, compared with the ALA-PTX solution. (B) Endocytosis inhibitors co-incubated with ALA-PTX NPs in MCF-7, MCF-7/ADR and HepG2 cell to explore the endocytosis pathways. **p<0.01, compared with the cellular uptake of the no inhibitor treatment group (control). |
Endocytosis Pathway of the ALA-PTX NPs
Identifying the NP cellular uptake pathway is important because the cellular uptake pathway can affect the function, intracellular fate and biological response of NPs.28 Commonly, cellular uptake pathways include clathrin-mediated endocytosis, caveolae-mediated endocytosis, clathrin- and caveolin-independent endocytosis, phagocytosis and macropinocytosis. We used pathway inhibitors to investigate the cellular uptake pathway of ALA-PTX NPs in MCF-7, MCF-7/ADR and HepG2 cells. The results showed that ALA-PTX NPs were taken up by these three cell lines through clathrin-mediated endocytosis, as shown in Figure 3B.
Intracellular Behaviors of ALA-PTX NPs
The co-localization of ALA-PTX NPs with clathrin was investigated, as shown in Figure 4A. The results of immunofluorescence labeling experiments showed that the co-localization of ALA-PTX NPs with clathrin was observed, indicating that ALA-PTX NPs were internalized through the clathrin-mediated endocytosis pathway. Caveolin-1 labeling immunofluorescence results indicated that colocalization of ALA-PTX NPs with caveolin-1 was not observed (Figure 4B). These results indicated that the cellular uptake pathway of ALA-PTX NPs was independent of caveolin-mediated endocytosis.
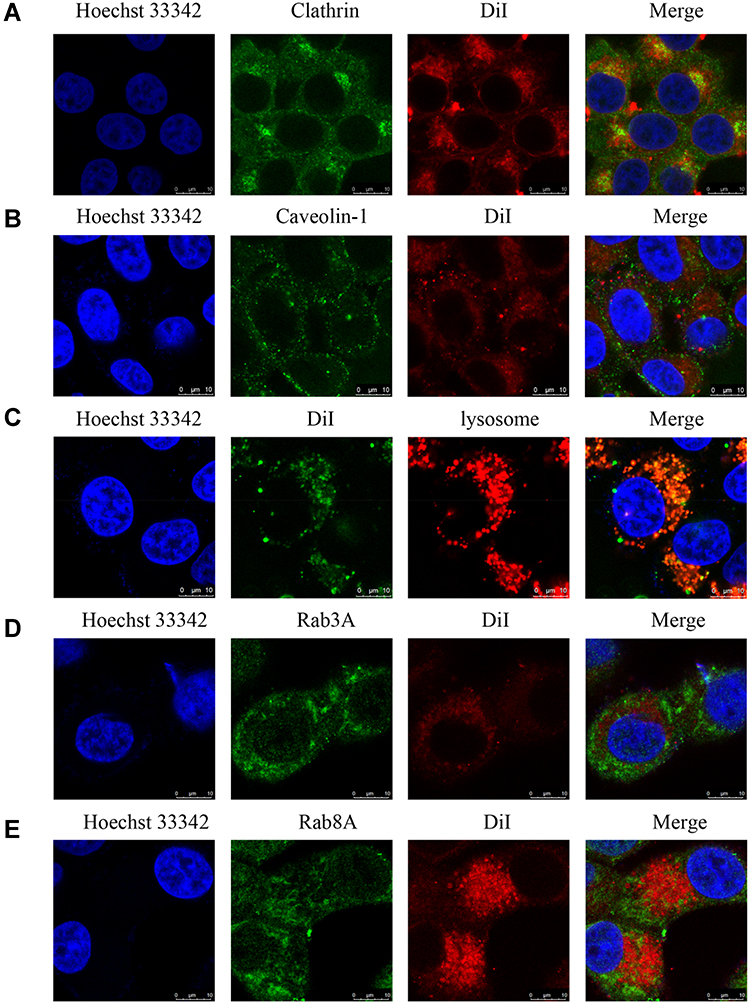 |
Figure 4 Intracellular behavior of ALA-PTX NPs in HepG2 cells. (A) Co-localization of ALA-PTX NPs and clathrin analyzed by CLSM. (B) Co-localization of ALA-PTX NPs and caveolin-1 analyzed by CLSM. (C) Co-localization of ALA-PTX NPs and lysosome analyzed by CLSM. (D) Co-localization of ALA-PTX NPs and Rab3A analyzed by CLSM. (E) Co-localization of ALA-PTX NPs and Rab8A analyzed by CLSM. |
Reportedly, NPs that enter cells through clathrin-mediated pathways eventually fuse with lysosomes.28 To investigate the intracellular transport behavior of ALA-PTX NPs, lysosomes were labeled with LysoTracker red, and the colocalization of ALA-PTX NPs with lysosomes was investigated. As shown in Figure 4C, ALA-PTX NPs extensively colocalized with lysosomes, indicating that the ALA-PTX NPs taken up by cells will be delivered into lysosomes.
In addition, the efflux of ALA-PTX NPs was also investigated. Our results indicated that classic secretory vesicles (marked by Rab3) and GLUT4 translocation vesicles (marked by Rab8)29 had almost no effect on ALA-PTX NP exocytosis, as shown in Figure 4D–E.
Intracellular Dissolution of ALA-PTX NPs
NPs need to dissolve within the cell to release free drugs to be effective. Therefore, the dissolution of NPs is very important for antitumor efficacy. The intracellular dissolution of ALA-PTX NPs was investigated by Förster resonance energy transfer (FRET) effect. The dyes DiI and DiO with FRET effect were wrapped in ALA-PTX NPs to prepare DiO&DiI-ALA-PTX NPs (Figure S1). When DiO&DiI-ALA-PTX NPs remained intact without dissolution, NPs were excited at 488 nm (excitation wavelength of DiO), and the maximum emission wavelength appeared at 565 nm. If NPs dissolved, when excited with 488 nm, the FRET effect was weakened, the emission light at 501 nm was enhanced, and the emission light at 565 nm was weakened.
As shown in Figure 5A, the water dilution of DiO&DiI-ALA-PTX NPs was excited at 488 nm, the maximum emission wavelength was at 570 nm, and there was a strong FRET effect. The FRET ratio of 0, 12, and 24 h at room temperature was 0.91. Therefore, DiO and DiI in DiO&DiI-ALA-PTX NPs would not dissolve in advance within 24 h. DiO&DiI-ALA-PTX NPs completely dissolved after being diluted with ethanol, and the FRET effect disappeared. Under the excitation of 488 nm, the maximum emission wavelength appeared at 502 nm, and the FRET ratio was 0.11. This showed that the FRET ratio of DiO&DiI-ALA-PTX NPs will decrease after the dissolution of DiO&DiI-ALA-PTX NPs. The FRET ratio can be used to qualitatively describe the intracellular dissolution of DiO&DiI-ALA-PTX NPs.
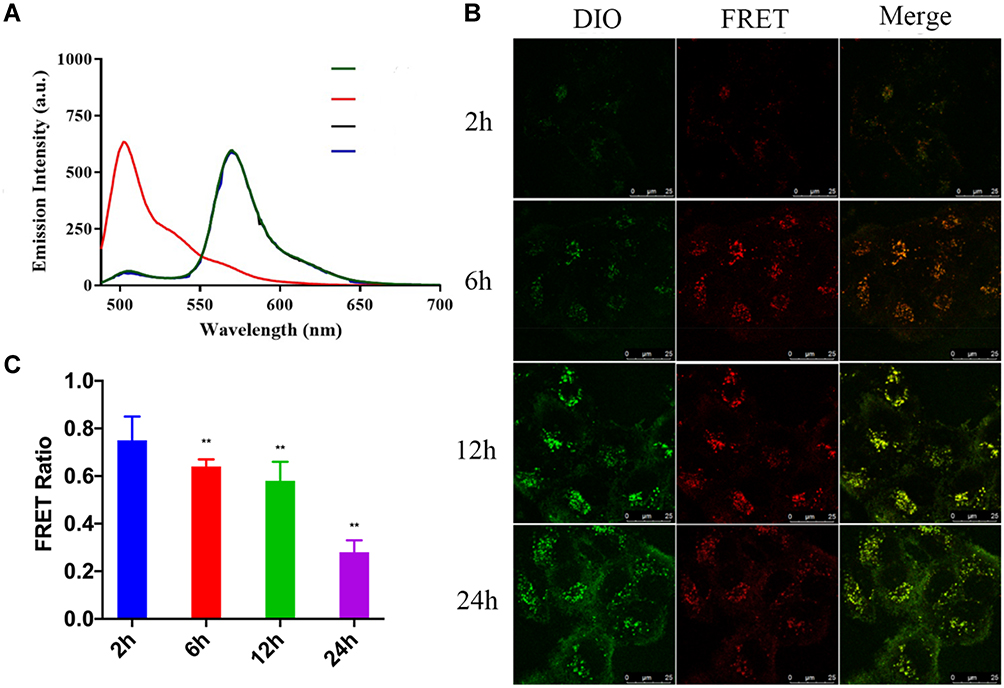 |
Figure 5 Intracellular dissolution of ALA-PTX NPs. (A) Fluorescence emission spectra of DiO&DiI-ALA-PTX NPs. Green line: ALA-PTX NPs dissolved in water at 0h; Black line: ALA-PTX NPs dissolved in water at 12h; Blue line: ALA-PTX NPs dissolved in water at 24h; Red line: ALA-PTX NPs dissolved in ethanol at 0h. (B) FRET images of MCF-7/ADR cells incubated with DiO&DiI-ALA-PTX NPs for 2, 4, 6, 12, 24h. (C) FRET ratio of MCF-7/ADR cells incubated with DiO&DiI-ALA-PTX NPs for 2, 4, 6, 12, 24h. **p<0.01, compared with the 2h group. |
DiO&DiI-ALA-PTX NPs and cells were incubated for 2, 6, 12, and 14 h, and the intracellular dissolution of NPs was observed by a confocal microscope. As shown in Figure 5B, the green fluorescence was the emission channel of DiO, which represented dissolved NPs, and the red fluorescence was the emission channel of FRET, which represented the completed NPs. At 6h, the fluorescence intensity of the FRET channel was increased compared to 2h, and the difference between 12–24h and 6h was not much. The results showed that the uptake of NPs increased first and then tended to level off. With the extension of the incubation time, the fluorescence intensity of the DiO channel continued to increase, which proved that more and more NPs were dissolved. As shown in Figure 5C, we counted the FRET ratios at different time points, and the results showed that the FRET ratio at 2, 6, 12 and 24 h was respectively 0.75±0.10, 0.64±0.03, 0.58±0.08 and 0.28±0.05. With the extension of the incubation time, the FRET ratio decreased significantly (p<0.01), especially at 24 hours. This was because the red fluorescence of the FRET channel remained unchanged and the green fluorescence intensity of DiO increased, so the FRET ratio decreased. Therefore, DiO&DiI-ALA-PTX NPs gradually dissolved after cellular uptake, and a large number of disbanded within 24 hours.
In vitro Cytotoxicity
The in vitro cytotoxicity of ALA-PTX NPs was evaluated in MCF-7/ADR and HepG2 cells, as shown in Table 1. Although the values of IC50 of ALA-PTX NPs were slightly higher than that of PTX injection, the results indicated that there was no significant difference between the IC50 of ALA-PTX NPs and PTX injection in MCF-7/ADR cells and HepG2 cells (P>0.05).
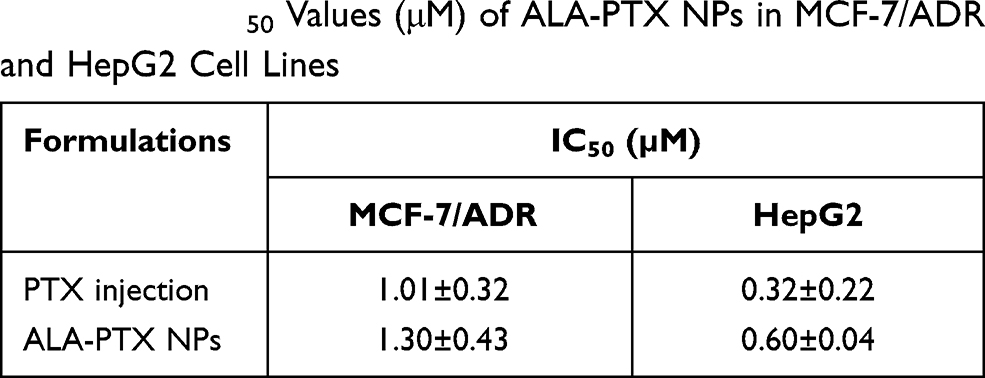 |
Table 1 The IC50 Values (μM) of ALA-PTX NPs in MCF-7/ADR and HepG2 Cell Lines |
The in vivo Antitumor Activity of ALA-PTX NPs in HepG2 Cell Tumor- and MCF-7/ADR Cell Tumor-Bearing BALB/c-Nude Mice
The in vivo antitumor activity of ALA-PTX NPs was investigated in MCF-7/ADR cell tumor-bearing nude mice. As shown in Figure 6A, tumor growth was significantly inhibited in the PTX injection and ALA-PTX NP groups compared with the control group (p<0.01). The antitumor activity in the ALA-PTX NP group was significantly higher than that in the PTX injection group (p<0.01). The individual tumor growth curve of MCF-7/ADR cell tumor-bearing BALB/c-nude mice was shown in Figure 6D. The mean tumor size 39 days after implantation in the ALA-PTX NP group and PTX injection group was 439 ± 174 and 679 ± 63 mm3, respectively, compared with 1266 ± 278 mm3 in the control group. The corresponding tumor growth inhibition in the ALA-PTX NP group and PTX injection group was 61.3% and 28.9% compared with the control group, respectively. Additionally, no obvious weight loss was observed in the mice in the treatment groups and the control group, as shown in Figure 6B. The overall survival of MCF-7/ADR tumor-bearing BALB/c-nude mice in the ALA-PTX NP treatment group was higher than that in PTX injection treatment groups, as shown in Figure 6C.
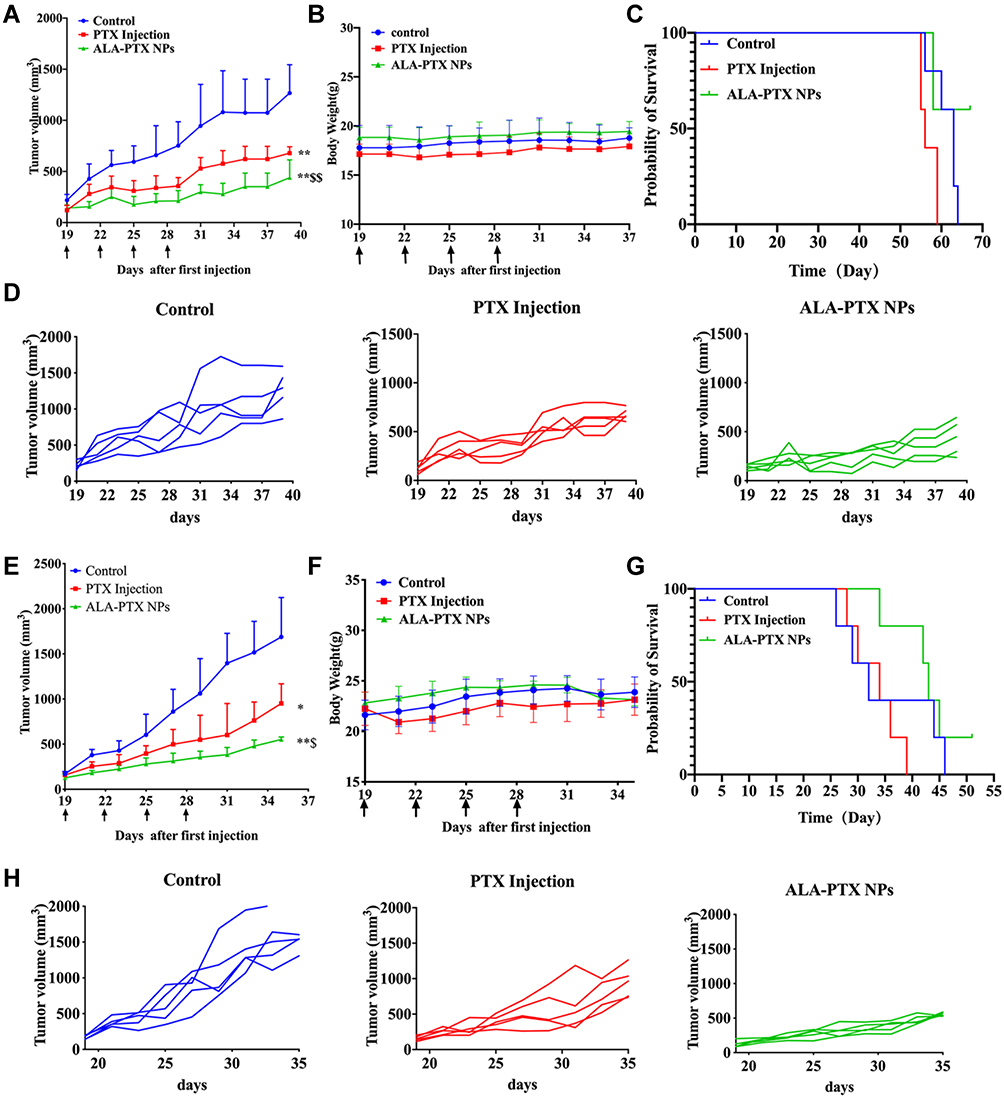 |
Figure 6 In vivo antitumor activity of ALA-PTX NPs in MCF-7/ADR cell tumor- and HepG2 cell tumor-bearing BALB/c-nude mice treated with 5% glucose injection, PTX injection (10 mg/kg of PTX, iv, q3d × 4) or ALA-PTX NPs (40 mg/kg of ALA-PTX, iv, q3d × 4). (A) Tumor growth curve during treatments of MCF-7/ADR tumor-bearing BALB/c-nude mice. (B) Body weight of MCF-7/ADR tumor-bearing BALB/c-nude mice during treatment. (C) Survival curves of MCF-7/ADR tumor-bearing BALB/c-nude mice. (D) Individual tumor growth curve of MCF-7/ADR tumor-bearing BALB/c-nude mice. (E) Tumor growth curve during the treatments of HepG2 tumor-bearing BALB/c-nude mice. (F) Body weight of HepG2 tumor-bearing BALB/c-nude mice during treatment. (G) Survival curves of HepG2 tumor-bearing BALB/c-nude mice. (H) Individual tumor growth curve of HepG2 tumor-bearing BALB/c-nude mice. *p<0.05, **p<0.01, compared with the control group; $p<0.05, $$p<0.01, compared with the PTX injection group. |
Similar results were also observed in the HepG2 cell tumor-bearing nude mouse experiments. As shown in Figure 6E, tumor growth was significantly inhibited in the PTX injection and ALA-PTX NPs treatment groups compared with the control group (p<0.05 or p<0.01). The antitumor activity in the ALA-PTX NP treatment group was significantly higher than that in the PTX injection treatment group (p<0.05). The individual tumor growth curve of the HepG2 cell tumor-bearing BALB/c-nude mice was shown in Figure 6H. The mean tumor size at day 35 after implantation in the ALA-PTX NP and PTX injection treatment groups was 552 ± 28 and 951 ± 217 mm3, respectively, compared with 1688 ± 437 mm3 in the control group. The corresponding tumor growth inhibition in the ALA-PTX NP and PTX injection treatment groups was 67.3% and 43.6% compared with the control group, respectively. Additionally, no obvious weight loss was observed in the mice in the treatment groups and the control group, as shown in Figure 6F. The overall survival of HepG2 tumor-bearing BALB/c-nude mice in the ALA-PTX NP treatment group was higher than that in PTX injection treatment groups, as shown in Figure 6G.
Discussion
Due to the large demand for exogenous α-linolenic acid (ALA) by tumor cells, we used ALA as a small molecule functional material to modify the chemotherapeutic drug PTX and synthesized ALA-PTX. Then we prepared and characterized ALA-PTX NPs. The cellular uptake of ALA-PTX NPs was significantly higher than that of PTX injection by MCF-7, MCF-7/ADR and HepG2 cells. The in vitro and in vivo antitumor activity of ALA-PTX NPs was confirmed in MCF-7/ADR and HepG2 cell models and tumor-bearing nude mouse models.
In our previous study, we proposed the prediction criterions which PTX conjugates could form stable NPs in water: when the XlogP value of PTX conjugates increased more than 1.0-fold compared to PTX, the conjugate could form stable NPs; otherwise, if its δh and/or δp decreased more than 10%, the conjugate could also form stable NPs.25 We calculated the ΔXlogP value of ALA-PTX to be 1.82. The XlogP value of ALA-PTX increased more than 1.0-fold compared to PTX. Therefore, we predicted that ALA-PTX could form stable NPs.
Successfully, we prepared ALA-PTX NPs with high drug loading (approximately 90%), and without adding a surfactant and organic solvent. Compared with PTX injection, they have higher safety and have bright application prospects. In in vitro anti-tumor efficacy experiments, our results showed that IC50 of ALA-PTX NPs was similar with that of PTX injection. We investigated the maximal tolerable dose (MTD) of ALA-PTX NPs in healthy ICR mice. The results indicated that the MTD of ALA-PTX NPs was up to 270 mg/kg (equimolar to 200mg/kg PTX). It was almost eight fold than that of PTX injection (25mg/kg), indicating that the safety of ALA-PTX NPs was significantly enhanced compared to PTX injection. Therefore, in in vivo experiments we selected high doses of ALA-PTX NPs (equivalent with 40 mg/kg of PTX), highlighting that ALA-PTX NPs can be administered at high doses and the advantage of high security. Due to the poor safety, PTX injection could not be administered at a dose of 40 mg/kg. So, we selected the administrated dose of PTX injection was 10 mg/kg of PTX.
The cellular uptake of ALA-PTX NPs, ALA-PTX solution, and PTX injection was evaluated in MCF-7, MCF-7/ADR and HepG2 cell lines. The results showed that the uptake of ALA-PTX solution was better than that of PTX injection in these three cell lines, indicating that ALA’s chemical modification of PTX could improve cellular uptake. The uptake of ALA-PTX NPs is greater than that of ALA-PTX solution and PTX injection in these three cell lines, which showed that NP dosage forms had advantages in cellular uptake. Recent studies have found that omega-3 PUFAs have the property of crossing the blood-brain barrier through specific receptors.20 We speculated that ALA, which is easily transmembrane mediated by receptors, was the reason for the high uptake of ALA-modified chemotherapeutic drugs and their NPs in tumor cells.
After cell internalization, NPs are transported and transported to various intracellular destinations.28 If NP cellular uptake occurs via endocytic pathways, NPs are confined within a membrane-lined vesicle, such as an endosome.30 These vesicles are transported throughout the cell in complex modes of transport.28 Therefore, it is important to understand and explore intracellular behaviors for NPs. We conducted a relatively complete investigation of the intracellular behaviors of ALA-PTX NPs in the aspects of co-localization with endocytic proteins, intracellular transport, and efflux pathways. Our results showed that ALA-PTX NPs were ingested through clathrin-mediated endocytosis, then transferred to lysosomes, and did not excrete cells through classic secretory vesicles and GLUT4 translocation vesicles. ALA-PTX NPs could dissolve in cells to play an antitumor activity.
Conclusion
In summary, we synthesized and characterized ALA-PTX, and then prepared and characterized ALA-PTX NPs. The cellular uptake of ALA-PTX NPs was significantly higher than that of PTX injection by MCF-7, MCF-7/ADR and HepG2 cells. ALA-PTX NPs were ingested through clathrin-mediated endocytosis, then transferred to lysosomes. ALA-PTX NPs could dissolve in cells to play an antitumor activity. The in vitro and in vivo antitumor activity of ALA-PTX NPs was confirmed in MCF-7/ADR and HepG2 cell models and tumor-bearing nude mouse models. In the future, we will study the in vivo behavior of ALA-PTX NPs, such as experiments on the concentration of nanoparticles in tumor tissue and healthy tissue. ALA-PTX NPs developed in this study could provide a new method for the preparation of nano-delivery systems suitable for antitumor therapy that could increase tumor cellular uptake and enhance antitumor activity. The NPs which do not have surfactants and organic solvents, are expected to be clinically converted.
Acknowledgments
Authors gratefully acknowledge the financial support from the National Key Research and Development Program of China (2017YFA0205600), the National Natural Science Foundation of China (No. 81573360), the Major New Drug Innovation and Development Program of China (2018ZX09721003-004), and the Innovation Team of the Ministry of Education (No. BMU2017TD003).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shi J, Kantoff PW, Wooster R, et al. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017;17:20–37. doi:10.1038/nrc.2016.108
2. Pellico J, Gawne PJ, de Rosales TM. Radiolabelling of nanomaterials for medical imaging and therapy. Chem Soc Rev. 2021;50:3355–3423.
3. Peer D, Karp JM, Hong S, et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–760. doi:10.1038/nnano.2007.387
4. Bahrami B, Hojjat-Farsangi M, Mohammadi H, et al. Nanoparticles and targeted drug delivery in cancer therapy. Immunol Lett. 2017;190:64–83.
5. Xin Y, Huang Q, Tang J-Q, et al. Nanoscale drug delivery for targeted chemotherapy. Cancer Lett. 2016;379:24–31. doi:10.1016/j.canlet.2016.05.023
6. Sreekanth V, Bajaj A. Recent advances in engineering of lipid drug conjugates for cancer therapy. ACS Biomater Sci Eng. 2019;5:4148–4166. doi:10.1021/acsbiomaterials.9b00689
7. Karaosmanoglu S, Zhou M, Shi B, et al. Carrier-free nanodrugs for safe and effective cancer treatment. J Control Release. 2021;329:805–832. doi:10.1016/j.jconrel.2020.10.014
8. Zheng Y, Ying X, Su Y, et al. Kinetically-stable small-molecule prodrug nanoassemblies for cancer chemotherapy. Int J Pharm. 2021;597:120369. doi:10.1016/j.ijpharm.2021.120369
9. Wang H, Xie H, Wang J, et al. Self-assembling prodrugs by precise programming of molecular structures that contribute distinct stability, pharmacokinetics, and antitumor efficacy. Adv Funct Mater. 2015;25:4956–4965. doi:10.1002/adfm.201501953
10. Fang T, Dong Y, Zhang X, et al. Integrating a novel SN38 prodrug into the PEGylated liposomal system as a robust platform for efficient cancer therapy in solid tumors. Int J Pharm. 2016;512:39–48. doi:10.1016/j.ijpharm.2016.08.036
11. Zhang T, Li M, Yang R, et al. Therapeutic efficacy of lipid emulsions of docetaxel-linoleic acid conjugate in breast cancer. Int J Pharm. 2018;546:61–69. doi:10.1016/j.ijpharm.2018.05.032
12. Zeng J, Li C, Duan X, et al. PEGylation of lipophilic SN38 prodrug with DSPE-mPEG (2000) versus cremophor EL: comparative study for intravenous chemotherapy. Drug Deliv. 2019;26:354–362. doi:10.1080/10717544.2019.1587045
13. Schulze M, Minihane A, Saleh R, et al. Intake and metabolism of omega-3 and omega-6 polyunsaturated fatty acids: nutritional implications for cardiometabolic diseases. Lancet Diabetes Endocrinol. 2020;8:915–930. doi:10.1016/S2213-8587(20)30148-0
14. Panagiotopoulos AA, Kalyvianaki K, Castanas E, et al. Eicosanoids in prostate cancer. Cancer Metastasis Rev. 2018;37:237–243. doi:10.1007/s10555-018-9750-0
15. Ke XY, Zhao BJ, Zhao X, et al. The therapeutic efficacy of conjugated linoleic acid - paclitaxel on glioma in the rat. Biomaterials. 2010;31:5855–5864. doi:10.1016/j.biomaterials.2010.03.079
16. Zhong T, Yao X, Zhang S, et al. A self-assembling nanomedicine of conjugated linoleic acid-paclitaxel conjugate (CLA-PTX) with higher drug loading and carrier-free characteristic. Sci Rep. 2016;6:36614. doi:10.1038/srep36614
17. Golanski J, Szymanska P, Rozalski M. Effects of omega-3 polyunsaturated fatty acids and their metabolites on haemostasis-current perspectives in cardiovascular disease. Int J Mol Sci. 2021;22(5):2394. doi:10.3390/ijms22052394
18. Zaloga G. nNarrative review of −3 polyunsaturated fatty acid supplementation upon immune functions, resolution molecules and lipid peroxidation. Nutrients. 2021;13(2):662. doi:10.3390/nu13020662
19. Macaron T, Giudici K, Bowman G, et al. Associations of Omega-3 fatty acids with brain morphology and volume in cognitively healthy older adults: a narrative review. Ageing Res Rev. 2021;67:101300. doi:10.1016/j.arr.2021.101300
20. Cater R, Chua G, Erramilli S, et al. Structural basis of omega-3 fatty acid transport across the blood-brain barrier. Nature. 2021;595:315–319. doi:10.1038/s41586-021-03650-9
21. Gu Z, Suburu J, Chen H, et al. Mechanisms of omega-3 polyunsaturated fatty acids in prostate cancer prevention. Biomed Res Int. 2013;2013:824563. doi:10.1155/2013/824563
22. Truan JS, Chen JM, Thompson LU. Flaxseed oil reduces the growth of human breast tumors (MCF-7) at high levels of circulating estrogen. Mol Nutr Food Res. 2010;54:1414–1421. doi:10.1002/mnfr.200900521
23. Abouelela SH, Prasse KW, Farrell RL, et al. Effects of D,L-2-difluoromethylornithine and indomethacin on mammary tumor promotion in rats fed high n-3 and/or n-6 fat diets. Cancer Res. 1989;49:1434–1440.
24. Brown MD, Hart CA, Gazi E, et al. Promotion of prostatic metastatic migration towards human bone marrow stoma by Omega 6 and its inhibition by Omega 3 PUFAs. Br J Cancer. 2006;94:842–853. doi:10.1038/sj.bjc.6603030
25. Zhong T, Hao YL, Yao X, et al. Effect of XlogP and Hansen solubility parameters on small molecule modified paclitaxel anticancer drug conjugates self-assembled into nanoparticles. Bioconjug Chem. 2018;29:437–444. doi:10.1021/acs.bioconjchem.7b00767
26. Duan XC, Yao X, Zhang S, et al. Antitumor activity of the bioreductive prodrug 3-(2-nitrophenyl) propionic acid-paclitaxel nanoparticles (NPPA-PTX NPs) on MDA-MB-231 cells: in vitro and in vivo. Int J Nanomedicine. 2019;14:195–204. doi:10.2147/IJN.S186556
27. Du R, Zhong T, Zhang WQ, et al. Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid-paclitaxel (CLA-PTX) on B16-F10 melanoma. Int J Nanomedicine. 2014;9:3091–3105.
28. Donahue ND, Acar H, Wilhelm S. Concepts of nanoparticle cellular uptake, intracellular trafficking, and kinetics in nanomedicine. Adv Drug Deliv Rev. 2019;143:68–96.
29. Ding L, Zhu X, Wang Y, et al. Intracellular fate of nanoparticles with polydopamine surface engineering and a novel strategy for exocytosis-inhibiting, lysosome impairment-based cancer therapy. Nano Lett. 2017;17:6790–6801. doi:10.1021/acs.nanolett.7b03021
30. Kim CS, Li X, Jiang Y, et al. Cellular imaging of endosome entrapped small gold nanoparticles. MethodsX. 2015;2:306–315. doi:10.1016/j.mex.2015.06.001
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.