")
Back to Journals » Drug Design, Development and Therapy » Volume 13
Antiproliferative activity and possible mechanism of action of certain 5-methoxyindole tethered C-5 functionalized isatins
Authors Almutairi MS, Hassan ES, Keeton AB, Piazza GA, Abdelhameed AS , Attia MI
Received 10 March 2019
Accepted for publication 19 August 2019
Published 27 August 2019 Volume 2019:13 Pages 3069—3078
DOI https://doi.org/10.2147/DDDT.S208241
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sukesh Voruganti
Maha S Almutairi,1 Eman S Hassan,2 Adam B Keeton,3 Gary A Piazza,3 Ali S Abdelhameed,1 Mohamed I Attia1,4
1Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, Riyadh 11451, Saudi Arabia; 2Department of Medical Laboratory Sciences, Al-Ghad International Medical Sciences College, Female Section, Riyadh 13315, Saudi Arabia; 3Department of Oncologic Sciences and Pharmacology, Drug Discovery Research Center, Mitchell Cancer Institute, University of South Alabama, Mobile, AL 36604-1405, USA; 4Medicinal and Pharmaceutical Chemistry Department, Pharmaceutical and Drug Industries Research Division, National Research Centre (ID: 60014618), Giza 12622, Egypt
Correspondence: Mohamed I Attia
Department of Pharmaceutical Chemistry, College of Pharmacy, King Saud University, P.O. Box. 2457, Riyadh 11451, Saudi Arabia
Tel +966 1 467 7337
Fax +966 1 467 6220
Email [email protected]
Background: Cancer is one of the most dreaded human diseases, that has become an ever-increasing health problem and is a prime cause of death globally. The potential antiproliferative activity of certain indole–isatin molecular hybrids 5a-w was evaluated in vitro against three human cancer cell lines.
Methods: Standard protocols were adopted to examine the antiproliferative potential and mechanisms of compounds 5a-w. Western blot analysis was carried out on compound 5o.
Results: Compounds 5a-w demonstrated in vitro antiproliferative activity in the range of 22.6–97.8%, with compounds 5o and 5w being the most active antiproliferative compounds with IC50 values of 1.69 and 1.91 μM, which is fivefold and fourfold more potent than sunitinib (IC50=8.11 μM), respectively. Compound 5o was selected for in-depth pharmacological testing to understand its possible mechanism of antiproliferative activity. It caused a lengthening of the G1 phase and a reduction in the S and G2/M phases of the cell cycle and had an IC50 value of 10.4 μM with the resistant NCI-H69AR cancer cell line. Moreover, compound 5o significantly decreased the amount of phosphorylated Rb protein in a dose-dependent fashion, which was confirmed via Western blot analysis.
Conclusion: The current investigation highlighted the potential antiproliferative activity of compounds 5a-w as well as the antiproliferative profile of compound 5o. These compounds can be harnessed as new lead antiproliferatives in the preclinical studies of cancer chemotherapy.
Keywords: isatin, indole, synthesis, antiproliferative, apoptosis
Introduction
Cancer is one of the most terrifying diseases of humanity and has become a fundamental health problem and a principal cause of death globally. One in four deaths in the United States is a result of cancer.1 More than ten million new cases of cancer occur every year, approximately half of which occur in developed countries, with the disease causing more than six million deaths every year.2–4 A molecularly targeted approach has recently been utilized for the management of disseminated cancer which depends on the study of oncogenes and tumor suppressors which are involved in the emergence of human cancers.5 Consequently, there has been an advancement in the specificity of cancer management, progressing from the use of general cytotoxic agents such as nitrogen mustard in the 1940s, the development of chemotherapeutic agents such as anthracyclines and Vinca alkaloids from natural resources in the 1960s and finally the use of specific monoclonal antibodies6 and specific chemotherapeutic agents which inhibit protein tyrosine kinases (PTKs) as advanced approaches.7–9 These targeted chemotherapeutic agents usually attenuate signaling pathways which control the cancer cell cycle and alter its microenvironment, blocking tumor cell proliferation, cell apoptosis and/or hindering tumor mass growth.10 These developments led to a reduction of anticancer side effects and ameliorated the response rate. Therefore, the study of the mechanisms by which cancers resist chemotherapeutic agents gave rise to a deep understanding of the reasons for the failure of cancer therapies.
Indole (I, Figure 1) is a privileged bicyclic structure which was first synthesized in 1866. The indole scaffold is incorporated into a large number of biologically active molecules endowed with a wide range of bioactivities and is naturally occurring in Vinca and ergot alkaloids, fungal metabolites and marines.11 In recent years, indole and its functionalized derivatives have been embedded in myriad bioactive pharmaceuticals including anti-inflammatories, analgesics, antimicrobials and antitumors.12–18 Furthermore, 5-methoxyindole is the fundamental fragment in the natural hormone melatonin (MLT, II, Figure 1). MLT and its derivatives have a broad spectrum of pharmaceutical applications, particularly for the treatment of headache, depression and sleep disorders, and for the management of certain types of cancer.19–21
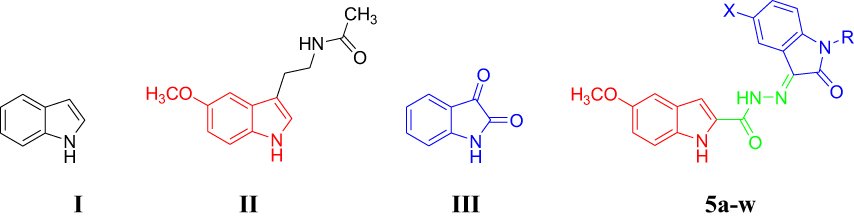 |
Figure 1 Chemical structures of compounds I-III and 5a-w. |
On the other hand, isatin (2,3-dioxindole, III) is considered an oxidized form of indole and has been recognized to be an endogenous multifunctional molecule in human beings and other mammals.22 The special electronic properties of isatin along with its proper molecular size give rise to several different valuable biological characteristics. Therefore, isatin was embedded into the backbone of various bioactive molecules including anticonvulsants,23 antifungals,24 antibacterials,25 anti-HIV agents24,26 and anticancer agents.27–31
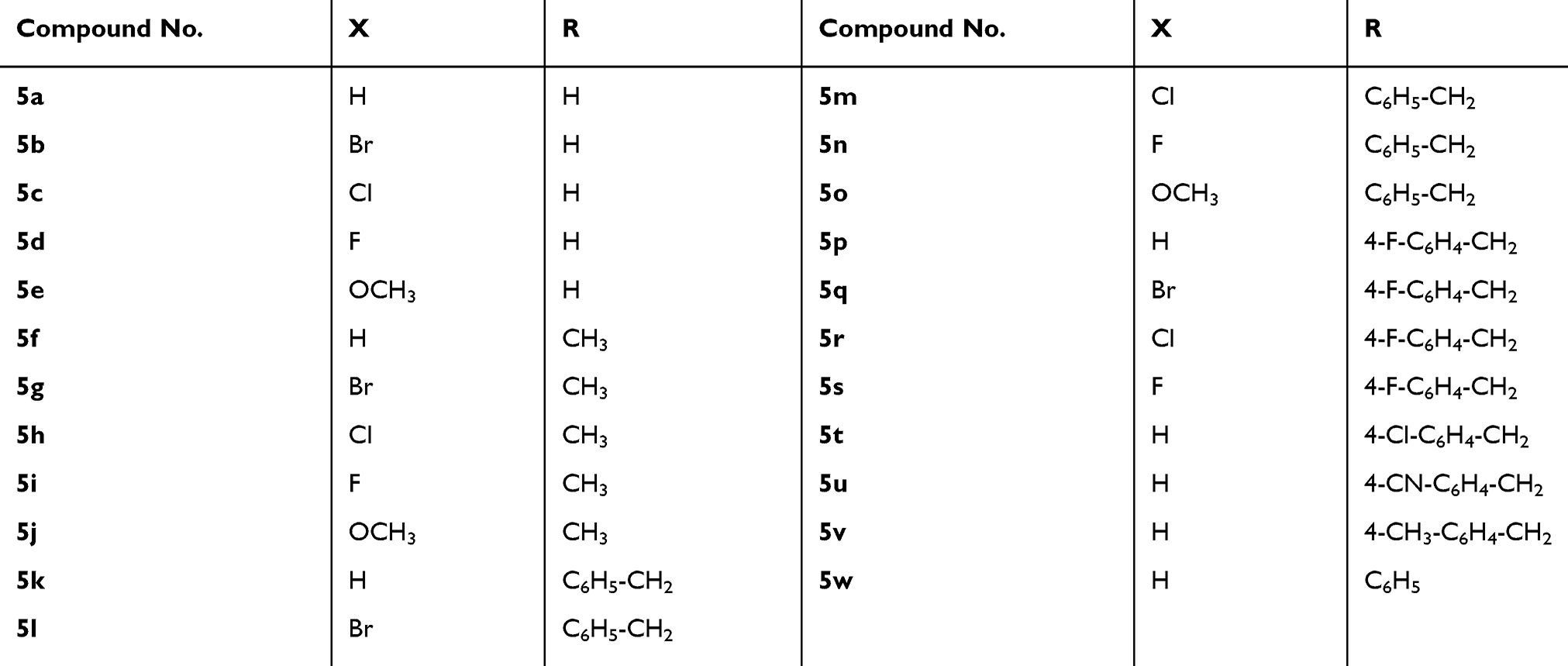
According to the aforementioned premises, it was our interest to prepare the indole–isatin conjugates 5a-w as hybrid molecules tailored from indole and isatin pharmacophore fragments for biological evaluation. The isatin moiety of the target conjugates 5a-w is functionalized on their C-5 position and bears various N-aralkyl substitutions that alter the electronic and lipophilic environment, allowing us to explore their impact on the biological activity of compounds 5a-w. Compounds 5a-wdisplayed moderate antimicrobial potential.32,33 The current report deals with the assessment of their in vitro antiproliferative potential. The most active antiproliferative candidates were subjected to deep pharmacological testing to gain insight into the possible mechanism of their antiproliferative activity.
Materials and methods
Chemistry
5-Methoxy-1H-indole-2-carbohydrazide (3) – The acid hydrazide 3 was prepared from the corresponding ester 234 using the documented method.32 It has a melting point (m.p.) of 266–268°C.
General method for the preparation of 5-methoxy-1H-indole-2-carbo hydrazide derivatives 5a-w
Glacial acetic acid (catalytic amount) was added to a mixture of the proper isatin derivative 4a-n (1 mmol) and the acid hydrazide 3 (1 mmol) in absolute ethyl alcohol (15 mL). The reaction mixture was then stirred under reflux for 4 hrs. The precipitated solid was filtered while hot, and the obtained solid was recrystallized from an ethyl alcohol/dimethylformamide mixture (3:1) to furnish the corresponding compounds 5a-w in 43–94% yields. The analytical data of compounds 5a-w are previously documented.32,33
Pharmacological evaluation
Pharmacological assessment of the title compounds including antiproliferative activity, selectivity, cell cycle effects and quantitative immunofluorescence of 5a-w was performed with previously documented methods.29 Western blot analysis of total cellular proteins enabled detection of P-Rb and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) using antibodies obtained from Cell Signaling Technology (Boston, MA, USA). Western blots were imaged by direct imaging of chemiluminescent blots (ChemiDoc Imaging System; BioRad, Hercules, CA, USA). Quantitation was carried out using NIH ImageJ public domain image analysis software. The cell lines were purchased commercially from the American Type Culture Collection (ATCC).
Results and discussion
Chemistry
Compounds 5a-w were prepared as illustrated in Scheme 1. Thus, the commercially available 5-methoxy indole-2-carboxylic acid (1) was esterified in absolute methanol and a catalytic amount of concentrated sulfuric acid, followed by hydrazinolysis, to prepare the hydrazide 3. Subsequently, compound 3 was reacted with the isatin derivatives 4a-n33 to achieve the respective title compounds 5a-w in moderate yields.
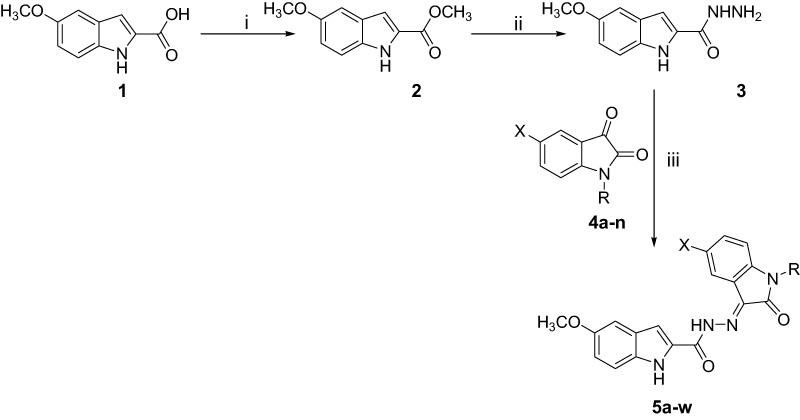 |
Scheme 1 Synthesis of compounds 5a-w. Reagents and conditions: (I) absolute methanol, H2SO4 (few drops), reflux, 4 hrs; (ii) absolute methanol, H2N-NH2.H2O, reflux, 2 hrs; (iii) absolute ethanol, acetic acid (few drops), reflux, 4 hrs. |
 |
|
Pharmacological investigations
Antiproliferative activity
The isatin nucleus is incorporated into various anticancer candidates.28,29,31,35,36 The preliminary antiproliferative potential of compounds 5a-w was tested using A-549 (lung), HT-29 (colon) and ZR-75 (breast) human cancer cell lines, and the obtained data are presented in Table 1. Sunitinib was used against the same human cancer cell lines as a reference drug for the experiments. The results are expressed as an average percent growth inhibition at 30 µM concentration for each compound tested in quadruplicate. The title compounds 5a-w exhibited an average growth inhibition of 22.6–97.8% in the antiproliferative assay against the tested human cancer cell lines, except for compound 5e, which stimulated the growth of the ZR-75 cell line. It seems that the N-unsubstituted isatin (compounds 5b and 5c), N-methyl (compounds 5g-i), N-benzyl (compound 5o) or N-phenyl (compound 5w) moieties are the preferred fragments at the isatin nitrogen, as they induced average growth inhibition of 96.0, 91.3, 94.5, 95.3, 91.8, 97.8 and 97.6%, respectively. Also, halogen substitution at isatin C-5 is the favored substituent, except for compounds 5o and 5w which bear methoxy and hydrogen functionalities, respectively.
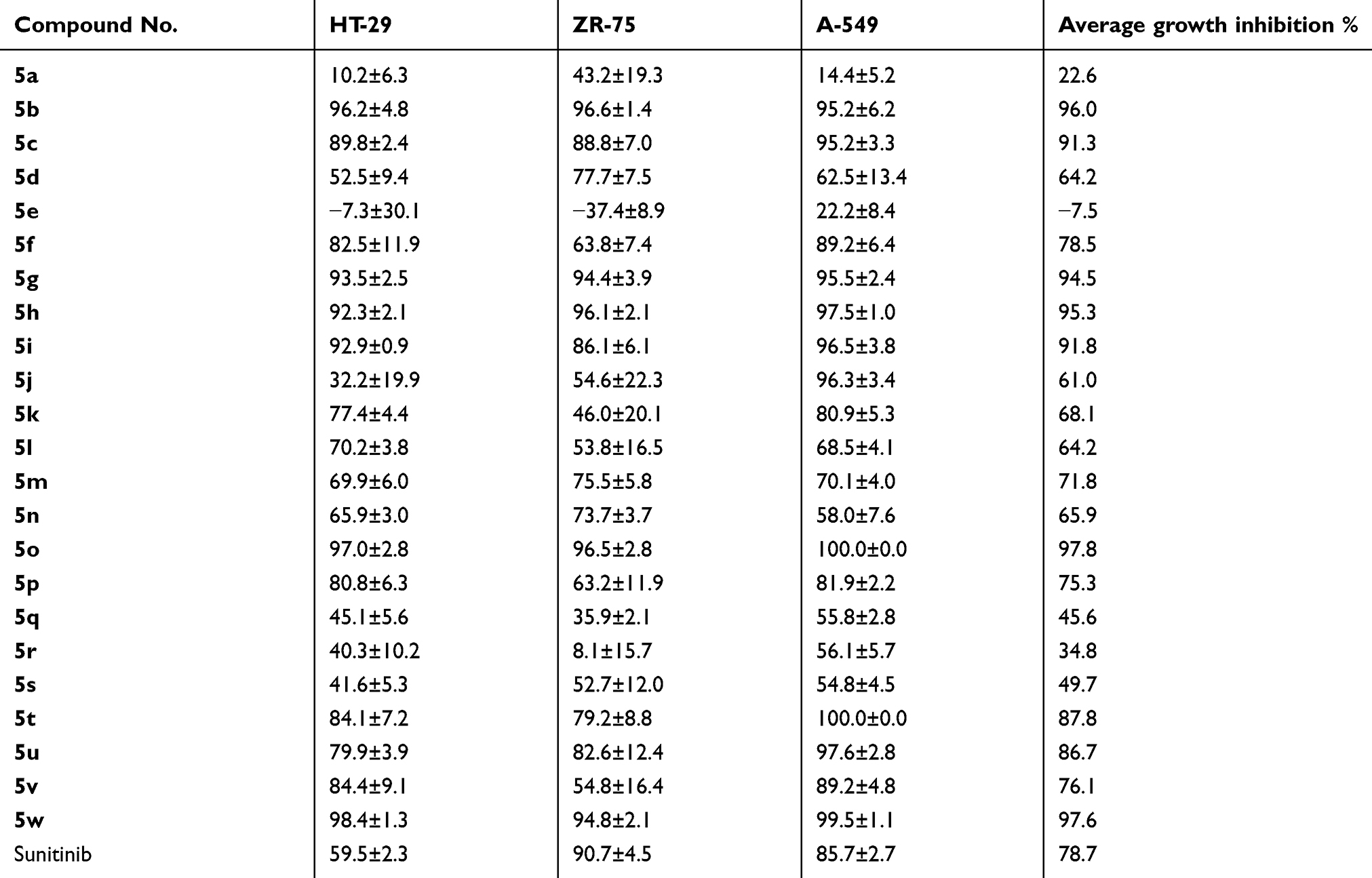 |
Table 1 In vitro antiproliferative potential of compounds 5a-w and sunitinib against HT-29, ZR-75 and A-549 cell lines |
Compounds displaying an average growth inhibition of more than 90% toward ZR-75, HT-29, and A-549 cell lines were subjected to median growth inhibitory concentration (IC50) determination. Table 2 illustrates the IC50 values of compounds 5b, 5c, 5g-i, 5o, 5w and sunitinib toward ZR-75, HT-29 and A-549 cell lines. The most active candidates are 5o (bearing an N-benzylisatin moiety) and 5w (bearing an N-phenylisatin moiety) with IC50 values of 1.69 and 1.91 µM, respectively, which are about fivefold and fourfold more potent than sunitinib (IC50=8.11 µM). Therefore, detailed pharmacological studies were carried on compound 5o, aiming to gain insight into the integrated pharmacological profile of this compound, as a representative for compounds 5a-w.
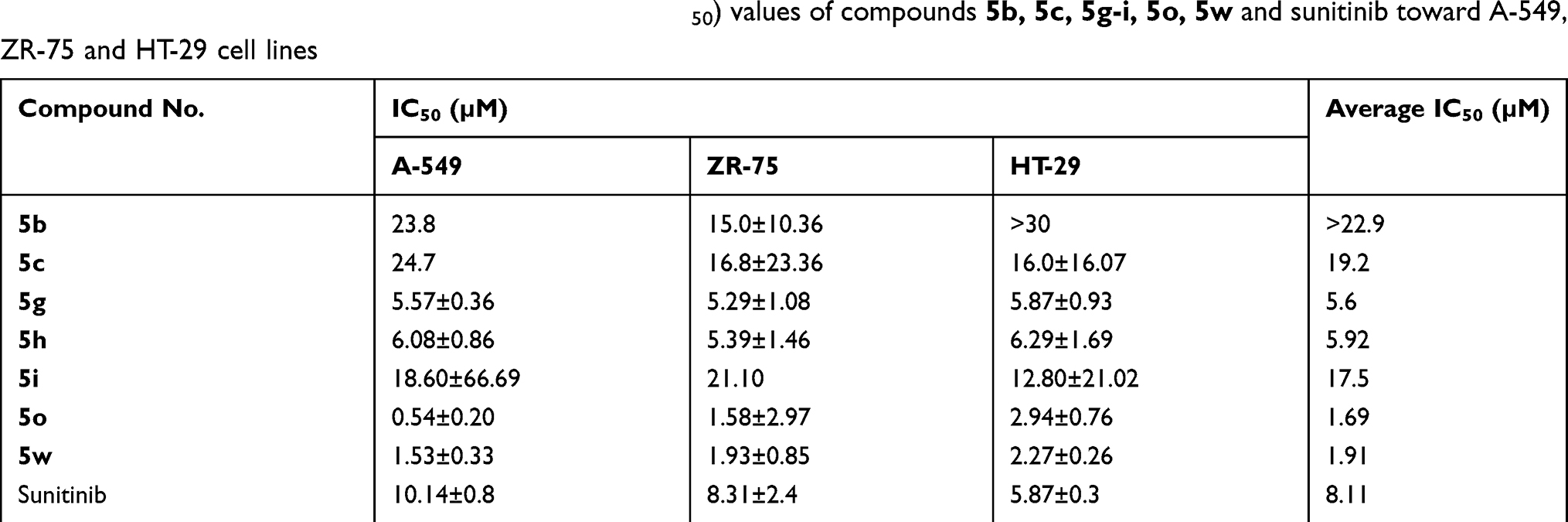 |
Table 2 Antiproliferative inhibitory concentration 50% (IC50) values of compounds 5b, 5c, 5g-i, 5o, 5w and sunitinib toward A-549, ZR-75 and HT-29 cell lines |
Caspase 3/7 activity
The A-549 cell line was utilized to assess the apoptosis-inducing potential of compound 5o. Activity assessment of compound 5o was carried out at concentrations equal to its IC50 for growth inhibition and at threefold above this concentration over a 2–48 hr time course. Compound 5o did not induce any substantial rise in caspase 3/7 activity at any concentration or time point tested.
Cell cycle influences
The A-549 cell line was used to examine the influence of compound 5o on different features of the cell cycle progression. Activity assessment of compound 5o was conducted using immunofluorescent imaging of phosphorylated Rb protein as well as by quantification of the total DNA content of each cell to ascertain the phase of the cell cycle. Concentrations of less than 100 µM to 50 nM of compound 5o were utilized to assess its capability to influence cell cycle distribution as well as Rb phosphorylation. Figure 2 and Table 3 indicated that the total cell number was reduced with an IC50 value of 2.20 µM after a 48 h treatment. Also, the levels of phosphorylated Rb protein were substantially decreased in a dose-dependent manner (Figure 3A).
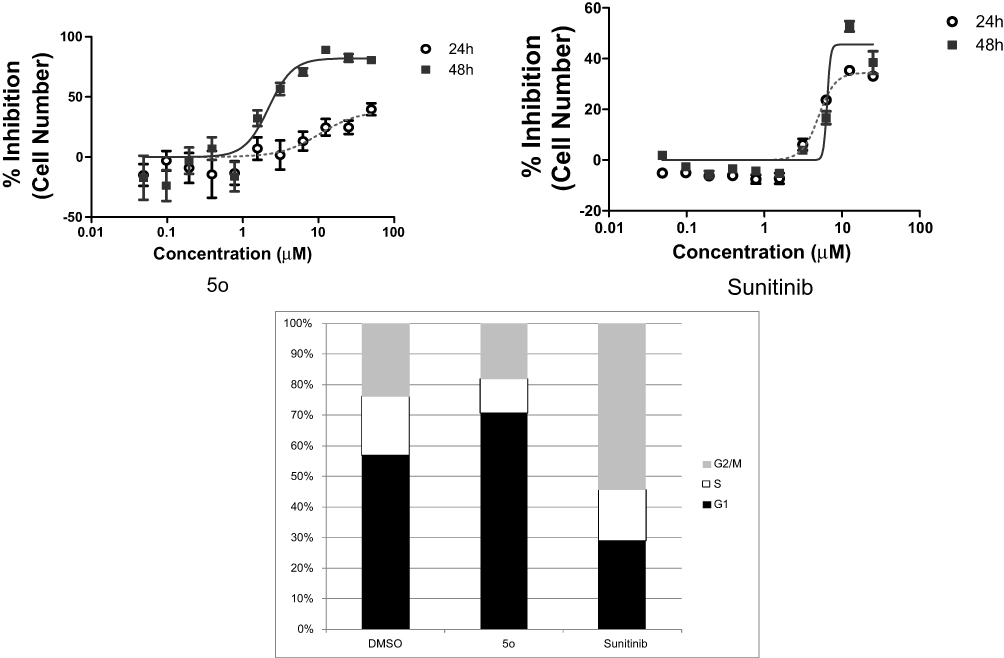 |
Figure 2 Cell cycle influences of compound 5o after incubation for 24 and 48 hrs. |
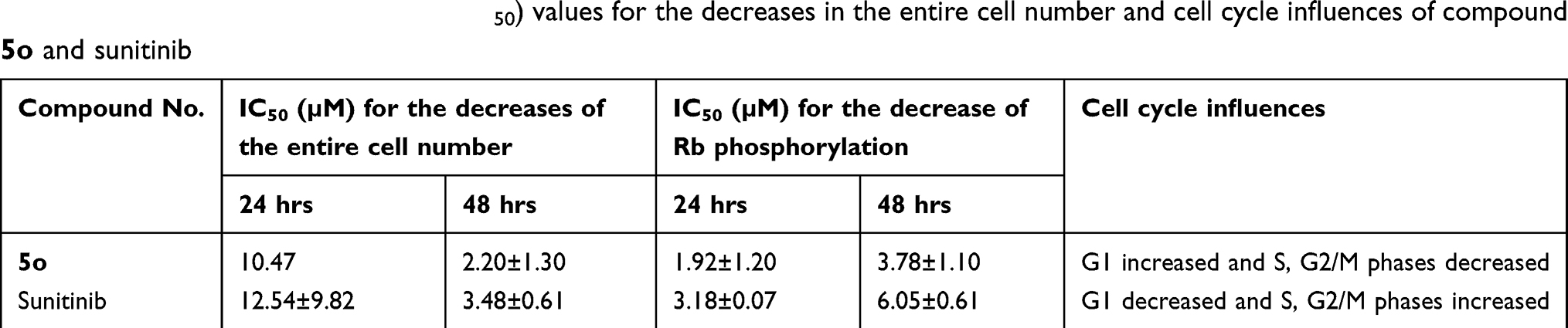 |
Table 3 Inhibitory concentration 50% (IC50) values for the decreases in the entire cell number and cell cycle influences of compound 5o and sunitinib |
Independent experiments confirmed the effect of 5o to reduce Rb phosphorylation by Western blot analysis (Figure 3B). Moreover, compound 5o induced a reduction in the percentage of cells in the S and G2/M phases of the cell cycle, with a concomitant rise in the G1 phase. These results suggest that part of the growth inhibition effect of compound 5o could be attributed to decreases in the progression rate of the cell cycle, with a concomitant reduction in proliferation. On the contrary, sunitinib showed an increase in the percentage of cells in the S or G2/M phases of the cell cycle, with a concomitant reduction in the G1 phase. Mitotic catastrophe followed by programmed death of cells containing aberrant or multiple nuclei may result from mitotic arrest due to arrest in the G2 phase of the cell cycle, which might represent a checkpoint blockade.
It should be mentioned that both compound 5o and sunitinib substantially reduced the extent of phosphorylated Rb protein in a dose-dependent fashion (Figure 3). Compound 5o exhibited IC50 values of 3.78 and 1.92 µM after 48 and 24 hrs, respectively, which was roughly twofold more potent than sunitinib (Table 3). This may advance the assumption that the growth inhibitory potential of 5o could be attributed, in part, to its ability to inhibit cyclin-dependent kinases.
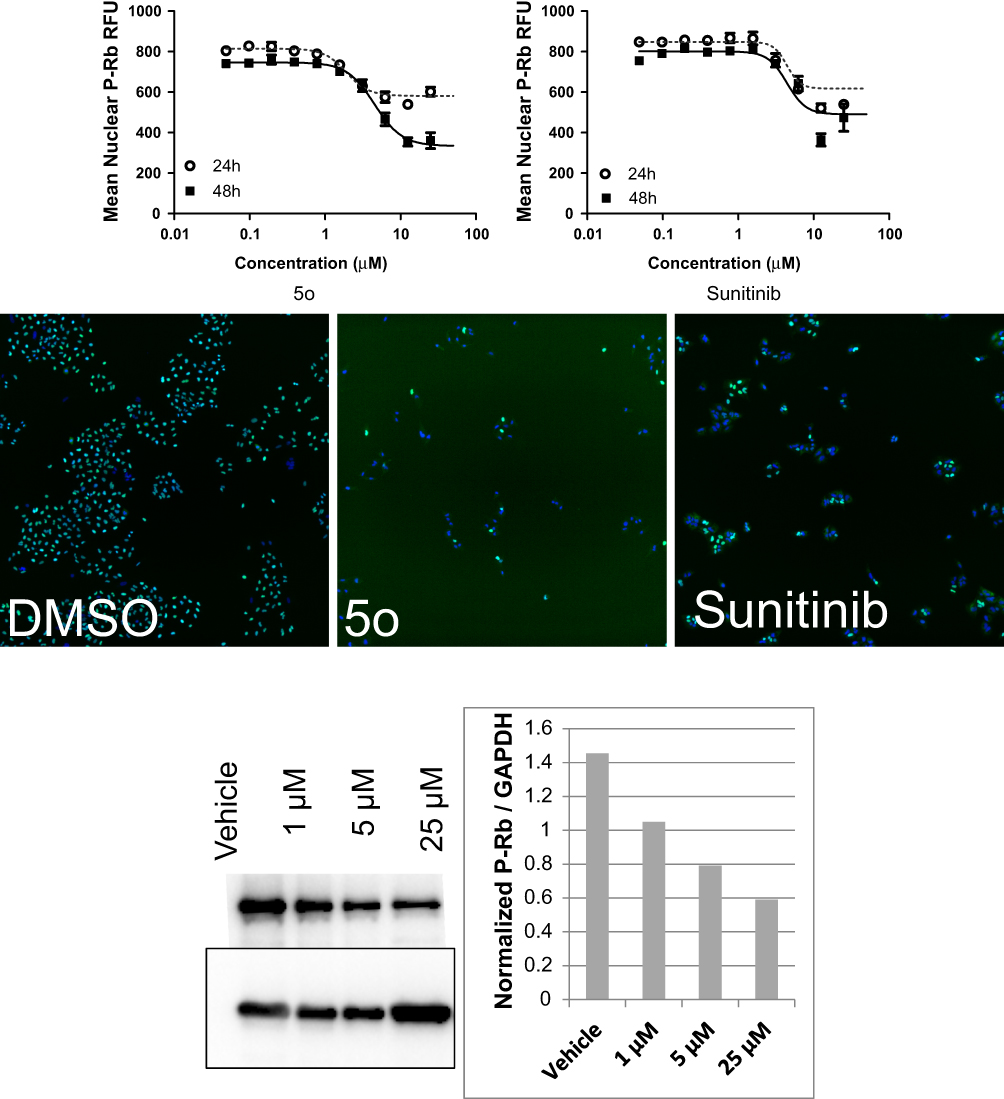 |
Figure 3 (A). Reduction of phosphorylated Rb protein by compound 5o and sunitinib. Levels of P-Rb in the nuclei were shown by immunofluorescence in cells treated with vehicle, 5o or sunitinib. Automated image analysis (Molecular Devices) was used to quantitate P-Rb changes and these are presented in the dose–response graphs for each compound after 24 hr or 48 hr treatment. (B) Western blot analysis of A-549 NSCLC cells treated with compound 5o shows the effect of on total cellular levels of P-Rb after 24 hr treatment (left). Densitometric analysis of P-Rb normalized to GAPDH loading control is presented (right).Abbreviations: GAPDH, glyceraldehyde-3-phosphate dehydrogenase; NSCLC, non-small cell lung cancer. |
Selectivity
Three nontumorigenic cell lines (Table 4) were utilized to examine the growth inhibitory selectivity of compound 5o: IEC-6 cells which show morphologic and karyotypic characteristics of normal rat intestinal epithelial cells,37 MCF-10A cells which feature the characteristics of primary cultures of breast tissue with a dome formation38 and Swiss 3t3 fibroblasts derived from mice embryonic tissue which are both contact inhibited and nontumorigenic.39 A human non-small cell lung cancer (NSCLCA-549) cell line was used for comparison. Compound 5o was tested in quadruplicate at a maximum concentration of 25 µM and 10 subsequent serially diluted concentrations.
 |
Table 4 Selectivity of 5o and sunitinib against nontumorigenic and tumor cell lines |
Figure 4 and Table 4 indicate that compound 5o was able to inhibit cell growth in both tumor and normal cells. However, it showed threefold selectivity, while sunitinib displayed 1.4-fold selectivity.
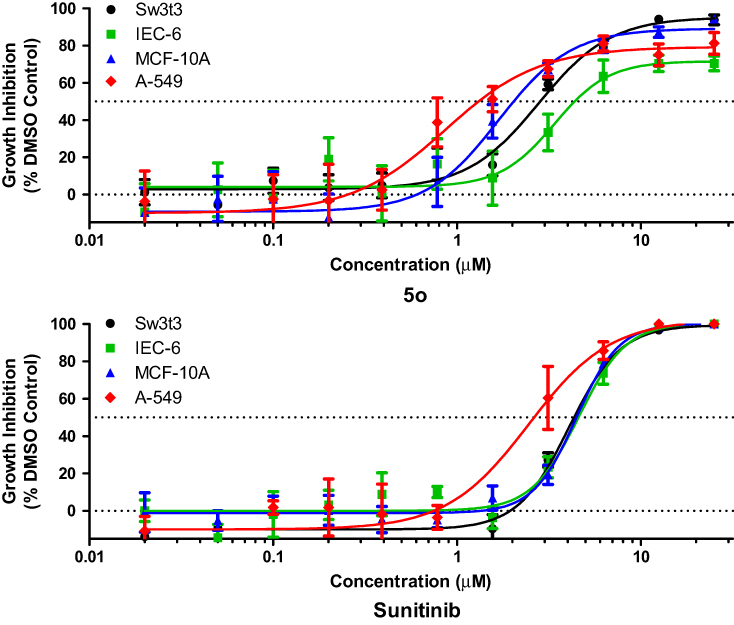 |
Figure 4 Selectivity characteristics of compound 5o and sunitinib. |
Activity against multidrug-resistant cancer cell line
The growth inhibitory potential of compound 5o was tested against the sensitive lung cancer cell line NSCLC A-549 and the multidrug-resistant lung cancer cell line NCI-H69AR which expresses the ABCC1 efflux pump protein. Compound 5o was tested in quadruplicate at a maximum concentration of 25 µM and 10 subsequent serially diluted concentrations.
Figure 5 and Table 5 indicate that compound 5o induced growth inhibition in both lung cancer cell lines, with an IC50 value of 0.9 µM in A-549 cells, and being about 12-fold less sensitive toward the NCI-H69AR cell line. This result indicates that compound 5o might undergo efflux by the ABCC1 efflux pump protein. In contrast, sunitinib was only 1.9-fold less potent toward the NCI-H69AR cell line.
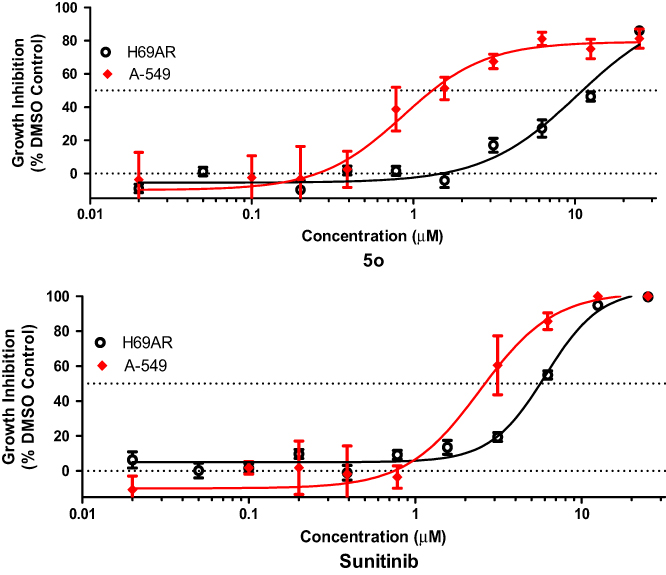 |
Figure 5 Activity of compound 5o and sunitinib against A-549 and NCI-H69AR cell lines. |
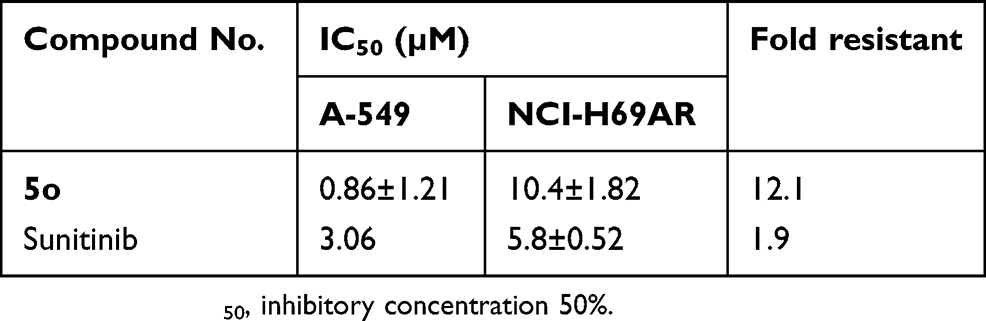 |
Table 5 Cancer cell growth inhibitory activity of compound 5o and sunitinib toward sensitive (A-549) and resistant (NCI-H69AR) cell lines |
Conclusion
The molecular hybrids 5a-w were evaluated as new antiproliferative conjugates. Compounds 5o (bearing an N-benzylisatin moiety) and 5w (bearing an N-phenylisatin moiety) were the most active antiproliferative candidates, with IC50 values of 1.69 and 1.91 µM, respectively, being about fivefold and fourfold more potent than sunitinib (IC50=8.11 µM).
Detailed pharmacological studies were conducted on compound 5o, a promising antiproliferative candidate, for a better understanding of its pharmacological properties. Compound 5o did not show any significant rise in caspase 3/7 activity at any concentration or time point tested. Moreover, it exhibited an increase in the G1 phase and a reduction in the S and G2/M phases of the cell cycle, and it presented an IC50value of 10.4 μM toward the resistant NCI-H69AR cancer cell line. Furthermore, the extent of phosphorylated Rb protein was substantially decreased in a dose-dependent fashion by compound 5o which was further confirmed via Western blot analysis. This promotes the assumption that inhibition of cyclin-dependent kinases by compound 5o plays a role in its growth inhibitory potential.
Overall, the current investigation indicates that the new antiproliferative potential of the chemical entities 5a-w, compound 5o in particular, can support the development of new antiproliferative leads to be harnessed in preclinical studies of cancer chemotherapy.
Acknowledgment
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of this research through the Research Group Project no. RGP-196.
Disclosure
Dr Adam B Keeton is a shareholder for ADT Pharmaceuticals, LLC, outside the submitted work. Prof. Dr. Gary A Piazza is a co-founder, shareholder, and Chief Scientist for ADT Pharmaceuticals LLC and founder and president of PDEi Pharmaceuticals LLC. The authors report no other conflicts of interest in this work.
References
1. Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58(2):71–96. doi:10.3322/CA.2007.0010
2. Sinha R, El-Bayoumy K. Apoptosis is a critical cellular event in cancer chemoprevention and chemotherapy by selenium compounds. Curr Cancer Drug Targets. 2004;4(1):13–28. doi:10.2174/1568009043481614
3. Cozzi P, Mongelli N, Suarato A. Recent anticancer cytotoxic agents. Curr Med Chem-Anti-Cancer Agents. 2004;4(2):93–121.
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
5. Barinaga M. From bench top to bedside. Science. 1997;278(5340):1036–1039.
6. Nabholtz J-M, Slamon D. New adjuvant strategies for breast cancer: meeting the challenge of integrating chemotherapy and trastuzumab (Herceptin). Semin Oncol. 2001;28(3):1–12.
7. Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Eng J Med. 2001;344(14):1038–1042.
8. Padma VV. An overview of targeted cancer therapy. BioMedicine. 2015;5(4):1–6.
9. Broekman F, Giovannetti E, Peters GJ. Tyrosine kinase inhibitors: multi-targeted or single-targeted? World J Clin Oncol. 2011;2(2):80–93.
10. Topcul M, Cetin I. Endpoint of cancer treatment: targeted therapies. Asian Pac J Cancer Prev. 2014;15(11):4395–4403. doi:10.7314/APJCP.2014.15.11.4395
11. Srivastava A, Pandeya S. Indole a versatile nucleus in pharmaceutical field. Int J Curr Pharm Rev Res. 2011;1(11):1–17.
12. Black W, Bayly C, Belley M, et al. From indomethacin to a selective COX-2 inhibitor: development of indolalkanoic acids as potent and selective cyclooxygenase-2 inhibitors. Bioorg Med Chem Lett. 1996;6(6):725–730. doi:10.1016/0960-894X(96)00100-X
13. Flynn BL, Hamel E, Jung MK. One-pot synthesis of benzo[b]furan and indole inhibitors of tubulin polymerization. J Med Chem. 2002;45(12):2670–2673. doi:10.1021/jm020077t
14. Leboho TC, Michael JP, van Otterlo WA, et al. The synthesis of 2- and 3-aryl indoles and 1,3,4,5-tetrahydropyrano[4,3-b]indoles and their antibacterial and antifungal activity. Bioorg Med Chem Lett. 2009;19(17):4948–4951. doi:10.1016/j.bmcl.2009.07.091
15. Rapolu M, Kumanan R, Duganath N, et al. Synthesis, characterization and pharmacological screening of 2-methyl-1H-indole-3-carboxylic acid [2-(2-substituted phenyl)-4-oxothiazolidin-3-yl]amide derivatives. Int J Chem Sci Appl. 2011;2(1):91–99.
16. Gollapalli M, Taha M, Ullah H, et al. Synthesis of Bis-indolylmethane sulfonohydrazides derivatives as potent α-Glucosidase inhibitors. Bioorg Chem. 2018;80:112–120. doi:10.1016/j.bioorg.2018.06.001
17. Khan KM, Salar U, Afzal S, et al. Schiff bases of tryptamine as potent inhibitors of nucleoside triphosphate diphosphohydrolases (NTPDases): structure-activity relationship. Bioorg Chem. 2019;82:253–266. doi:10.1016/j.bioorg.2018.10.046
18. Anouar EH, Moustapha ME, Taha M, et al. Synthesis, molecular docking and β-glucuronidase inhibitory potential of indole base oxadiazole derivatives. Molecules. 2019;24(5):963. doi:10.3390/molecules24050963
19. Attia MI, Witt-Enderby PA, Julius J. Synthesis and pharmacological evaluation of pentacyclic 6a,7-dihydrodiindole and 2,3-dihydrodiindole derivatives as novel melatoninergic ligands. Bioorg Med Chem. 2008;16(16):7654–7661. doi:10.1016/j.bmc.2008.07.012
20. Markl C, Attia MI, Julius J, et al. Synthesis and pharmacological evaluation of 1,2,3,4-tetrahydropyrazino[1,2-a]indole and 2-[(phenylmethylamino)methyl]-1H-indole analogues as novel melatoninergic ligands. Bioorg Med Chem. 2009;17(13):4583–4594. doi:10.1016/j.bmc.2009.04.068
21. Markl C, Clafshenkel WP, Attia MI, et al. N-Acetyl-5-arylalkoxytryptamine analogs: probing the melatonin receptors for MT(1) -selectivity. Arch Pharm. 2011;344(10):666–674.
22. Pandeya SN, Smitha S, Jyoti M, Sridhar SK. Biological activities of isatin and its derivatives. Acta Pharm. 2005;55(1):27–46.
23. Bhattacharya SK, Chakrabarti A. Dose-related proconvulsant and anticonvulsant activity of isatin, a putative biological factor, in rats. Indian J Exp Biol. 1998;36(1):118–121.
24. Pandeya SN, Sriram D, Nath G, DeClercq E. Synthesis, antibacterial, antifungal and anti-HIV activities of Schiff and Mannich bases derived from isatin derivatives and N-[4-(4ʹ-chlorophenyl)thiazol-2-yl]thiosemicarbazide. Eur J Pharm Sci. 1999;9(1):25–31.
25. Sridhar SK, Saravanan M, Ramesh A. Synthesis and antibacterial screening of hydrazones, Schiff and Mannich bases of isatin derivatives. Eur J Med Chem. 2001;36(7–8):615–625.
26. Banerjee D, Yogeeswari P, Bhat P, et al. Novel isatinyl thiosemicarbazones derivatives as potential molecule to combat HIV-TB co-infection. Eur J Med Chem. 2011;46(1):106–121.
27. Vine KL, Indira Chandran V, Locke JM, et al. Targeting urokinase and the transferrin receptor with novel, anti-mitotic N-alkylisatin cytotoxin conjugates causes selective cancer cell death and reduces tumor growth. Curr Cancer Drug Targets. 2012;12(1):64–73.
28. Attia MI, Eldehna WM, Afifi SA, et al. New hydrazonoindolin-2-ones: synthesis, exploration of the possible anti-proliferative mechanism of action and encapsulation into PLGA microspheres. PLoS One. 2017;12(7):e0181241.
29. Abdel-Aziz HA, Eldehna WM, Keeton AB, et al. Isatin-benzoazine molecular hybrids as potential antiproliferative agents: synthesis and in vitro pharmacological profiling. Drug Des Devel Ther. 2017;11:2333–2346.
30. Abdelhameed A, Bakheit A, Mohamed M, et al. Synthesis and biophysical insights into the binding of a potent anti-proliferative non-symmetric bis-isatin derivative with bovine serum albumin: spectroscopic and molecular docking approaches. Appl Sci. 2017;7(6):617.
31. Eldehna WM, Al-Wabli RI, Almutairi MS. Keeton AB, et al. Synthesis and biological evaluation of certain hydrazonoindolin-2-one derivatives as new potent anti-proliferative agents. J Enzym Inhib Med Chem. 2018;33(1):867–878.
32. Almutairi MS, Zakaria AS, Ignasius PP, et al. Synthesis, spectroscopic investigations, DFT studies, molecular docking and antimicrobial potential of certain new indole-isatin molecular hybrids: experimental and theoretical approaches. J Mol Struct. 2018;1153:333–345.
33. Al-Wabli R, Zakaria A, Attia M. Synthesis, spectroscopic characterization and antimicrobial potential of certain new isatin-indole molecular hybrids. Molecules. 2017;22(11):1958.
34. Almutairi MS, Xavier S, Sathish M, et al. Spectroscopic (FT-IR, FT-Raman, UV, 1H and 13C NMR) profiling and computational studies on methyl 5-methoxy-1H-indole-2-carboxylate: a potential precursor to biologically active molecules. J Mol Struct. 2017;1133:199–210.
35. Eldehna WM, EL-Naggar DH, Hamed AR, et al. One-pot three-component synthesis of novel spirooxindoles with potential cytotoxic activity against triple-negative breast cancer MDA-MB-231 cells. J Enzym Inhib Med Chem. 2018;33(1):309–318.
36. Eldehna WM, Abo-Ashour MF, Ibrahim HS, et al. Novel [(3-indolylmethylene) hydrazono] indolin-2-ones as apoptotic anti-proliferative agents: design, synthesis and in vitro biological evaluation. J Enzym Inhib Med Chem. 2018;33(1):686–700.
37. Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80(2):248–265.
38. Soule HD, Maloney TM, Wolman SR, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50(18):6075–6086.
39. Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.