")
Back to Journals » International Journal of Nanomedicine » Volume 14
Antileishmanial activity and cytotoxicity of ZnO-based nano-formulations
Authors Nazir S, Rabbani A, Mehmood K , Maqbool F, Shah GM, Khan MF, Sajid M
Received 21 April 2019
Accepted for publication 17 July 2019
Published 25 September 2019 Volume 2019:14 Pages 7809—7822
DOI https://doi.org/10.2147/IJN.S203351
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Samina Nazir,1 Atiya Rabbani,2 Khalid Mehmood,3 Farhana Maqbool,4 Ghulam Mujtaba Shah,4 Muhammad Fiaz Khan,5 Muhammad Sajid2
1Department of Chemistry, College of Science, King Faisal University, Al-Ahsa, Saudi Arabia; 2Department of Biochemistry, Hazara University, Mansehra, Pakistan; 3Medical Centre, Quaid-e-Azam University, Islamabad, Pakistan; 4Department of Microbiology, Hazara University, Mansehra, Pakistan; 5Department of Zoology, Hazara University, Mansehra, Pakistan
Correspondence: Muhammad Sajid
Department of Biochemistry, Hazara University, Mansehra, Pakistan
Tel +92 333 507 1757
Fax +92 99 741 4111
Email [email protected]
Introduction: Nanoparticles (NPs) can be toxic due to their nano-range sizes. Zinc oxide (ZnO) has good biocompatibility and is commercially used in cosmetics. Moreover, ZnO NPs have potential biomedical uses, but their safety remains unclear.
Methods: A range of doped ZnO NPs was evaluated for antileishmanial activity and in vitro toxicity in brine shrimp and human macrophages, and N-doped ZnO NPs were evaluated for in vivo toxicity in male BALB/C mice. N-doped ZnO NPs were administered via two routes: intra-peritoneal injection and topically as a paste. The dosages were 10, 50, and 100 mg/kg/day for 14 days.
Results: Topical administration was safe at all dosages, but intra-peritoneal injection displayed toxicity at higher doses, namely, 50 and 100 mg/kg/day. The pathological results for the i.p. dose groups were mild to severe degenerative changes in parenchyma cells, increases in Kupffer cells, disappearance of hepatic plates, increases in cell size, ballooning, cytoplasmic changes, and nuclear pyknosis in the liver. Kidney histology was also altered in the i.p. administration group (dose 100 mg/kg/day), with inflammatory changes in the focal area. We associate pathological abnormalities with the presence of doped ZnO NPs at the diseased site, which was verified by PIXE analysis of the liver and kidney samples of the treated and untreated mice groups.
Conclusion: The toxicity of the doped ZnO NPs can serve as an essential determinant for the effects of ZnO NPs on environmental toxicity and can be used for guidelines for safer use of ZnO-based nanomaterials in topical treatment of leishmaniasis and other biomedical applications.
Keywords: ZnO, nanoparticles, doped-ZnO, NP toxicity, in vivo toxicity
Introduction
Innovation in biomedical nanotechnology is expected to lead to many exciting applications.1 Nanoparticles (NPs) have different properties, which depend on their size, shape, and morphology and allow them to interact with microorganisms, plants, and animals.2 The unique properties of nanomaterials are being exploited for multiple biological and therapeutic applications. To date, the integration of nanomaterials with biology has expedited advancements in analytical tools, diagnostic devices, contrast agents, physical therapy, and drug delivery vehicles. Further advances in nano-medicine are expected to lead to significant research tools and clinically helpful devices within a brief span of time.3 ZnO NPs have acquired enormous importance in nano-medicine and have valuable biomedical applications due to their low cost, high stability, wide band gap, and inherent photoluminescence properties, which are beneficial for biomolecule sensing and for understanding photocatalytic (PC) activity and the production of reactive oxygen species (ROS).4–6 ZnO-based nanomaterials exhibit antibacterial, antifungal, anticancer, and antidiabetic activity.7–11
Leishmania is a genus of parasite protozoa in the order Trypanosomatida that causes leishmaniasis.12 Leishmania parasites are currently living in about 12 million individuals in 88 countries, and 350 million more are at risk of contracting leishmaniasis. L. tropica is strictly restricted to human beings and the lesions that it causes can persist for long periods (6–15 months). Current antileishmanial drugs have some limitations, including toxicity, cost, and being vulnerable to resistance by the parasite.13 Hence, there is an immediate need to develop new antileishmanial compounds. Metallic compounds are considered to be successful enzyme inhibitors of trypanothione metabolism, which plays an essential role in the survival of Leishmania parasites.14 Caballero et al15 examined the antileishmanial adequacy of metals and produced promising results. In another study, the clinical utilization of zinc sulfate showed high cure rates (>96%) against cutaneous leishmaniasis when it was orally applied at a concentration of 10 mg/kg for 45 days.16 Likewise, the antileishmanial efficacies of gold (III) and rhenium (V) have also been verified: they hinder the cysteine protease enzymes of parasites.17
We previously exploited the PC activity of ZnO-based nanomaterials and proposed their use in the photodynamic therapy (PDT) of cancers and Leishmania lesions.18–22 This study evaluated the in vivo toxicity of materials that can be successfully manipulated for the treatment of leishmaniasis. We exploited doped ZnO-based PC nanomaterials PC1–PC4 (ZnO, ZnO:N, ZnO:C, ZnO:C:N) for treating Leishmania promastigotes and analyzed them for in vitro toxicity, while N-doped ZnO NPs (PC2) were tested for in vivo toxicity. PC2 displayed pronounced antileishmanial activity, and it was found to be quite benign when administered topically to mice in high doses for 15 consecutive days. Nevertheless, intraperitoneal injections with the same dose produced toxicity in male BALB/C mice. This study is a step toward the development of a formulation for the PDT of cutaneous and sub-cutaneous leishmaniasis under sunlight.
Materials and methods
Synthesis of doped Zno nanomaterials
Nitrogen- and carbon-doped ZnO NPs were prepared using a wet chemical method (co-precipitation) with zinc acetate (99.9%) and sodium hydroxides as precursors and Tween 80 as a stabilizing agent, following standard protocols.23 Briefly, a 50 mM solution of Zn(Ac)2 was titrated against 100 mM NaOH, with a drop rate of 1 drop per 15 s. Titration was continued with constant stirring until the pH reached 12. The precipitated ZnO NPs were filtered and washed three or four times with deionized water and then with ethanol until the pH became neutral (pH=7). The filtered precipitates were first dried at 80 °C, and then solid phase N, C, and N+ C doping was carried out by stirring ZnO NPs in separate ethanol solutions of urea (3 mol%), glucose (3 mol%), and urea + glucose (3 mol% each). Annealing was carried out afterwards at 400 °C.
The structural morphology, optical properties, band gap, purity, and composition of the NPs, among other aspects, were characterized using powder X-ray diffraction, UV-Vis spectroscopy (UV-Vis), diffuse-reflectance spectroscopy, and Rutherford backscattering spectroscopy. The average crystallite size of all synthesized NPs was calculated using Scherrer’s formula.24 The sizes of the ZnO, ZnO:N, ZnO:C, and ZnO:N:C were 6.9 nm, 9.8 nm, 7.4 nm, and 6.9 nm, respectively. (For analysis, see Table S1 and Figures S1–S3, in the Supplementary Material)
In vitro photocatalytic antileishmanial activity
The PC antileishmanial activity of ZnO NPs (PC1), nitrogen-doped ZnO NPs (PC2), carbon-doped ZnO NPs (PC3), and carbon + nitrogen-doped ZnO NPs (PC4) against L. promastigotes was studied.
Leishmania tropica promastigotes were grown at 24 °C in medium199 pH 7.2 supplemented with 10% (v/v) heat-inactivated fetal bovine serum, 25 mM HEPES, and 0.1 µg/mL streptomycin and penicillin.25 The numbers of parasites were determined via direct counting in a Neubauer chamber.
PDT efficacy of PCs 1–4 against promastigotes
Promastigotes of L. tropica (clinical isolates) were harvested and washed twice with HBSS buffer. They were seeded in 96-well titer plates containing fresh medium in the presence of different concentrations of PCs 1–4 for 24 h. The plates were kept under a light source, except for the control. Cell proliferation was verified by counting the viable cells in a Neubauer chamber. IC50 was calculated using GraphPad Prism 5 software. Sunlight was used to check the PDT efficiency of PCs 1–4. The experiments were performed in triplicate.
Trypan blue assay
Promastigotes of L. tropica (2×106 cells/mL) were treated with or without different concentrations of PCs 1–4 for 24 h. Cells were harvested and resuspended in HBSS. Harvested cells were incubated using Trypan Blue (0.4% w/v) for 15 min at 26 °C. Cell proliferation was verified by counting the viable cells, which were clear through their blue color, in a Neubauer chamber under a microscope.
Promastigote membrane permeabilization studies
Promastigotes were treated with the PCs for 24 h and incubated with 1 μM SYTOX Green 5 min prior to being placed on slides. The increase in fluorescence caused by the binding of the dye to intracellular nucleic acids was checked using a Leica fluorescent microscope with a Canon camera, with 485 nm and 530 nm filters for the excitation and emission wavelengths, respectively. Full permeabilization (100%) was considered to be that achieved after the addition of 0.1% Triton X-100.
In vitro cytotoxicity of PCs 1–4
The in vitro cytotoxicity of PC1–PC4 was measured in human macrophages and brine shrimp.
Brine shrimp assay
Artificial seawater was prepared by dissolving 3.8 g sea salt per liter of water, and filtered. The seawater was placed in a small unequally divided tank, and shrimp eggs were added to the tank’s larger compartment, which was darkened using an aluminum foil cover. The illuminated compartment attracted shrimp larvae (nauplii) through perforations in the compartments. The shrimp were allowed to hatch and mature for 2 days at room temperature (22–29 °C). Each fraction of PC1–PC4 was tested initially at concentrations of 100, 50, and 10 μg/mL. Tests were carried out in triplicate. After 2 days (when the shrimp larvae were mature), we added 5 mL seawater to each vial and transferred 20 shrimp to each vial, using a Pasteur pipette. The vials were maintained under sunlight illumination. After 24 h, the surviving shrimp were counted, and the number was recorded using a 3× magnifying glass. The data were analyzed using Biostat 2009 with Probit analysis to determine LD50 values and 95% confidence intervals.
Cytotoxicity in human macrophages
Fresh human macrophages (approved by the Bioethical Committee of the Biochemistry Department, Hazara University, Mansehra, Pakistan; the subjects also provided written informed consent) were isolated from human blood using a Ficoll–Hypaque gradient (density =1.070 g/mL) and were purified using a Percoll gradient (density =1.064 g/mL). The percentage of monocytes after the Percoll gradient was greater than 90%. Cells were suspended in RPMI medium at 37 °C, supplemented with 10% fetal bovine serum, 25 mM HEPES, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. The cells were viable and functional. They were seeded into a 96-well plate at a density of 105 cells, and they were differentiated and incubated with PCs for 24 h. Viability was then checked using a Trypan Blue assay. The numbers of viable cells were counted, and the LD50 was calculated using Biostat 2009 software. The results were confirmed using SYTOX green dye staining.
In vivo toxicity of PC2
The in vivo application of PC2 was studied using two administrative routes, topical and intraperitoneal. The topical application was an aqueous cream-based nanocomposite ointment; for the intraperitoneal route of administration, a nanocomposite injection was used. All of the in vivo studies were approved by the Advanced Studies and Research Board of Hazara University. The Ethical Committee of Hazara University approved the animal studies, following the ethical guidelines of the university (HU/E.C./Biochem/207).
Preparation of aqueous cream-based nanocomposite ointment
An aqueous paste of PC2 at three different doses (10, 50, 100 mg/kg) was prepared by dissolving 0.05, 0.25, and 0.5 g PC2 in separate 3.5 mL solutions of distilled water and PEG 400 solution (0.5 mL PEG 400 in 3 mL distilled water). The solution was sonicated until it completely dissolved the nanocomposite and produced a clear solution. All steps of the preparation process were performed in the dark.
Each solution was separately added slowly into the emulsifying ointments (1.5 g/solution) in the dark for the formation of three doses of nanocomposite aqueous paste. The three mixtures were stirred for 2 h to achieve complete homogenization.
Preparation of nanocomposite injections
PC2 NPs were dissolved in a solution of starch and sterilized distilled water (5 w% soluble starch in 15 mL distilled water) in the three concentrations of 0.05, 0.25, and 0.5 g for the formation of three separate doses. The solutions were sonicated overnight to obtain a completely dispersed solution of NPs. In contrast with conventional chemicals, NPs in a liquid medium agglomerate due to their higher ratio of surface area to volume. To avoid this, soluble starch was used. Agglomeration increases with increasing concentrations of NP suspension, which increases the hydrodynamic size of the NP. In PC2, the 2000 and 5000 mg/kg doses show extensive agglomeration.26 The maximum dose used in our experiments was 100 mg/kg.
Experimental animals and treatment
Unchallenged BALB/C male mice, 100–120 g and 7–8 weeks old, were provided by NIH Islamabad and housed under standard environment conditions at an ambient temperature of 25 °C. The animals were humanely cared for and supplied with food and water ad libitum. After 1 week of acclimation, the mice were randomly divided into eight groups (four animals in each group), made up of two control groups (one for injection and one for paste) and six experimental groups. For concentration-dependent toxicological experiments, the topical and intraperitoneal modes of administration were selected. The mice were administered 10, 50, and 100 mg/kg PC2 using both techniques. The two control groups were P4 (administered an aqueous paste vehicle without PC2) and I4 (administered a starch and water injection without PC2). For topical administration, 0.1 g/day/mouse paste was applied to the base of the mouse tail for better absorption and penetration, and for intraperitoneal administration, 0.3 mL/day/mouse containing a required concentration of PC2 was administered intraperitoneally. The same amount of control vehicle (P4 and I4) was administered to two control groups. After regular administration, the mice were weighed and observed for illness, activity, and behavioral changes on a daily basis for 15 days. Data on mice that died during the experiment were also registered. On day 15, all mice still living were sacrificed under anesthesia with 10% chloroform, using bioethical and sterile techniques. Whole liver and kidney samples were taken for histopathology and PIXE analysis. Before sacrifice, blood was taken from each experimental group for hematological and biochemical analyses.
Hematology and biochemistry analysis
Using a standard saphenous vein blood collection technique, blood was collected in a potassium EDTA collection tube for hematology analysis and in a lithium–heparin collection tube for serum biochemistry analysis. The levels of standard hematological parameters such as white blood cells (WBCs), lymphocytes (LYMs), red blood cells (RBCs), hematocrit (HCT), hemoglobin (Hb), platelets (PLTs), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), and mean corpuscular hemoglobin concentration (MCHC) were determined. To determine the serum levels of biochemical markers such as alanine aminotransferase (ALT), alkaline phosphatase (ALP), blood urea nitrogen (BUN), creatinine (CREA), and total bilirubin (TBIL), whole blood was centrifuged at 3000 rpm for 15 min and analyzed with an automatic biochemical analyzer.
Histopathological observations
At 15 days of treatment, the mice were sacrificed after isoflurane anesthesia and exsanguinated using an angiocatheter. All liver and kidney samples from each treatment group were fixed with 10% formalin for later use.
Small pieces of liver and kidney were fixed in 10% formalin and then embedded into paraffin, divided into 5–6 mm sections, and mounted on glass microscope slides using standard histopathological techniques. The sections were stained with hematoxylin and then eosin, and they were examined by light microscopy.
Particle-induced x-ray emission analysis
Particle-induced X-ray emission (PIXE) analysis is becoming increasingly common as an analytical technique for the determination of trace elements in biological materials. For PIXE analysis, the livers and kidneys of mice were prepared into pellets following a previously described procedure.27 Samples from BALB/C mice were dried at 37 °C in an oven for 24 h and then freeze-dried and ground with a mortar and pestle into a homogeneous powder mixture. This mixture was hard pressed into a 2 cm by 1–2 mm pellet.
A proton (H+) beam of 2.5 MeV was generated in a 5-UDH-Pelletron Tendam Accelerator at a current of 2 nA, and the charge was 0.5 Uc. An SiLi detector was used with a resolution of 138 eV, and a Mylar filter of 100 µm was used. The SRM used in this study was bovine liver (SRM 1577).
Statistical analysis
All data are given as means ± SDs across at least four independent experiments. For comparison among groups, one-way analysis of variance (ANOVA) and a student's two-tailed t-test were used, with the assistance of the software Graph Pad Prism 5 and Excel 2010. The results were considered significant at p<0.05.
Results and discussion
In vitro antileishmanial activity
Taking information from the literature on the concentrations of PCs, the types of negative control and reference drugs used in this study were adjusted, and the optimization experiments were performed for the in vitro antileishmanial and cytotoxicity investigation of PC1–PC4 at concentrations of 100, 10, 1, 0.1, 0.01, 0.1x10−2, 0.1x10−3, and 0.1×10−4 µg/mL. Promastigote forms of clinical isolates of L. tropica were exposed to PC1–PC4 for 24 h. PC1–PC4 showed inhibition in both time- and dose-dependent manners. At higher concentrations (100 µg/mL, 10 µg/mL, and 1 µg/mL) and in the presence of sunlight, the killing was rapid, with a complete killing within 2 h. No promastigotes survived even at lower concentrations (0.1 and 0.01 µg/mL). The viability of the promastigotes was investigated with both Trypan Blue dye and SYTOX green dye by observing the motility of the L. tropica parasites. Trypan Blue is a negative dye and gives a blue color to non-viable cells. The IC50 values were calculated using the software GraphPad Prism 5 and are shown in Figure 1. The IC50 of PC1 was 0.084±0.004 µg/mL and 0.012±0.002 µg/mL for PC2 (ZnO:N), 0.016±0.003 for ZnO:C, and 0.016±0.002 µg/mL for ZnO:C:N (Figure 2). The tested PCs are significantly more effective than the standard drug Amphotericin B, with an IC50 value of 0.34±0.06 µg/mL under the same set of conditions. The inhibition of the parasite is due to the formation of ROS by PC1–PC4 in visible light. These ROS are toxic to cells and cause cell death by disturbing the normal morphology of the cell membrane and increasing its porosity.18,19 Damage to the cell membrane caused by the PCs was confirmed using a vital SYTOX green fluorescent dye (Figure S4, Supplementary Material). N-doped PC2 was considered the most potent for the photodynamic killing of the promastigotes. This increased efficacy of PC2 may be due to the enhanced electrons and hole splitting on the NP surface, leading to increased ROS production.20 The cytotoxic potentials of PC1–PC4 were evaluated through toxicity assays of brine shrimp and macrophages.
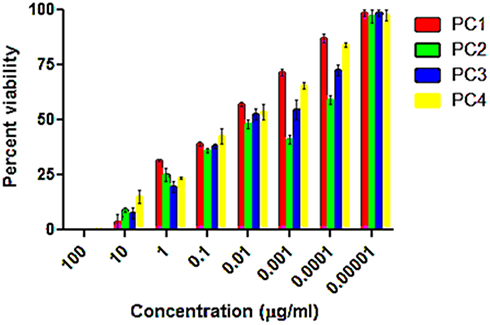 |
Figure 1 Percent survival of promastigotes after treating with ZnO NP photocatalysts 1–4 in direct sunlight at different concentrations. The data show high statistical significance, p<0.0001, determined by two-way ANOVA using the software Graph Pad Prism 5. |
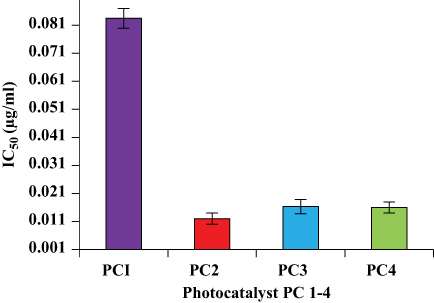 |
Figure 2 IC50 values for photocatalytic killing of promastigotes by PC1–PC4. PC-treated promastigotes were exposed for 15 min to sunlight (168 W/m2). The IC50 of amphotericin B was found to be 0.34±0.06. |
In vitro cytotoxicity
The cytotoxicities of PC1–PC4 were measured using the traditional brine shrimp assay and human macrophages. The data were analyzed using Biostat 2009 with Probit analysis to determine LD50. The LD50 of PC 1–PC4 obtained from brine shrimp assay is shown in Figure 3. The LD50 values indicate that PC2 (ZnO:N) was significantly less toxic than PC1 (ZnO), PC3 (ZnO:C), and PC4 (ZnO:C:N). PC3 showed higher toxicity than the others. PC2 and PC4 showed lower toxicity even at higher concentrations, which indicates that N-doping is less toxic than C-doping of ZnO NPs.
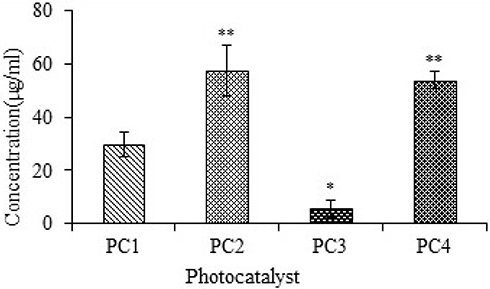 |
Figure 3 Studies of brine shrimp lethality for PC1–PC4, given as LD50 values. Bars represent means ± standard deviations. Data were analyzed using Student’s t-test. *Represents a significant difference from the base line control group for p<0.05 and **Represents significant difference from the baseline control group for p<0.001 (n=5). |
Cytotoxicity was also calculated using human macrophages, which were treated with PC1–PC4 for 24 h. Their viability was checked using the Trypan Blue assay. The number of viable cells was counted, and the LD50 was calculated using Biostat 2009 software. The viabilities of the macrophages treated with PC2 and PC4 were higher than those of macrophages treated with PC1 and PC3 at all dilutions. The LD50 values of PC1–PC4 against human macrophages are shown in Figure 4. The LD50 increased in the case of ZnO:N and ZnO:N:C NPs. The LD50 of ZnO:N was significantly higher than that of ZnO NPs (p<0.01). The decreased macrophage lethality of PC2 and PC4 again shows that N-doping is comparatively more biocompatible than C-doping and could be safely used in further studies.
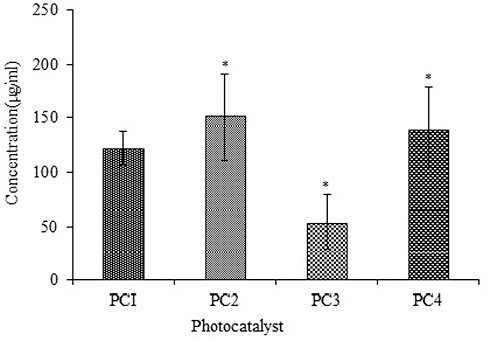 |
Figure 4 Toxicity against macrophages of PC1–PC4 as LD50 values. Bars represent means ± standard deviations. Data were analyzed by t-test. *Represents a significant difference from the baseline control group for p<0.05. |
In vivo toxicity
Our results indicate that PC2 (ZnO:N) exhibited the highest antileishmanial activity, lowest brine shrimp lethality, and lowest human macrophage cytotoxicity. Hence, we assessed the in vivo toxicity of PC2 to explore the possibility of its use in biomedical applications.
Our in vivo toxicity experiment was designed for 14 days of consecutive administration of PC2 via two different routes: intraperitoneal and topical. Both routes were designed to evaluate the possible toxicity of PC2 in treating cutaneous and sub-cutaneous leishmaniasis. Higher doses of intraperitoneal exposure could serve as a borderline value for accessing nano ZnO safety in a wide range of biomedical and other applications. Currently, there are no recommendations or standard methodologies for the in vivo toxicity assessment of NPs, so we chose to work within the dose limit recommended by OECD guideline 420 for investigating the oral toxicity of any new substance.28 The animal survival, hematology, biochemistry, histology, and percent elemental composition of the liver and kidney were characterized for the doses of 10 mg/kg, 50 mg/kg, and 100 mg/kg PC2 in the two routes of administration.
Hematological and biochemical investigations
For hematological analysis we selected the standard hematology markers WBCs, LYMs, RBCs, HCT, Hb, PLTs, MCV, MCH, and MCHC. The results are presented in Figures 5 and 6. The LYM, RBC, Hb, and HCT levels decreased at higher i.p. doses, and the PLT level increased at higher i.p. doses. A similar set of trends was reported by Wang et al for blood parameters at 200 μg/kg and 400 μg/kg intra-tracheal installation of ZnO NPs.29 No significant changes in toxicity were observed for topical application at any dose.
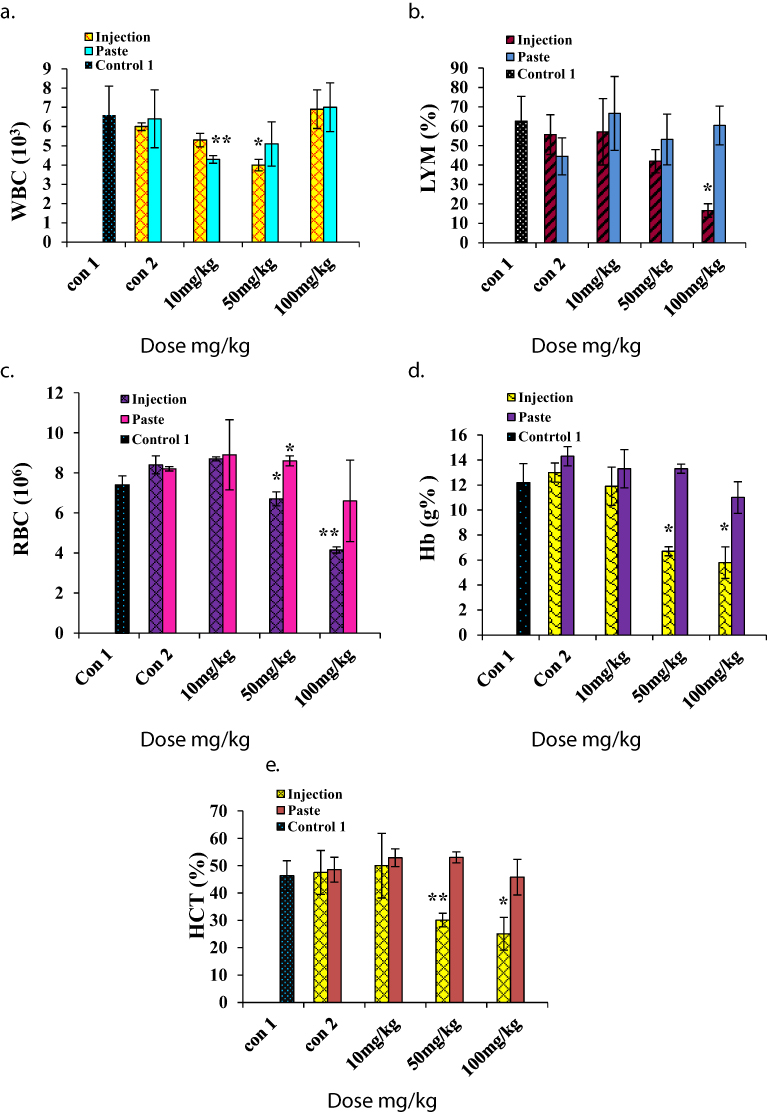 |
Figure 5 Hematology results for mice treated with ZnO NPs and the two control groups (baseline and injection/paste) 14 days after intraperitoneal injection/topical paste at doses of 10, 50, and 100 mg/kg. These results show means and standard deviations for (A) white blood cells, (B) lymphocytes, (C) red blood cells, (D) hemoglobin, and (E) hematocrit. Bars represent means ± standard deviations. Data were analyzed using a t-test. *Represents a significant difference from the baseline control group for p<0.05, and **represents a significant difference from the baseline control group for p<0.001 (n=5). Abbreviations: WBC, white blood cell; LYM, lymphocytes; RBC, red blood cell; Hb, hemoglobin; HCT, hematocrit. |
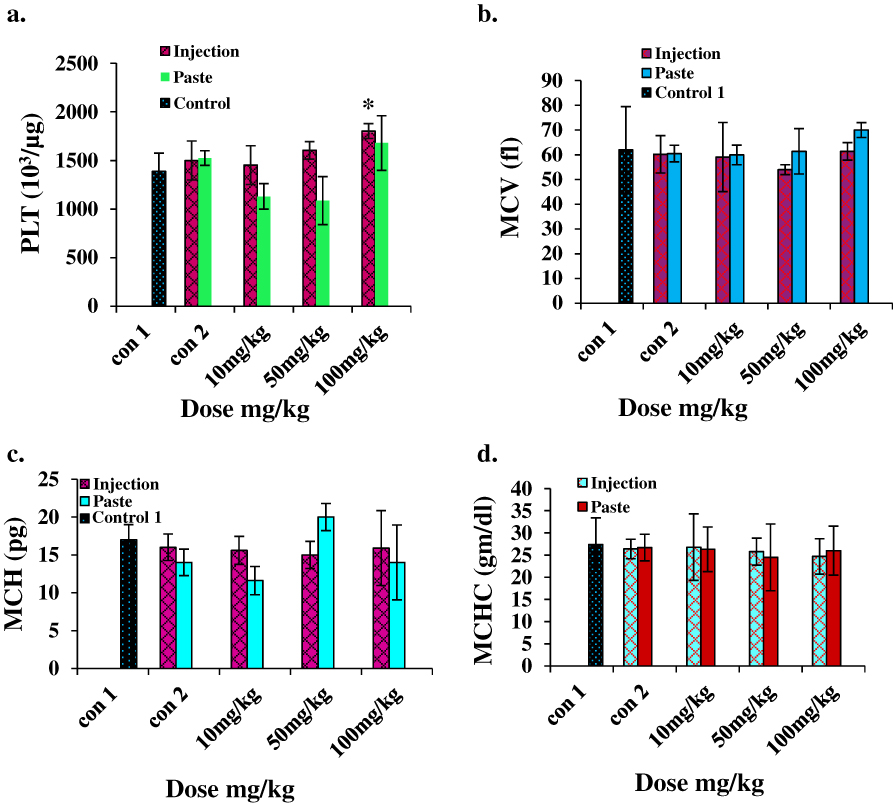 |
Figure 6 Hematology results for mice treated with ZnO NPs and the two control groups (baseline and injection/paste) 14 days after intraperitoneal injection/topical paste at a dose of 10, 50, and 100 mg/kg. (A) Platelet, (B) mean corpuscular volume, (C) mean corpuscular hemoglobin, and (D) mean corpuscular hemoglobin concentration. Bars represent means ± standard deviations. Data were analyzed using the t-test. *Represents a significant difference from the baseline control group for p<0.05. Abbreviations: PLT, platelet; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration. |
For biochemical analysis we assessed the levels of ALT, ALP, BUN, CREA, and TBIL. The results are shown in Figure 7. The ALT level increased significantly (p<0.001) in the i.p. mice group treated with higher doses, indicating the inflammatory condition of the liver; on the other hand, no significant changes were noted in any mice groups for topical administration. A significant decrease (p<0.05) in the ALP level was observed in i.p. mice groups. Kidney injury or dysfunction is characterized by an elevated BUN level. No notable change in the BUN level was detected in any of the topical administration groups. The CREA levels in mice treated with a higher dose of ZnO by i.p. decreased sharply (p<0.05), indicating poor liver function. CREA levels of topically administered mice showed no significant change. TBIL also showed no significant change in both i.p. or topical administration groups. Several in vivo studies have suggested that the chief distribution site and target organs for NP exposure are the liver and kidney.29–31 Our results clearly show that ZnO:N NPs are highly toxic to the liver and produce clear signs of liver dysfunction when administered i.p. at doses of 50 mg/kg and 100 mg/kg. Nonetheless, this is completely safe for the liver and kidney when applied topically at doses of 10 mg/kg, 50 mg/kg, and 100 mg/kg. ZnO:N NPs are nephrotoxic at i.p. doses of 50 mg/kg and 100 mg/kg, but they are safe for the kidney at a 10 mg/kg i.p. dose. The kidney is more involved in intracellular catabolism than the liver. Therefore, the physicochemical features of NP clearance in these organs are of great concern. However, ZnO:N NPs were found to be non-toxic at all doses when used topically.
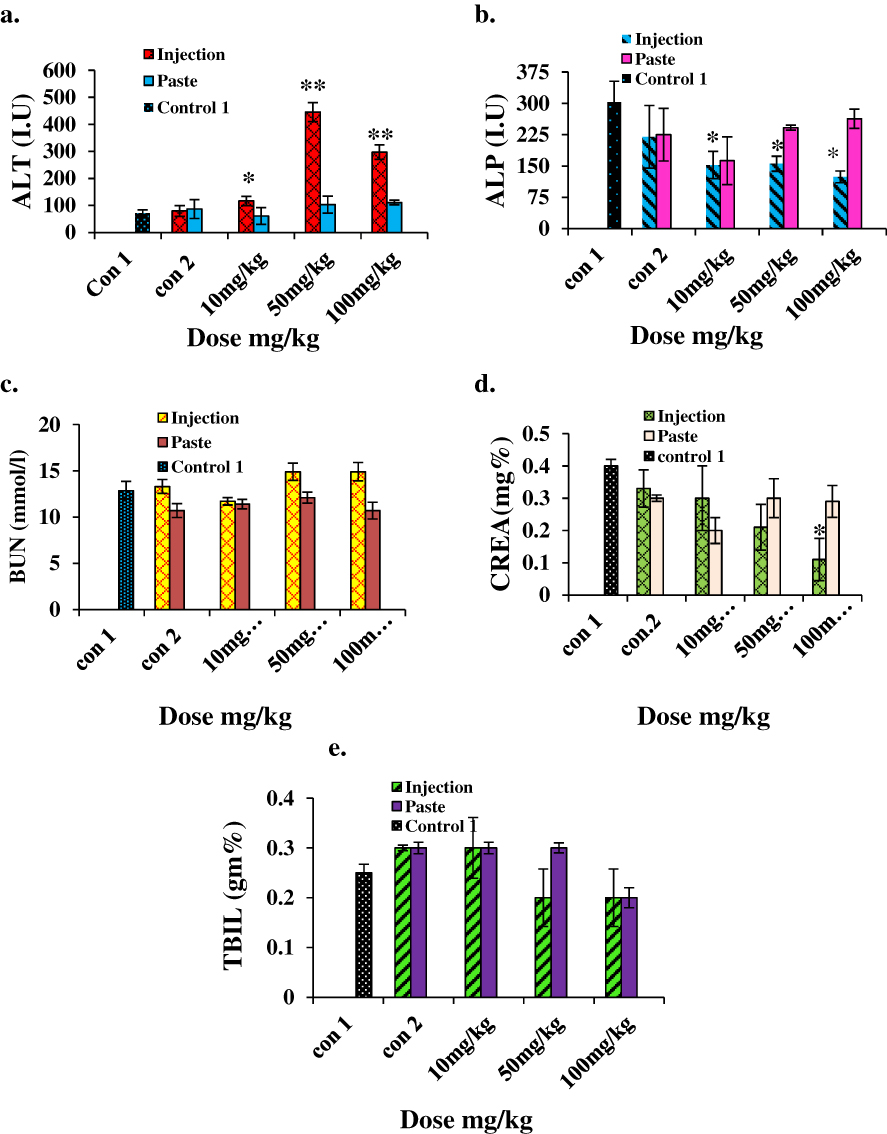 |
Figure 7 Biochemical results for mice treated with ZnO NPs and the two control groups 14 days after intraperitoneal injection/topical paste at a dose of 10, 50, and 100 mg/kg. These results show means and standard deviations for (A) alanine transaminase, (B) alkaline phosphatase, (C) blood urea nitrogen, (D) creatinine, and (E) total bilirubin. Bars represent means ± standard deviations. Data were analyzed using Student’s t-test. *Represents a significant difference from the base line control group for p<0.05, and **represents a significant difference from the base line control group for p<0.001 (n=5). Abbreviations: ALT, alanine transaminase; ALP, alkaline phosphatase; BUN, blood urea nitrogen; CREA, creatinine; TBIL, total bilirubin. |
Histopathology results
The histopathological liver and kidney specimens of treated mice were cut into longitudinal and sagittal sections, respectively. Changes in color and volume were noted.
Gross histopathology showed no color change visible to the naked eye in the external surface or on the longitudinally and sagittally cut surfaces of any samples, except for the i.p. mice group that received 100 mg/kg, in which the liver color changed from light brown to dark brown, and the volume also expanded significantly (p<0.05). The kidney specimens showed no significant change in color relative to baseline, but kidney volume significantly shrunk (p<0.05). The volume changes are shown in Table 1.
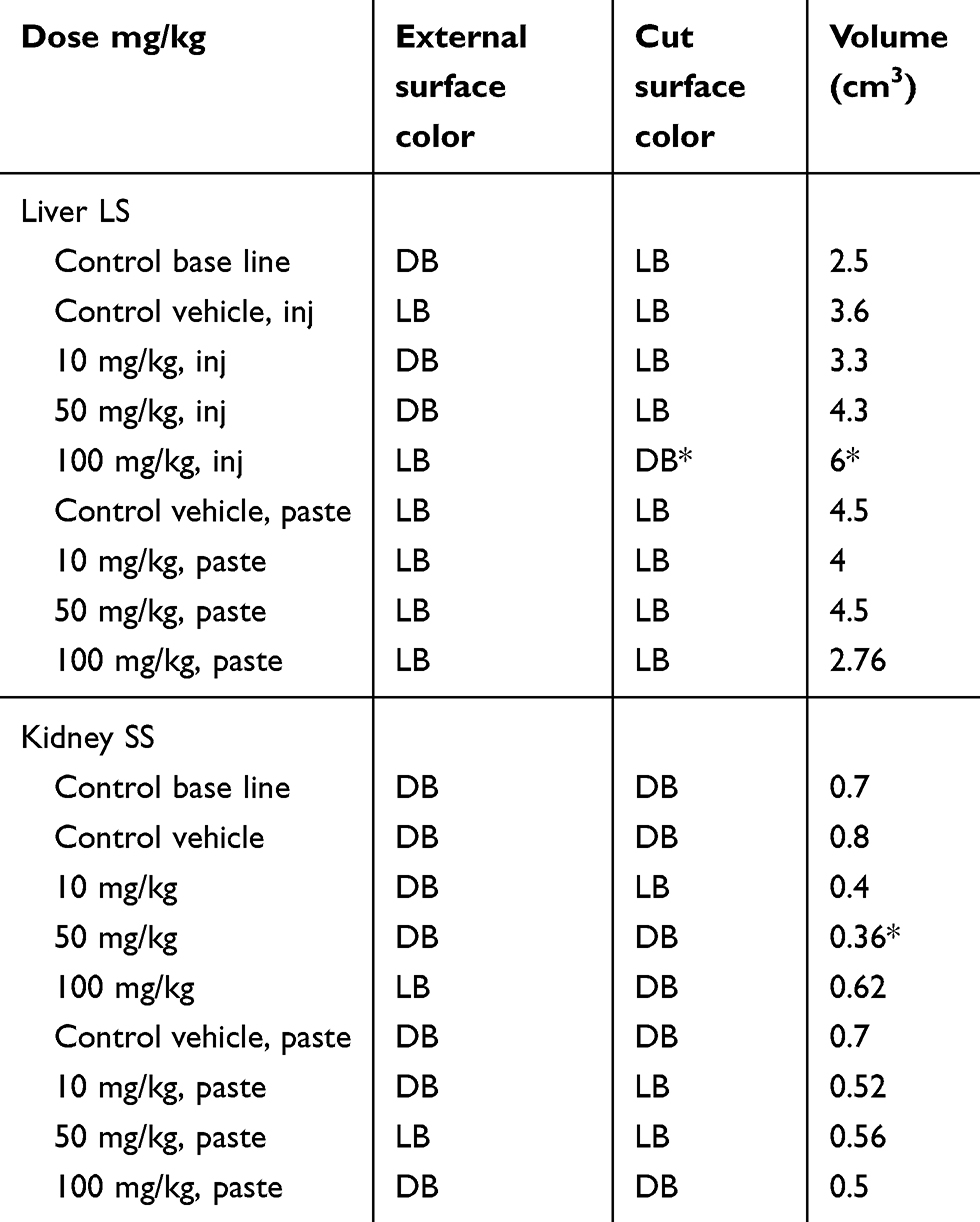 |
Table 1 Gross changes after 14 days in i.p. and dermal administration |
The histopathological observation under a microscope revealed that the control, the injection vehicle, and the paste vehicle groups had normal liver tissue, with a slight inflammatory response but no specific injury, as shown in Figure 8. Polygonal cells had prominent round nuclei, eosinophilic cytoplasm, and a few spaced hepatic sinusoids arranged among hepatic cord cells, with a fine arrangement of Kupfer cells. The pathological results of the i.p. dose groups represented mild to severe degenerative changes. The i.p. dose group of 10 mg/kg demonstrated normal histology with no obvious inflammatory changes (Figure 8D); the i.p. dose group of 50 mg/kg showed normal bile duct and slight periportal inflammation and no degenerative changes (Figure 8E); the i.p. dose group of 100 mg/kg demonstrated liver dysfunction, as revealed by early degenerative changes in parenchyma cells, disappearing hepatic plates, cell size increase, ballooning, cytoplasmic changes, and nuclear pyknosis (Figure 8F); and the topical group (Figure 8G) showed normal histopathology, similar to that of the control group. The 50 mg/kg and 100 mg/kg i.p. dose groups exhibited liver dysfunction according to liver histology and blood biochemistry results.
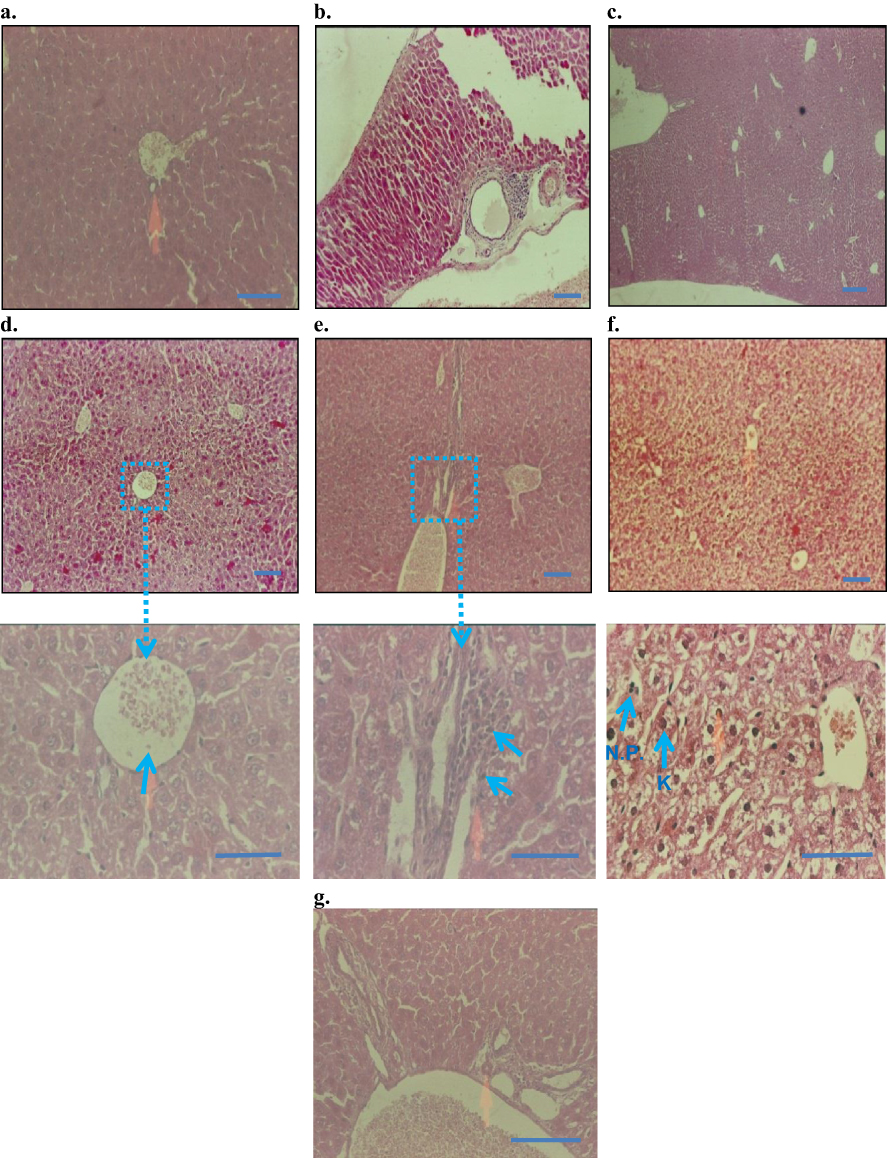 |
Figure 8 Histopathology of liver (H&E staining, 10× and 50×): (A) control baseline, (B) control injection vehicle, no parenchymal change with very slight periportal inflammation in 10× (Starch-i.p., inj 5 v% for 14 days), (C) no degenerative changes (control paste vehicle), (D) i.p. at 10 mg/kg NP, showing normal histology, (E) i.p. 50 mg/kg NP, slight peritoneal inflammation, (F) i.p.100 mg/kg NP, severe inflammation and liver parenchyma cell death, nuclear pyknosis (arrows), G) topical application 100 mg/kg NP, normal histology, and no signs of inflammation. Note: Small arrows in (B) and (C) indicate increased amounts of Kupffer cells (K) and nuclear pyknosis (NP). Boxes and connected arrows show the magnification of selected area. Bars 50 µm. |
Kidney histology did not significantly differ among groups. According to kidney section slides (Figure 9), glomeruli and tubules had normal morphology and there was no evidence of interstitial lymphocytic infiltration. In the i.p. 100 mg/kg dose group, there was mild inflammatory focal interstitial changes (Figure 9F). The three topical groups showed normal histopathological findings. An image of the sagittal section of a kidney at the 100 mg/kg topical dose is given in Figure 9G.
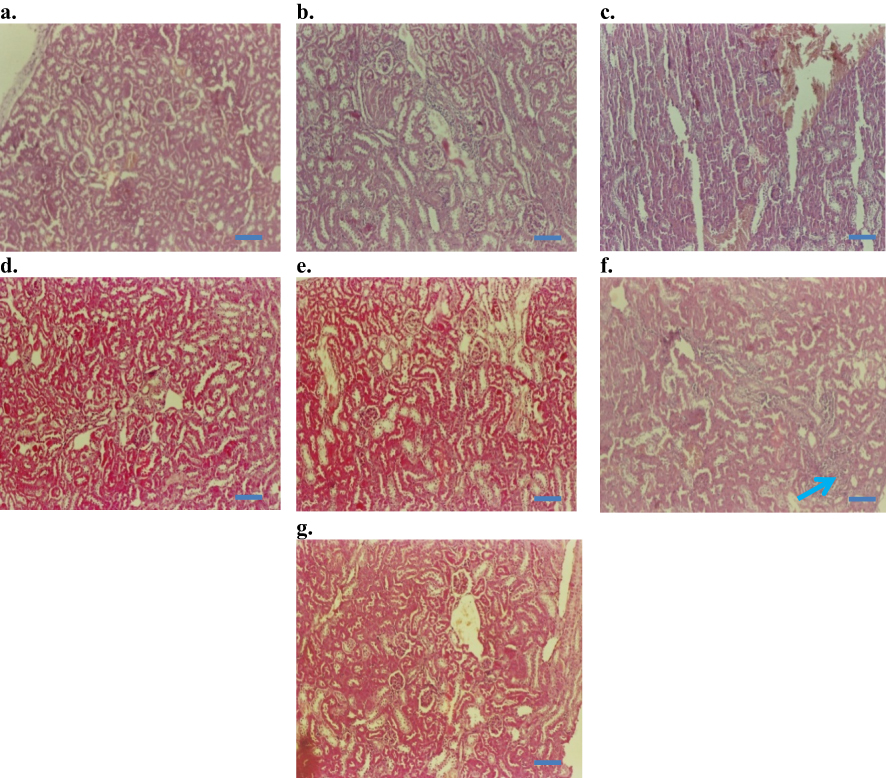 |
Figure 9 Histopathology of kidney (H&E staining, 200×): (A) Control baseline, (B) control injection vehicle, (C) control paste vehicle, (D) i.p. at 10 mg/kg NP, (E) i.p. 50 mg/kg NP, (F) i.p.100 mg/kg NP, and (G) topical dose at 100 mg/kg NP. Note: Arrow in (F) indicates mild inflammatory changes after 100 mg/kg NP dose, given as i.p. injection for 14 days. Bars 50 µm. |
Particle-induced x-ray emission spectroscopy
PIXE is the most subtle and advanced analytical technique used for trace element detection. The logarithmic PIXE spectra of the livers and kidneys of mice showed no significant changes in the Zn peak for the topical route of administration but strong peaks in the i.p. groups, specifically at the doses of 50 mg/kg and 100 mg/kg in both tissues (Figure 10).
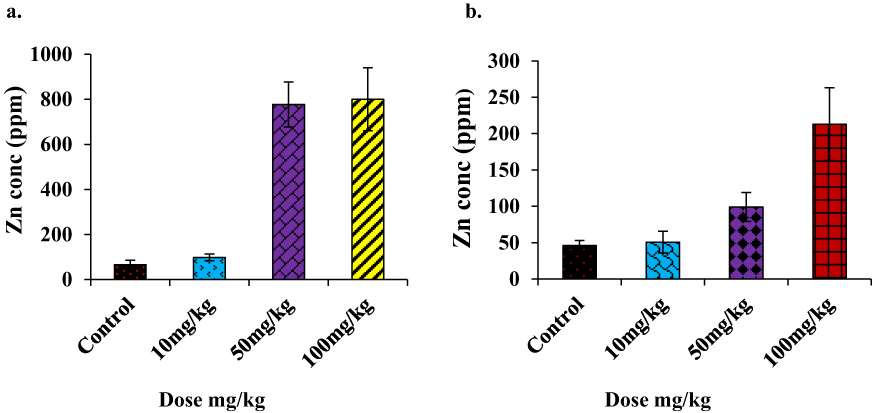 |
Figure 10 (A) Concentration (ppm) of Zn found in PIXE analysis of liver, (B) concentration (ppm) of Zn found in PIXE analysis of kidney (i.p. injections administered mice group only). The Zn concentration in the liver (an average of four animal samples) reached a maximum of 800 ppm, and the average was 220 ppm in the kidney at the highest dose. |
ZnO is generally recognized as safe (GRAS), according to the FDA. However, the title GRAS usually regards materials in the micron to larger range. In the nanophase, such materials can display toxicity.32 Moreover, the Scientific Committee on Cosmetic Products and Non-Food Products (SCCNFP) reported that the LD50 of normal ZnO for rats is greater than 5 g/kg BW, and ZnO is a non-toxic chemical (in a single oral dose). However, they (SCCNFP, 2003) have also stated that a suitable safety dossier on micro-sized ZnO, including probable pathways of cutaneous penetration and systemic exposure, is still needed.33 Furthermore, the cytotoxicity of ZnO NPs depends on NP uptake, accompanied by intracellular disintegration into Zn2+.34–36 This has been observed in several in vitro studies in diverse biological systems, including bacteria and mammalian cells.37–39 In mammalian cells, the toxic effects of ZnO NPs on phenomena such as the inflammatory response, membrane injury, DNA injury, and apoptosis have been determined.40–42 Earlier studies of in vitro toxicity have included assessments of the cytotoxic behavior of ZnO NPs in various cell lines, such as RAW 264.7 cells, BEAS-2B cells, human monocytes, cancerous T cells, and macrophages.28,43,44 Some in vivo studies have indicated that the liver, heart, spleen, pancreas, and bone are all target sites for ZnO NPs in mice, and potent but reversible pulmonary inflammation can be produced by the inhalation of these particles in rats.29 We also demonstrated that the liver and kidneys are the target organs for in vivo toxicity. Furthermore, the toxicology and pharmacokinetics of drugs or biomaterials depend on their route of administration.29 In one previous study, lung sections from mice instilled with different doses of un-doped ZnO NPs, such as 200 μg/kg, 400 μg/kg, and 800 μg/kg for 14 days, resulted in mild to severe fibrosis and chronic inflammation, while the oral route was found to be safe.29 In our case, the i.p. route was found to be slightly toxic at 50 mg/kg/day and 100 mg/kg/day, while topical application at the same dose was found to be safe. We found that male BALB/C mice had liver dysfunction at doses of 50 mg/kg or higher while there was no liver damage at 10 mg/kg N-doped ZnO NP. Liver dysfunction (as indicated by elevated ALP levels) was found for i.p. doses in male ICR mice for un-doped ZnO NP at 100 μg/mL in a previous study.31 Thus, N-doped ZnO NPs seem to be less toxic and more benign than ZnO NPs.
Conclusion
This study showed that ZnO nanocomposites (ZnO:N) have remarkable antileishmanial activity and less in vitro toxicity, suggesting that they could serve as a possible future treatment of cutaneous leishmaniasis. We investigated the toxicological aspects of ZnO NPs in male BALB/C mice, considering both i.p. and topical routes of administration and three doses. The results suggest that the liver and kidney are the target organs for ZnO NPs and that the toxicity of ZnO nanocomposite relates not only to its physiological properties but also to the particular route of administration. The topical route seems to be much safer than i.p; it was found to be safe at all dosages. Thus, this may be the best possible means of treatment for cutaneous leishmaniasis.
Disclosure
The authors report no conflicts of interest related to this work.
References
1. Alexis F, Pridgen EM, Langer R, Farokhzad OC. Nanoparticle technologies for cancer therapy, drug delivery. Handb Exp Pharmacol. 2010;197:55–86.
2. Siddiqi KS, Husen A, Rao RAK. A review on biosynthesis of silver nanoparticles and their biocidal properties. J Nanobiotechnol. 2018;16:14.
3. Wagner V, Dullaart A, Bock A-K, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24(10):1211–1218. doi:10.1038/nbt1006-1211
4. Ahmad R, Tripathy N, Hahn YB. Wide linear-range detecting high sensitivity cholesterol biosensors based on aspect-ratio controlled ZnO nanorods grown on silver electrodes. Sens Actuator B-Chem. 2012;169:382–386. doi:10.1016/j.snb.2012.05.027
5. Chen Z, Zhang N, Xu YJ. Synthesis of graphene–ZnO nanorod nanocomposites with improved photoactivity and anti-photocorrosion. Cryst Eng Comm. 2013;15:3022–3030. doi:10.1039/c3ce27021a
6. Jiang J, Pi J, Cai J. The advancing of zinc oxide nanoparticles for biomedical applications. Bioinorg Chem Appl. 2018;2018:1062562.
7. Sirelkhatim A, Mahmud S, Seeni A, et al. Review on zinc oxide nanoparticles: antibacterial activity and toxicity mechanism. Nano-Micro Lett. 2015;7:219–242. doi:10.1007/s40820-015-0040-x
8. Dananjaya SHS, Kumar RS, Yang M, Nikapitiya C, Lee J, De Zoysa M. Synthesis, characterization of ZnO-chitosan nanocomposites and evaluation of its antifungal activity against pathogenic Candida albicans. Int J Biol Macromol. 2018;108:1281–1288. doi:10.1016/j.ijbiomac.2017.11.046
9. Bisht G, Rayamajhi S. ZnO nanoparticles: a promising anticancer agent. Nanobiomedicine. 2016;3(9):1–11.
10. Wang J, Lee JS, Kim D, Zhu L. Exploration of zinc oxide nanoparticles as a multitarget and multifunctional anticancer nanomedicine. ACS Appl Mater Interfaces. 2017;9(46):39971–39984. doi:10.1021/acsami.7b11219
11. El-Gharbawy RM, Emara AM, Abu-Risha SES. Zinc oxide nanoparticles and a standard antidiabetic drug restore the function and structure of beta cells in type-2 diabetes. Biomed Pharmacother. 2016;84:810–820. doi:10.1016/j.biopha.2016.09.068
12. James WD, Berger T, Elston D. Andrew’s Diseases of the Skin: Clinical Dermatology.
13. Verma N, Dey C. The anti-leishmanial drug miltefosine causes insulin resistance in skeletal muscle cells in vitro. Diabetologia. 2006;49(7):1656–1660. doi:10.1007/s00125-006-0260-1
14. Navarro M, Gabbiani C, Messori L, Gambino D. Metal-based drugs for malaria, trypanosomiasis and leishmaniasis: recent achievements and perspectives. Drug Discov Today. 2010;15(23–24):1070–1078. doi:10.1016/j.drudis.2010.10.005
15. Caballero AB, Salas JM, Sánchez-Moreno M. Metal-based therapeutics for leishmaniasis, In: Claborn D, editor. Leishmaniasis: trends in epidemiology, diagnosis, and treatment. London: InTechOpen. 2014;465–493.
16. Minodier P, Parola P. Cutaneous leishmaniasis treatment. Travel Med Infect Di. 2007;5(3):150–158. doi:10.1016/j.tmaid.2006.09.004
17. Fricker SP, Mosi RM, Cameron BR, et al. Metal compounds for the treatment of parasitic diseases. J Inorg Biochem. 2008;102(10):1839–1845. doi:10.1016/j.jinorgbio.2007.08.010
18. Nadhman A, Nazir S, Khan MI, et al. PEGylated silver doped zinc oxide nanoparticles as novel photosensitizers for photodynamic therapy against Leishmania. FRBM. 2014;77(2014):230–238. doi:10.1016/j.freeradbiomed.2014.09.005
19. Nadhman A, Nazir S, Khan MI, et al. Visible light responsive ZnCuO nanoparticles: benign photodynamic killers of infectious protozoan. Int J Nanomed. 2015;10(1):6891–6903.
20. Nadhman A, Khan MI, Nazir S, et al. Annihilation of Leishmania by daylight responsive ZnO nanoparticles: a temporal relationship of reactive oxygen species-induced lipid and protein oxidation. Int J Nanomedicine. 2016;11:2451–2461. doi:10.2147/IJN.S105195
21. Nadhman A, Sirajuddin M, Nazir S, Yasinzai M. Photo-induced Leishmania DNA degradation by silver-doped zinc oxide nanoparticle: an in-vitro approach. IET Nanobiotechnol. 2016;10(3):129–133. doi:10.1049/iet-nbt.2015.0015
22. Arooj S, Nazir S, Nadhman A, et al. Novel ZnO:Ag nanocomposites induce significant oxidative stress in human fibroblast malignant melanoma (HT144) cells. Beilstein J Nanotechnol. 2015;6:570–582. doi:10.3762/bjnano.6.59
23. Tang E, Tian B, Zheng E, Fu C, Cheng G. Preparation of zinc oxide nanoparticle via uniform precipitation method and its surface modification by methacryloxypropyltrimethoxysilane. Chem Eng Commun. 2008;195(5):479–491. doi:10.1080/00986440701707834
24. Singhal S, Kaur J, Namgyal T, Sharma R. Cu-doped ZnO nanoparticles: synthesis, structural and electrical properties. Physica B: Condens Matter. 2012;407:1223–1226. doi:10.1016/j.physb.2012.01.103
25. Ali SA, Khalil NY, Iqbal J, Yasinzai MM. In vitro maintenance of Leishmania promastigote in an egg based biphasic culture medium. Methods Cell Sci. 1997;19(2):107–110. doi:10.1023/A:1009786915090
26. Botham P. Acute systemic toxicity—prospects for tiered testing strategies. Toxicol In Vitro. 2004;18(2):227–230. doi:10.1016/S0887-2333(03)00143-7
27. Lozano O, Mejia J, Masereel B, Toussaint O, Lison D, Lucas S. Development of a PIXE analysis method for the determination of the biopersistence of SiC and TiC nanoparticles in rat lungs. Nanotoxicology. 2012;6(13):263–271. doi:10.3109/17435390.2011.572301
28. Lanone S, Rogerieux F, Geys J, et al. Comparative toxicity of 24 manufactured nanoparticles in human alveolar epithelial and macrophage cell lines. Part Fibre Toxicol. 2009;6(14):1–12. doi:10.1186/1743-8977-6-14
29. Wang D, Li H, Liu Z, Zhou J, Zhang T. Acute toxicological effects of zinc oxide nanoparticles in mice after intratracheal instillation. Int J Occup Environ Health. 2017;23(1):11–19. doi:10.1080/10773525.2016.1278510
30. Sharma V, Singh P, Pandey AK. Dhawan A. Induction of oxidative stress, DNA damage and apoptosis in mouse liver after sub-acute oral exposure to zinc oxide nanoparticles. Mutat Res Genet Toxicol Environ Mutagen. 2012;745(1–2):84–91. doi:10.1016/j.mrgentox.2011.12.009
31. Hong T-K, Tripathy N, Son H-J, Ha K-T, Jeong H-S, Hahn Y-B. A comprehensive in vitro and in vivo study of ZnO nanoparticles toxicity. J Mater Chem B. 2013;1(23):2985–2992. doi:10.1039/c3tb20251h
32. Rasmussen JW, Martinez E, Louka P, Wingett DG. Zinc oxide nanoparticles for selective destruction of tumor cells and potential for drug delivery applications. Expert Opin Drug Deliv. 2010;7(9):1063–1077. doi:10.1517/17425247.2010.502560
33. Schilling K, Bradford B, Castelli D, et al. Human safety review of “nano” titanium dioxide and zinc oxide. Photochem Photobiol Sci. 2010;9:495–509. doi:10.1039/b9pp00180h
34. Gilbert B, Fakra SC, Xia T, Pokhrel S, Madler L, Nel AE. The fate of ZnO nanoparticles administered to human bronchial epithelial cells. ACS Nano. 2012;6(6):4921–4930. doi:10.1021/nn300425a
35. Buerki-Thurnherr T, Xiao L, Diener L, et al. In vitro mechanistic study towards a better understanding of ZnO nanoparticle toxicity. Nanotoxicology. 2013;7(4):402–416. doi:10.3109/17435390.2012.666575
36. Kim YH, Fazlollahi F, Kennedy IM, et al. Alveolar epithelial cell injury due to zinc oxide nanoparticle exposure. Am J Respir Crit Care Med. 2010;182(11):1398–1409. doi:10.1164/rccm.201002-0185OC
37. Miller RJ, Lenihan HS, Muller EB, Tseng N, Hanna SK, Keller AA. Impacts of metal oxide nanoparticles on marine phytoplankton. Environ Sci Technol. 2010;44(19):7329–7334. doi:10.1021/es100247x
38. Sinha R, Karan R, Sinha A, Khare S. Interaction and nanotoxic effect of ZnO and Ag nanoparticles on mesophilic and halophilic bacterial cells. Bioresour Technol. 2011;102(2):1516–1520. doi:10.1016/j.biortech.2010.07.117
39. Wang HJ, Growcock AC, Tang T-H, O’Hara J, Huang Y-W, Aronstam RS. Zinc oxide nanoparticle disruption of store-operated calcium entry in a muscarinic receptor signaling pathway. Toxicol In Vitro. 2010;24(7):1953–1961. doi:10.1016/j.tiv.2010.08.005
40. Jeng HA, Swanson J. Toxicity of metal oxide nanoparticles in mammalian cells. J Environ Sci Health A Tox Hazard Subst Environ Eng. 2006;41(12):2699–2711. doi:10.1080/10934520600966177
41. Osman IF, Baumgartner A, Cemeli E, Fletcher JN, Anderson D. Genotoxicity and cytotoxicity of zinc oxide and titanium dioxide in HEp-2 cells. Nanomedicine. 2010;5(8):1193–1203. doi:10.2217/nnm.10.2
42. Sharma V, Singh SK, Anderson D, Tobin DJ, Dhawan A. Zinc oxide nanoparticle induced genotoxicity in primary human epidermal keratinocytes. J Nanosci Nanotechnol. 2011;11(5):3782–3788. doi:10.1166/jnn.2011.4250
43. Xia T, Kovochich M, Liong M, et al. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano. 2008;2(10):2121–2134. doi:10.1021/nn800511k
44. Hanley C, Layne J, Punnoose A, et al. Preferential killing of cancer cells and activated human T cells using ZnO nanoparticles. Nanotechnology. 2008;19(29):295103. doi:10.1088/0957-4484/19/29/295103
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.