")
Back to Journals » Journal of Experimental Pharmacology » Volume 7
Antihyperalgesic effects of ProTx-II, a Nav1.7 antagonist, and A803467, a Nav1.8 antagonist, in diabetic mice
Authors Tanaka K, Sekino S, Ikegami M, Ikeda H, Kamei J
Received 29 December 2014
Accepted for publication 9 April 2015
Published 24 June 2015 Volume 2015:7 Pages 11—16
DOI https://doi.org/10.2147/JEP.S79973
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Ken-ichiro Tanaka,1,2, Shota Sekino,1 Megumi Ikegami,1 Hiroko Ikeda,1 Junzo Kamei1
1Department of Pathophysiology and Therapeutics, School of Pharmacy and Pharmaceutical Sciences, Hoshi University, Tokyo, Japan; 2Department of Endocrinology and Metabolism, Graduate School of Medicine, Yokohama City University, Yokohama, Japan
Abstract: The present study investigated the effects of intrathecal administration of ProTx-II (tarantula venom peptide) and A803467 (5-[4-chloro-phenyl]-furan-2-carboxylic acid [3,5-dimethoxy-phenyl]-amide), selective Nav1.7 and Nav1.8 antagonists, respectively, on thermal hyperalgesia in a painful diabetic neuropathy model of mice. Intrathecal administration of ProTx-II at doses from 0.04 to 4 ng to diabetic mice dose-dependently and significantly increased the tail-flick latency. Intrathecal administration of A803467 at doses from 10 to 100 ng to diabetic mice also dose-dependently and significantly increased the tail-flick latency. However, intrathecal administration of either ProTx-II (4 ng) or A803467 (100 ng) had no effect on the tail-flick latency in nondiabetic mice. The expression of either the Nav1.7 or Nav1.8 sodium channel protein in the dorsal root ganglion in diabetic mice was not different from that in nondiabetic mice. The present results suggest that ProTx-II and A803467, highly selective blockers of Nav1.7 and Nav1.8 sodium channels, respectively, in the spinal cord, can have antihyperalgesic effects in diabetic mice.
Keywords: painful diabetic neuropathy, Nav1.7, Nav1.8, ProTx-II, A803467
Introduction
Painful neuropathy is one of the most frequent complications of diabetes mellitus. Streptozotocin (STZ)-induced diabetes in rodents has been extensively used to study neuropathic pain and to explore the efficacies of new potential therapeutic drugs through the use of sensory behavioral tests. Diabetic neuropathy can be easily identified in this model through associated sensory dysfunctions, including thermal hyperalgesia and mechanical allodynia in rodents.1–3
Sodium channel blockade is one of the best-known treatments for relieving diabetes-induced pain. Some sodium-channel blockers, such as lidocaine and mexiletine, effectively prevent the tactile allodynia associated with diabetes-induced neuropathy under clinical diabetic neuropathic pain conditions4–7 and in rodent models of painful diabetic neuropathy.8,9 At least nine different voltage-dependent sodium-channel subtypes (Nav1.1–1.9) have been identified in the nervous system. Each voltage-dependent sodium-channel subtype can be classified functionally as either tetrodotoxin (TTX)-sensitive (fast inactivating) (TTX-S) or -resistant (slow inactivating) (TTX-R).10–12 Seven of these subtypes are expressed in sensory neurons in the dorsal root ganglion (DRG), and peripheral nerve injury can alter the expression patterns of several types of voltage-gated sodium channels.13–15 Recently, voltage-gated sodium channels, especially Nav1.7 and Nav1.8, have been implicated in chronic neuropathic pain.16,17 These subtypes are expressed in small nociceptive fibers, and can be down- or upregulated under painful conditions.18,19 It is well recognized that Nav1.7 is a TTX-S channel and Nav1.8 is TTX-R.16,19 These channels are coexpressed in nociceptive neurons of the DRG.20 Nav1.7 may be involved in the initial phase of depolarization, and initiates action potentials.20 Action potentials are maintained by Nav1.8 and this is followed by hyperpolarization of the membrane.20
The precise roles of spinal Nav1.7 and Nav1.8 in the maintenance of thermal hyperalgesia in diabetic neuropathy are not clear. The present study investigated if intrathecal (IT) administration of ProTx-II (cystine knot peptides from tarantula venom) and A803467 (5-[4-chloro-phenyl]-furan-2-carboxylic acid [3,5-dimethoxy-phenyl]-amide), selective Nav1.7 and Nav1.8 antagonists, respectively, could reduce thermal hyperalgesia in a mouse model of diabetic neuropathy.21,22
Materials and methods
Animals
Male 4-week-old ICR mice (Tokyo Animal Laboratory Inc., Tokyo, Japan), weighing about 20 g at the start of the experiment, were used. The mice had free access to food and water and were housed five per cage in an animal room that was maintained at 24°C±1°C with a 12-hour dark/light cycle. All behavioral experiments were performed between 10 am and 7 pm each day. This study was carried out in accordance with the Declaration of Helsinki and/or according to the guidelines of the committee on the care and use of laboratory animals of Hoshi University, Tokyo, which is accredited by the Ministry of Education, Science, Sports and Culture.
Induction of diabetes with STZ
Mice were rendered diabetic by an intravenous injection of STZ (200 mg/kg) prepared in citrate buffer at pH 4.5. Age-matched animals were injected with vehicle alone. Experiments were conducted 2 weeks after the administration of STZ. Animals with a serum glucose level above 400 mg/dL were considered diabetic.
Assessment of thermal hyperalgesia
The antihyperalgesic response was evaluated using the tail-flick test (KN-205E Thermal Analgesimeter; Natume, Tokyo, Japan). Briefly, the mouse was gently restrained in a tube. The tail was positioned in a groove underneath a 50 W projection bulb with the dorsal part of the tail facing the light bulb. The light and timer were monitored with the same switch. Twitching or movement of the tail is a typical response elicited by heat. When this occurred, light reached a photocell and the light and timer were switched off. Latencies of such movement were determined as the mean of two trials. The voltage of a 50 W projection lamp was set to 50 V, which gave a baseline value in nondiabetic animals of 10–14 seconds.3 The cut-off time was set to 30 seconds to prevent injury to the tail. Tail-flick latencies were measured before and 15, 30, 60, 90, 120, 150, and 180 minutes after the injection of drugs.
IT injections
Sodium-channel blockers were administered by IT injection as described by Hylden and Wilcox23 using a 25 μL Gastight® syringe (Hamilton, Reno, NV USA) and a BD Precisionglide® 30G 1/2 inch needle (Becton Dickinson, Franklin Lakes, NJ, USA). The mouse was restrained manually and the needle was inserted between the L5 and L6 vertebrae. This site is near the end of the spinal cord and minimizes the risk of spinal damage.23 The drug solution was injected in a volume of 4 μL per mouse.
Western blotting
The DRG was quickly dissected 2 weeks after the administration of STZ. Western blotting was performed as described previously24 with some modifications. Briefly, the DRG was homogenized in ice-cold radioimmunoprecipitation assay buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% Triton X, 1 mM phenylmethylsulfonyl fluoride, 25 μg/mL leupeptin, 25 μg/mL aprotinin, 10 mM NaF, and 1 mM Na3VO4. The homogenate was then centrifuged at 20,000× g for 20 minutes at 4°C, and the supernatant was retained as total tissue lysate. The protein concentration was measured using a BCA assay kit (Thermo Fisher Scientific Inc., Waltham, MA, USA). Samples were diluted with radioimmunoprecipitation assay buffer to achieve the same concentration of protein (15 μg/5 μL). Samples were diluted with an equal volume of 2× electrophoresis sample buffer containing 2% SDS and 10% glycerol with 0.2 M dithiothreitol. Proteins were separated by SDS-PAGE (5%–20% gradient gel; Atto, Tokyo, Japan). After electrophoresis, proteins were transferred to a nitrocellulose membrane (Amersham Life Science, Arlington Heights, IL, USA) in Tris/glycine buffer containing 100 mM Tris, 192 mM glycine, and 5% methanol. The membrane was soaked in a blocking buffer (5% bovine serum albumin in Tris-buffered saline [TBS] [pH 7.6] containing 0.1% Tween-20 [TBS/T]) for 60 minutes at room temperature. The membrane was immunoblotted overnight at 4°C with rabbit antibodies against Nav1.7 (1:500; Alomone Labs, Jerusalem, Israel) and Nav1.8 (1:500; Sigma-Aldrich Co, St Louis, MO, USA). The membrane was then washed in TBS/T three times at 10-minute intervals and incubated in horseradish peroxidase-conjugated goat anti-rabbit IgG (1:20,000; Cell Signaling Technology, Beverly, MA, USA) for 90 minutes at room temperature. After the membranes were washed in TBS/T five times at 5-minute intervals and in TBS twice at 5-minute intervals, the antigen–antibody peroxidase complex was detected by enhanced chemiluminescence (Thermo Fisher Scientific) and immunoreactive bands were visualized by Light Capture (AE-6981C; Atto). The membranes were washed again in blocking buffer containing 0.1% sodium azide and incubated with antibody against GAPDH (1:100,000; Merck Millipore, Billerica, MA, USA) overnight at 4°C with gentle agitation. Membranes were incubated with second antibody, and image development was performed. The intensity of the band was analyzed and semiquantified by computer-assisted densitometry using a CS analyzer (Atto). Values for Nav1.7 and Nav1.8 in mice DRG were normalized by the respective value for GAPDH.
Drugs
Tarantula venom peptide ProTx-II, which selectively interacts with Nav1.7 sodium channel, was purchased from Peptide Institute Inc. (Ibaraki, Osaka, Japan). (5-[4-chloro-phenyl]-furan2-carboxylic acid [3,5-dimethoxy-phenyl]-amide (A803467), a selective Nav1.8 sodium channel blocker, and STZ were purchased from Sigma-Aldrich Co. ProTx-II was dissolved in saline. A803467 was dissolved in 30% dimethyl sulfoxide (DMSO) in saline. For vehicle-only control groups, equal volumes of DMSO or saline were injected.
Data analysis
The data are expressed as means ± standard error for ten mice in each group. The statistical significance of differences between groups was assessed with a two-way analysis of variance followed by the Bonferroni/Dunnett test.
Results
Thermal hyperalgesia in STZ- induced diabetic mice
To determine thermal hyperalgesia, all mice were examined with regard to their tail-flick responses to noxious thermal stimulation before and after STZ administration. Diabetic mice had lower nociceptive threshold values than did nondiabetic mice, as evidenced by a significant (P<0.05) difference in the tail-flick latency between the two groups (diabetic mice, 6.5±0.7 seconds; nondiabetic mice, 11.6±0.3 seconds).
Effects of IT ProTx-II and A803467 on thermal hyperalgesia in STZ- induced diabetic mice
The tail-flick latencies in unmanipulated age-matched nondiabetic and diabetic mice did not differ from those in vehicle-only control rats that received IT DMSO or saline. Treatment with either ProTx-II, A803467, or DMSO did not caused abnormal behaviors in mice. Furthermore, mice were fully awake, were responsive to stimuli, and retained the righting reflex after the administration of these drugs.
IT administration of ProTx-II at doses from 0.04 to 4 ng to diabetic mice dose-dependently and significantly increased the tail-flick latency. The antihyperalgesic effect of ProTx-II (4 ng) in diabetic mice peaked within 60 minutes after treatment (baseline, 6.6±0.2 seconds; 60 minutes, 15.2±0.6 seconds) and gradually returned to baseline within 150 minutes (Figure 1A). However, IT administration of ProTx-II to nondiabetic mice, even at a dose of 4 ng, did not significantly change the tail-flick latency (Figure 1B).
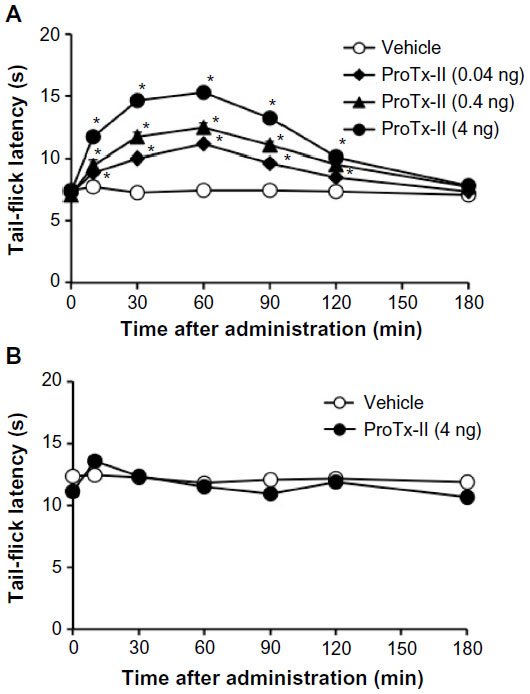 | Figure 1 Time course of the effect of ProTx-II on the thermal nociceptive threshold in diabetic (A) and nondiabetic (B) mice. |
After IT administration, A803467 at doses from 10 to 100 ng to diabetic mice also dose-dependently and significantly increased the tail-flick latency. The antihyperalgesic effect of A803467 (100 ng) in diabetic mice peaked within 30 minutes after treatment (baseline, 6.1±0.2 seconds; 30 minutes, 14.7±0.6 seconds) and gradually returned to baseline within 180 minutes (Figure 2B). However, IT administration of A803467 at a dose of 100 ng had no effect on the tail-flick latency in nondiabetic mice (Figure 2A).
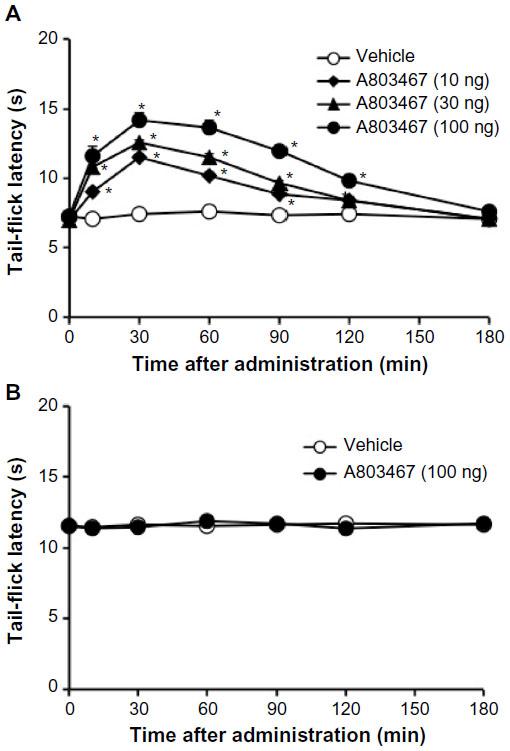 | Figure 2 Time course of the effect of A803467 on the thermal nociceptive threshold in diabetic (A) and nondiabetic (B) mice. |
Coadministration of ProTx-II (0.4 ng, IT) and A803467 (30 ng, IT) caused an additive antihyperalgesic effect in diabetic mice when the tail-flick latency was measured 30 minutes after administration (ProTx-II alone: baseline, 7.1±0.1 seconds; 30 minutes, 11.7±0.3 seconds, n=10; A803467 alone: baseline, 7.0±0.2 seconds; 30 minutes, 12.6±0.2 seconds, n=10; ProTx-II plus A803467: baseline, 6.3±0.3 seconds; 30 minutes, 16.8±0.5 seconds, n=10). However, IT coadministration of ProTx-II and A803467 had no effect on the tail-flick latency in nondiabetic mice (baseline, 11.9±0.3 seconds; 30 minutes, 12.4±0.2 seconds, n=10).
Effect of diabetes on the protein levels of Nav1.7 and Nav1.8 sodium channels in DRG
Expression of the Nav1.7 and Nav1.8 sodium-channel proteins was examined in the spinal cords of diabetic and nondiabetic mice. There was no difference in the expression of either the Nav1.7 or Nav1.8 sodium-channel protein in the DRG between diabetic and nondiabetic mice (Figure 3).
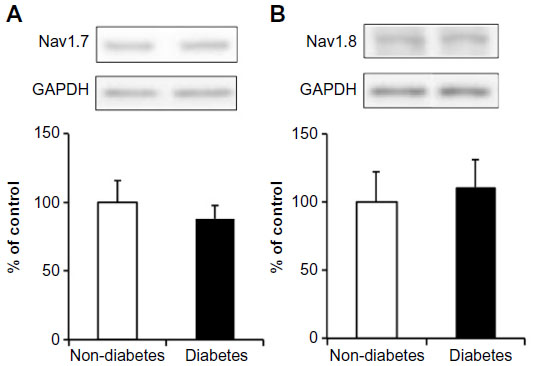 | Figure 3 The protein levels of Nav1.7 (A) and Nav1.8 (B) in the dorsal root ganglion of nondiabetic and diabetic mice. |
Discussion
Previously, we reported that the IT administration of sodium-channel opener fenvalerate produced characteristic behavioral responses which mainly consisted of hind limb scratching directed toward caudal parts of the body and biting or licking of the hind legs.25 The duration of this IT fenvalerate-induced characteristic behavioral response in diabetic mice was significantly longer than that in nondiabetic mice. Furthermore, pretreatment with intraperitoneal morphine (1–10 mg/kg) dose-dependently reduced fenvalerate-induced characteristic behavioral response.25 We also demonstrated that fenvalerate-induced characteristic behavioral response was reduced by mexiletine (10 and 30 mg/kg, intraperitoneal), a lidocaine-like sodium-channel blocker, in a dose-dependent manner.25 Therefore, we suggested that fenvalerate-induced behavior may be related to nociception caused by the activation of sodium channels. However, fenvalerate is known to modulate both TTX-R and TTX-S sodium channels in a similar direction.26 We recently demonstrated that the fenvalerate-induced nociceptive response was reduced by pretreatment with calphostin C, a selective protein kinase C inhibitor.25 On the other hand, the fenvalerate-induced nociceptive response was enhanced by pretreatment with protein kinase C activator, phorbol-12, 13-dibutyrate.25 Thio and Sontheimer reported that the activation of protein kinase C had different effects on TTX-S and TTX-R sodium currents.27 They reported that although phorbol 12-myristate 13-acetate reduced peak currents of TTX-S sodium channels, it potentiated peak current of TTX-R sodium channels.27 TTX-R current activation was faster and current inactivation changed from single- to bi-exponential after exposure to phorbol 12-myristate 13-acetate.27 In contrast, TTX-S current activation was unchanged, and current inactivation decreased by an average of 50% following exposure to phorbol 12-myristate 13-acetate.27 Similarly, it has been reported that the activation of protein kinase C increased TTX-R sodium current, whereas inhibitors of protein kinase C decreased TTX-R sodium current.28 These results suggest that the sensitization of TTX-R sodium channels, by the activation of protein kinase C, may play an important role in the enhancement of the duration of fenvalerate-induced nociceptive behavior in diabetic mice. Based on these results, we concluded that TTX-R sodium-channel function is altered in diabetes, which explains why neurons that participate in nociception are highly excitable in diabetic animals.29 On the other hand, it has been reported that changes in the activities of sodium channels, particularly TTX-R Nav1.8 sodium channels, can play an important role in the progression of diabetes-induced signs of pain, such as thermal hyperalgesia and mechanical allodynia.14,30 A803467 was 300- to 1,000-fold more potent at blocking Nav1.8 as compared with its ability to block Nav1.2, Nav1.3, Nav1.5, and Nav1.7.31 The present study demonstrated that IT administration of A803467, a specific Nav1.8 sodium-channel blocker, produced a dose-dependent antinociceptive effect in diabetic mice, but not in nondiabetic mice. This result supports the above hypothesis that TTX-R Nav1.8 sodium channels play an important role in the progression of painful diabetic neuropathy.
On the other hand, although several reports have indicated that animals with painful diabetic neuropathy exhibited an increase in protein levels of Nav1.7 sodium channel in DRG compared to lean controls with no pain,32–34 no previous study has indicated that Nav1.7 sodium-channel blockade has a therapeutic effect against painful diabetic neuropathy. Interestingly but unexpectedly, we observed that ProTx-II, a selective blocker of Nav1.7 sodium channels, has antinociceptive effects in diabetic mice with painful neuropathy. Although ProTx-II inhibits multiple sodium-channel subtypes (Nav1.1–1.8), it has been reported to be 100-fold more selective for Nav1.7.35,36 Thus, our data suggest that Nav1.7 sodium channels contribute to thermal hyperalgesia in diabetic mice with painful neuropathy.
The percentage of large-soma DRG neurons that express TTX-S Nav1.7 significantly increased in diabetic animals, suggesting that these alterations may underlie the increased sodium currents in diabetic rats. On the other hand, TTX-R Nav1.8 transcription and expression are decreased in diabetic rats30,37 with increased TTX-R sodium currents.30,38–40 In the present study, however, we observed that the expression of both Nav1.7 and Nav1.8 in the DRG of diabetic mice did not differ from those in nondiabetic mice. The phosphorylation of Nav1.7 and Nav1.8 appears to be increased in the DRG of diabetic rats, suggesting that changes in serine/threonine and tyrosine phosphorylation of voltage-gated sodium channels may play a role in the transmission of painful stimuli in diabetic neuropathy.30 Changes in the function of sodium channels, such as sodium-channel phosphorylation, but not the expression of channels, may contribute to painful diabetic neuropathy.
In summary, the present results suggest that ProTx-II and A803467, highly selective blockers of Nav1.7 and Nav1.8 sodium channels, respectively, in the spinal cord, can produce antihyperalgesic effects in diabetic mice. Furthermore, we observed that coadministration of ProTx-II (0.4 ng, IT) and A803467 (30 ng, IT) caused an additive antihyperalgesic effect in diabetic mice. These data firstly provide functional evidence that blockade of Nav1.7 and Nav1.8 sodium channels may be appropriate targets for treating the painful clinical signs of diabetes.
Disclosure
The authors report no conflicts of interest in this work.
References
Calcutt NA. Potential mechanisms of neuropathic pain in diabetes. Int Rev Neurobiol. 2002;50:205–228. | |
Kamei J, Ogawa M, Kasuya Y. Development of supersensitivity to substance P in the spinal cord of the streptozotocin-induced diabetic rats. Pharmacol Biochem Behav. 1990;35:473–475. | |
Ohsawa M, Kamei J. Role of intracellular calcium in thermal allodynia and hyperalgesia in diabetic mice. Brain Res. 1999;833:278–281. | |
Viola V, Newnham HH, Simpson RW. Treatment of intractable painful diabetic neuropathy with intravenous lignocaine. J Diabetes Complications. 2006;20:34–39. | |
Wolff RF, Bala MM, Westwood M, Kessels AG, Kleijnen J. 5% lidocaine medicated plaster in painful diabetic peripheral neuropathy (DPN): a systematic review. Swiss Med Wkly. 2010;140:297–306. | |
Dejgard A, Petersen P, Kastrup J. Mexiletine for treatment of chronic painful diabetic neuropathy. Lancet. 1988;1:9–11. | |
Oskarsson P, Ljunggren JG, Lins PE. Efficacy and safety of mexiletine in the treatment of painful diabetic neuropathy. The Mexilitine Study Group. Diabetes Care. 1997;20:1594–1597. | |
Kamei J, Saitoh A, Kasuya Y, Involvement of delta 1-opioid receptors in the antinociceptive effects of mexiletine in mice. Neurosci Lett. 1995;196:169–172. | |
Suzuki N, Hasegawa-Moriyama M, Takahashi Y, Kamikubo Y, Sakurai T, Inada E. Lidocaine attenuates the development of diabetic-induced tactile allodynia by inhibiting microglial activation. Anesth Analg. 2011;113:941–946. | |
Chahine M, Ziane R, Vijayaragavan K, Okamura Y. Regulation of Na v channels in sensory neurons. Trends Pharmacol Sci. 2005;26:496–502. | |
Devor M. Sodium channels and mechanisms of neuropathic pain. J Pain. 2006;7:S3–S12. | |
Zuliani V, Rivara M, Fantini M, Costantino G. Sodium channel blockers for neuropathic pain. Expert Opin Ther Pat. 2010;20:755–779. | |
Amaya F, Decosterd I, Samad TA, et al. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium-ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. | |
Liu M, Wood JN. The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med. 2011; 12 Suppl 3:S93–S99. | |
Novakovic SD, Tzoumaka E, McGivern JG, et al. Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci. 1998;18:2174–2187. | |
Cummins TR, Sheets PL, Waxman SG. The roles of sodium channels in nociception: Implications for mechanisms of pain. Pain. 2007;131:243–257. | |
Gardiner M. Molecular genetics of infantile nervous system channelopathies. Early Hum Dev. 2006;82:775–779. | |
Henry MA, Freking AR, Johnson LR, Levinson SR. Increased sodium channel immunofluorescence at myelinated and demyelinated sites following an inflammatory and partial axotomy lesion of the rat infraorbital nerve. Pain. 2006;124:222–233. | |
Rogers M, Tang L, Madge DJ, Stevens EB. The role of sodium channels in neuropathic pain. Semin Cell Dev Biol. 2006;17:571–581. | |
Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007;30:555–563. | |
Xiao Y, Blumenthal K, Jackson JO 2nd, Liang S, Cummins TR. The tarantula toxins ProTx-II and huwentoxin-IV differentially interact with human Nav1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol. 2010;78:1124–1134. | |
McGaraughty S, Chu KL, Scanio MJ, Kort ME, Faltynek CE, Jarvis MF. A selective Nav1.8 sodium channel blocker, A803467 [5-(4-chlorophenyl-N-(3,5-dimethoxyphenyl)furan-2-carboxamide], attenuates spinal neuronal activity in neuropathic rats. J Pharmacol Exp Ther. 2008;324:1204–1211. | |
Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. | |
Ohsawa M, Carlsson A, Asato M, et al. The dipeptide Phe-Phe amide attenuates signs of hyperalgesia, allodynia and nociception in diabetic mice using a mechanism involving the sigma receptor system. Mol Pain. 2011;7:85. | |
Kamei J, Sasaki M, Zushida K, Morita K, Tanaka S. Nociception and allodynia/hyperalgesia induced by intrathecal administration of fenvalerate. Jpn J Pharmacol. 2001;86:336–341. | |
Song JH, Nagata K, Tatebayashi H, Narahashi T. Interaction of tetramethrin, fenvalerate and DDT at the sodium channel in rat dorsal root ganglion neurons. Brain Res. 1996;708:29–37. | |
Thio CL, Sontheimer H. Differential modulation of TTX-sensitive and TTX-resistant Na+ channels in spinal cord astrocytes following activation of protein kinase C. J Neurosci. 1993;13:4889–4897. | |
Gold MS, Levine JD, Correa AM. Modulation of TTX-R INa by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci. 1998;18:10345–10355. | |
Kamei J, Iguchi E, Sasaki M, Zushida K, Morita K, Tanaka S. Modification of the fenvalerate-induced nociceptive response in mice by diabetes. Brain Res. 2002;948:17–23. | |
Hong S, Morrow TJ, Paulson PE, Isom LL, Wiley JW. Early painful diabetic neuropathy is associated with differential changes in tetrodotoxin-sensitive and -resistant sodium channels in dorsal root ganglion neurons in the rat. J Biol Chem. 2004;279:29341–29350. | |
Jarvis MF, Honore P, Shieh CC, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104:8520–8525. | |
Black JA, Frézel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain. 2012;8:82. | |
Galloway C, Chattopadhyay M. Increases in inflammatory mediators in DRG implicate in the pathogenesis of painful neuropathy in Type 2 diabetes. Cytokine. 2013;63:1–5. | |
Zhang JL, Yang JP, Zhang JR, et al. Gabapentin reduces allodynia and hyperalgesia in painful diabetic neuropathy rats by decreasing expression level of Nav1.7 and p-ERK1/2 in DRG neurons. Brain Res. 2013;1493:13–18. | |
Smith JJ, Cummins TR, Alphy S, Blumenthal KM. Molecular interactions of the gating modifier toxin ProTx-II with NaV 1.5: implied existence of a novel toxin binding site coupled to activation. J Biol Chem. 2007;282:12687–12697. | |
Schmalhofer WA, Calhoun J, Burrows R, et al. ProTx-II, a selective inhibitor of NaV1.7 sodium channels, blocks action potential propagation in nociceptors. Mol Pharmacol. 2008;74:1476–1484. | |
Craner MJ, Klein JP, Renganathan M, Black JA, Waxman SG. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol. 2002;52:786–792. | |
Shah BS, Gonzalez MI, Bramwell S, Pinnock RD, Lee K, Dixon AK. Beta3, a novel auxiliary subunit for the voltage gated sodium channel is upregulated in sensory neurones following streptozocin induced diabetic neuropathy in rat. Neurosci Lett. 2001;309:1–4. | |
Lampert A, O’Reilly AO, Reeh P, Leffler A. Sodium channelopathies and pain. Pflugers Arch. 2010;460:249–263. | |
Hirade M, Yasuda H, Omatsu-Kanbe M, Kikkawa R, Kitasato H. Tetrodotoxin-resistant sodium channels of dorsal root ganglion neurons are readily activated in diabetic rats. Neuroscience. 1999;90:933–939. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.