")
Back to Journals » Drug Design, Development and Therapy » Volume 11
Antiglioma effects of cytarabine on leptomeningeal metastasis of high-grade glioma by targeting the PI3K/Akt/mTOR pathway
Authors Zhao KH, Zhang C, Bai Y, Li Y, Kang X, Chen JX, Yao K, Jiang T, Zhong XS , Li WB
Received 27 February 2017
Accepted for publication 5 May 2017
Published 26 June 2017 Volume 2017:11 Pages 1905—1915
DOI https://doi.org/10.2147/DDDT.S135711
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Kai-Hong Zhao,1 Can Zhang,2 Yue Bai,1 Yan Li,1 Xun Kang,1 Jian-Xin Chen,1 Kun Yao,3 Tao Jiang,4 Xiao-Song Zhong,2 Wen-Bin Li1
1Department of Glioma, 2Clinical Center of Gene And Cell Engineering, Beijing Shijitan Hospital, 3Department of Neurosurgery, Beijing Tiantan Hospital, 4Department of Pathology, Sanbo Brain Hospital, Capital Medical University, Beijing, People’s Republic of China
Abstract: Leptomeningeal metastasis (LM) of high-grade glioma is a highly lethal disease requiring new effective therapeutic measures. For both de novo or relapsed glioma with LM, intrathecal cytarabine chemotherapy is not frequently used for first-line and relapse protocols. We encountered a clinical case demonstrating effective application of cytarabine in high-grade glioma with LM, prompting us to explore the effects of cytarabine on malignant glioma and molecular mechanisms of such effects through in vivo and in vitro experiments. The U87 cell line was selected to represent human glioma for studies. Cell viability was measured by MTT assay, plate colony formation assay, and trypan-blue dye exclusion test. Apoptosis was assessed by flow cytometry. Protein expression levels were detected by Western blot assay and immunohistochemistry. mRNA expression was examined by quantitative real-time reverse transcription polymerase chain reaction. Cytarabine inhibited tumor growth during the in vivo experiment. The present study confirmed that cytarabine inhibits proliferation and promotes apoptosis of U87 cells, and molecular analysis of this effect showed that cytarabine significantly reduces expression of phosphatidylinositol 3-kinase/serine/threonine kinase also known as the protein kinase B/mechanistic target of rapamycin (PI3K/Akt/mTOR) pathway, Ki-67, BCL2, and 4-1BB, and upregulates Bax and cleaved caspase-3. Our findings indicated that intrathecal administration of cytarabine manifests potential in prophylaxis and treatment of malignant glioma with LM. Effective medications for high-grade glioma with LM should contain cytarabine.
Keywords: leptomeningeal metastases, malignant glioma, cytarabine, PI3K/Akt/mTOR
Introduction
Leptomeningeal metastasis (LM) has been described with increasing frequency in glioma in recent years as a result of improving survival rates and survival times in patients with high-grade glioma. LM occurs in 20%–28%1–3 of supratentorial and infratentorial glioblastoma (GBM), as described previously by autopsy series. Noh et al4 reported that 23.4% of GMB patients (75 of 321) were diagnosed with LM; this observation was not significantly different from those in prior reports. Arita et al5 reported that 15 (15.6%) out of 97 patients with malignant gliomas developed LM at least 2 years after their initial treatment. Among patients in the age group less than 20 years, the incidence of LM was higher (46%) in comparison with older groups (12%), and all patients died 2–24 weeks after diagnosis of LM. Arita et al1 reported that 22 (14%) of 157 patients showed dissemination of malignant gliomas, and survival after dissemination was limited in all patients (mean, 19 weeks; range, 2–39 weeks). Prognosis in patients with LM is very poor and nearly always results in lethal outcomes. However, no consensus currently exists on treatment for LM in malignant glioma.
Risk factors of LM in patients with malignant gliomas include tumor location adjacent to the ventricular system,6,7 dispersion of tumor cells by recurrent operative treatment, ventricular entry during operation, and multiple resections.6 LM in malignant gliomas is increasingly considered as progression in management strategies to better control primary tumors and to improve survival. Clinicians should be familiar with the potential of LM as well as treatment modalities to prevent severe neurological deficits and to provide the most appropriate treatment to affected patients. Treatment options for LM are not very satisfactory and mainly palliative. LM is usually not amenable to surgery because of the diffuse nature of gliomas. Systemic chemotherapy penetration works poorly for cerebrospinal fluid (CSF) or periventricular regions adjacent to the CSF channels.8 One approach to targeting this region may be through CSF-directed chemotherapy. Thus, intracerebroventricular (ICV) or intracavitary (ICT) chemotherapy is an important and preferred method for directly addressing tumor cells.
Published studies showed activation or overexpression of p-serine/threonine kinase, also known as protein kinase B (Akt) and p-mechanistic target of rapamycin (mTOR), in human gliomas.9,10 The labeling index of the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway increased with increasing grade of malignancy, with stronger expression found in higher grade gliomas.9,10 The important role of the PI3K/Akt/mTOR pathway in angiogenesis was confirmed in various malignancies, where inhibition of PI3K by LY294002 and downregulation of PI3KC2α were shown to block tumor vascularization.11,12 In GBM, the PI3K/Akt/mTOR pathway plays a crucial role in the induction of invasion, angiogenesis, and expression of vascular endothelial growth factor in cells.13 Patients with GBM in whom the PI3K/Akt/mTOR pathway is activated show poorer prognosis than patients without oncogenic activation of the same pathway.14 Therefore, inhibitors targeting the PI3K/Akt/mTOR pathway emerge as a potential treatment for malignant gliomas.
Cytarabine (1-β-arabinofuranosylcytosine) is a pyrimidine nucleoside analog that inhibits synthesis of DNA at the S phase of the cell cycle. Intrathecal liposomal cytarabine is indicated to be effective for prophylaxis and treatment of meningeal leukemia, brain tumors (medulloblastoma, meningioma, neuro-ectodermal tumors, germ cell tumors, and oligodendroglioma), lymphoma, and disseminated malignancies (breast).15 Cytarabine manifests therapeutic efficiency in leukemic patients by inhibition of mTOR-dependent autophagic responses.16 Previous studies indicated that GBM-isolated CD133+ stem cells demonstrate resistance to many agents, including temozolomide, carboplatin, paclitaxel, etoposide, and carmustine,17,18 but show relative sensitivity to cytotoxic cytarabine.19,20 However, the inhibitory mechanism of action of cytarabine in GBM remains unclear.
In the present study, we observed a clinical case showing effective application of cytarabine for high-grade glioma with LM, prompting us to explore the effects of cytarabine on malignant glioma and the molecular mechanism of such effects through in vivo and in vitro experiments. We defined a novel mechanism underlying the inhibitory function of cytarabine in malignant gliomas.
Materials and methods
Ethics statement
As specified in the case report, the patient in our study provided written informed consent for publication of this article and the accompanying images. Animal experiments were approved by the Capital Medical University and Beijing Shijitan Hospital and performed in accordance with the ethical guidelines for animal experiments of the Beijing Shijitan Hospital Scientific Research Ethics Committee.
Patient and treatment
The patient was enrolled at Beijing Shijitan Hospital from October 2013 to March 2015. We collected information, such as age, sex, initial symptoms, diagnosis, treatment, efficacy, safety, and other data, from the electronic database of the Beijing Shijitan Hospital. Evidence of recurrence or progression was confirmed by evaluably enhancing the recurrent tumor on contrast-enhanced magnetic resonance imaging (MRI) and comparing laboratory data (mainly CSF protein levels) before and after treatments.
The patient was an 18-year-old male, admitted to the Beijing Shijitan Hospital between October 2013 and March 2015. Initial symptoms in this patient included neck pain and right limb weakness, and an MRI showed neoplasms in 3–6 cervical cord. Surgical resection was conducted in March 2013 with histologically confirmed World Health Organization grade III malignant glioma, anaplastic astrocytoma, and was followed by three cycles of oral chemotherapies with temozolomide (5/28 scheme). Severe headaches, projectile vomiting, motor aphasia, and diplopia were observed in September 2013. Meningeal dissemination was confirmed by MRI scans and CSF assays. Craniospinal irradiation combined with methotrexate (MTX) intrathecal chemotherapy was undertaken seven times from September to November 2013. No obvious improvement occurred according to evaluable contrast-enhanced MRI, symptoms, and laboratory data (mainly CSF protein levels). Then, cytarabine was intrathecally injected twice a week, six times, from December 2013 to April 2014 and monthly, three times, from April 2014 to July 2014.
Cell culture
Human glioma cell line U87 cells were obtained from the ATCC and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum and 1% antibiotic mixture (penicillin and streptomycin) at 37°C in 5% CO2. DMEM was used as the drug carrier for all cytarabine (C1768; Sigma Aldrich, St Louis, MO, USA) treatments. A PI3K inhibitor, LY294002 (L9908; Sigma Aldrich, St Louis, MO, USA), was used at a final concentration of 10 μM for several experiments. An mTOR inhibitor, rapamycin (37094; Sigma Aldrich, St Louis, MO, USA), was used at a final concentration of 10 ng/mL. Subsequently, Western blot (WB) assay was applied to both inhibitors after 30 min.
MTT assay
U87 cells were seeded in 96-well plates at 2,000 and 500 cells/well in 200 μL of medium. Cells were cultured for 24 h and allowed to sediment. Subsequently, different concentrations of cytarabine (2,000, 1,000, 500, 125, 62.5, 15.6, and 3.9 nM) were added, and the plates were incubated for 24, 48, 72, 96, and 120 h after addition of drugs. Exactly 10 μL of MTT (5 mg/mL) was added to each well. Plates were incubated at 37°C for 4 h. Then, 110 μL of dimethyl sulfoxide was added to each well. Plates were agitated on a plate shaker for 10 min. Optical density at 570 nm was read with an Multiskan Spectrum Microplate Reader (Thermo Fisher Scientifc, Waltham, MA, USA).
Plate colony formation assay
A total of 400 U87 cells were seeded in 6 cm dishes in complete medium. Cells were cultured for 24 h and allowed to sediment; then, various concentrations of cytarabine (2,000, 1,000, 500, 250, 125, 62.5, 15.6, and 3.9 nM) were added. After 3 weeks of growth, colonies could be counted by the naked eye. Cells were fixed with 4% paraformaldehyde, stained with crystal violet (0.1% w/v in 20 nM 4-morpholinepropanesulfonic acid; Sigma Aldrich), and washed twice with phosphate-buffered saline (PBS). Efficiency of plating was determined as the number of clearly visible colonies (diameter >50 μm).
Trypan-blue dye viability test
U87 cells were incubated with cytarabine (125, 500, and 1,000 nM), LY294002 (10 μM), and rapamycin (10 ng/mL) for 48 h, respectively. Exactly 100 μL of cell suspension was mixed with 100 μL 0.4% trypan blue solution, which was used to discriminate between viable and non-viable cells. Subsequently, cells were mixed intensively and incubated for 1 min at room temperature. A total of 10 μL mixed liquor was obtained, and the hemocytometer chamber was filled carefully. Cells were counted under the microscope in four 1×1 mm2 squares of one chamber, and the average number of cells per square was determined. Non-viable cells were blue, and viable cells remained unstained.
Apoptosis assay
Apoptotic cells were assessed using the PE Annexin V apoptosis detection kit (Becton, Dickinson and Company, NJ, USA). Cells in different groups were digested and resuspended in 100 μL of binding buffer containing 5 μL of PE Annexin V and 5 μL of 7-Amino-Actinomycin for 15 min at room temperature. Then, 400 μL of binding buffer was added for 1 h of incubation. Finally, cells were analyzed by flow cytometry on a FACSCalibur flow cytometer (Becton-Dickinson, Franklin-Lakes, NJ, USA).
Western blot analysis
U87 cells were incubated with cytarabine (15, 125, and 500 nM) for 0.5 h. Moreover, U87 cells were pretreated for 0.5 h with a PI3K inhibitor, LY294002 (10 μM), and the mTOR inhibitor, rapamycin (10 ng/mL). Cells were washed with PBS and lysed in Laemmli buffer to detect the PI3K/Akt/mTOR signal pathway. Akt (pan) (C67E7) Rabbit monoclonal antibody (mAb) (4691), Phospho-Akt (Ser473) (D9E) XP® Rabbit mAb (4060), mTOR (7C10) Rabbit mAb (2983), Phospho-mTOR (Ser2448) (D9C2) XP® Rabbit mAb (5536), p70S6 Kinase (49D7) Rabbit mAb (2708), and Phospho-p70S6 Kinase (Thr389) (108D2) Rabbit mAb (9234) were all acquired from Cell Signaling Technology (Danvers, MA, USA), and β-actin (A2228) was acquired from Sigma Aldrich (St Louis, MO, USA). Secondary antibodies were anti-rabbit antibodies (1:10,000; Cell Signaling Technology). Protein concentrations were detected by the Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. Forty microgram samples of each protein were loaded onto 10% sodium dodecyl sulfate-polyacrylamide gel. After electrophoresis, proteins were transferred to a hybridization nitrocellulose (NC) filter membrane (Millipore, Tullagreen, Carrigtwohill Co, Ireland). The NC membrane was blocked with blocking solution (Tris–HCl pH 7.4, NaCl, Tween 20, deionized water, and 5% bovine serum albumin [BSA]) for 1 h and reacted with antibodies overnight at 4°C. The membrane was then washed with blocking solution without skimmed milk and incubated with secondary antibodies for 1 h. After washing again, protein levels were analyzed by enhanced chemiluminescence (ECL) with an ECL and Western blotting detection system (Odyssey CLx; Gene Company Limited, Chai Wan, Hong).
Quantitative real-time reverse transcription polymerase chain reaction
Total RNA was extracted from cultured U87 cells using TRIzol reagent (Ambion; Thermo Fisher Scientific). High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA, USA) was used to reverse transcribe 1 μg of RNA. Samples were prepared with PerfeCTa SYBR Green FastMix (Thermo Fisher Scientific, Waltham, MA, USA) and primer sets hBCL2, sense GAGCCACGACCCTTCTTAAGACAT, antisense CAGGGGTCAATTAATCCATGACA; h4-1BB, sense ATGACAATAAGCCACGAGGTGCAG, antisense AAAGGGAGCAGGACAAAGGCAGAA; hBax, sense TCCACCAAGAAGCTGAGCGAG, antisense GTCCAGCCCATGATGGTTCT; and hβ-actin, sense TGACGTGGACATCCGCAAAG, antisense CTGGAAGGTGGACAGCGAGG. Each RNA level was normalized by comparison with the corresponding housekeeping RNA level in the same sample. Results were calculated using the 2ΔCt method.
In vivo treatment studies
Experiments were approved by the ethics committee. Six-week-old nonobese diabetic/severe combined immunodeficiency (NOD-SCID) mice (n=18) were housed in groups of two to four in cages. Indoor temperature was maintained at 20°C and mice were subjected to a 12 h light–12 h darkness cycle. A week later, 0.7×107 U87 cells were injected subcutaneously into the back of mice after their adaptation to the experimental environment. Mice were monitored by palpation every 2 days until tumors were palpable at the 14th day. All mice were randomly divided into three groups, and two groups were intraperitoneally and peritumorally injected with 60 mg/kg cytarabine, respectively. The control group was not treated. Mice received two-cycle injections continually, and each cycle included 10 consecutive days of treatment followed by 7 days of rest. The dose and treatment period in this study were selected on the basis of directions for use of clinical cytarabine (Cytosar). Tumor volumes and mice weights were measured at different time points using a caliper and an electronic scale. Tumor volumes (longest diameter × shortest diameter2)/2 were calculated. At the end of experiments, mice were narcotized by 10% chloral hydrate. Tumors were rapidly removed, and samples were preserved in formalin for subsequent hematoxylin and eosin staining for histopathological analysis and immunohistochemical (IHC) studies. Then, mice were sacrificed by cervical dislocation.
Immunohistochemistry and evaluation
Immunohistochemistry was performed, and diagnoses were confirmed. Percentages of positive cells were assessed and assigned to one of the following parameters: negative ≤10%, and ≥10% positive. Representative tissue blocks were cut at 4 mm thickness, deparaffinized with xylene rinse, and rehydrated with distilled water through graded alcohol. IHC staining was conducted on the other four slides using a two-step procedure. Slides were then incubated overnight at 4°C in humidified chambers with human Ki-67 (Abcam, Cambridge, UK; diluted 1:200), P-Akt (Cell Signaling Technology; diluted 1:200), and cleaved caspase-3 (Asp175; diluted 1:100; Cell Signaling Technology). Slides were washed thrice with PBS and further incubated with secondary antibodies for 30 min at room temperature. After washing with PBS, immunolabeled sections were incubated with biotin-conjugated secondary antibody for 20 min at room temperature, then with peroxidase-conjugated complex (Dako) for 20 min, visualized with 3,3′-diaminobenzidine, counterstained with hematoxylin, and then examined by light microscopy.
Statistical analysis
Each experiment was performed at least thrice, and representative data are presented as mean ± standard deviation. One-way analysis of variance was used to detect differences between control and experimental groups. Analyses were performed using SPSS 20.0 software. The standard value for statistical significance was P<0.05.
Results
Effects of treatment on patient
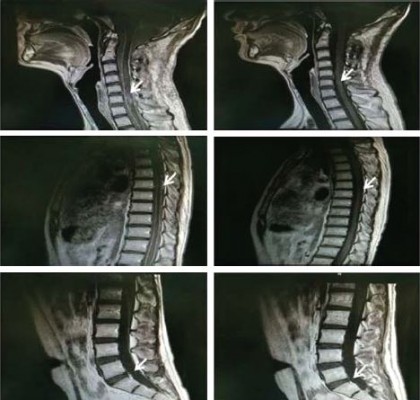
Tumor lesions in both cervical cord and meninges were eliminated in most parts according to radiographic assessment (Figure 1A). Levels of CSF protein gradually dropped to normal (Figure 1B). The patient remained stable until November 2014, and progression-free survival (PFS) was noted as 11 months.
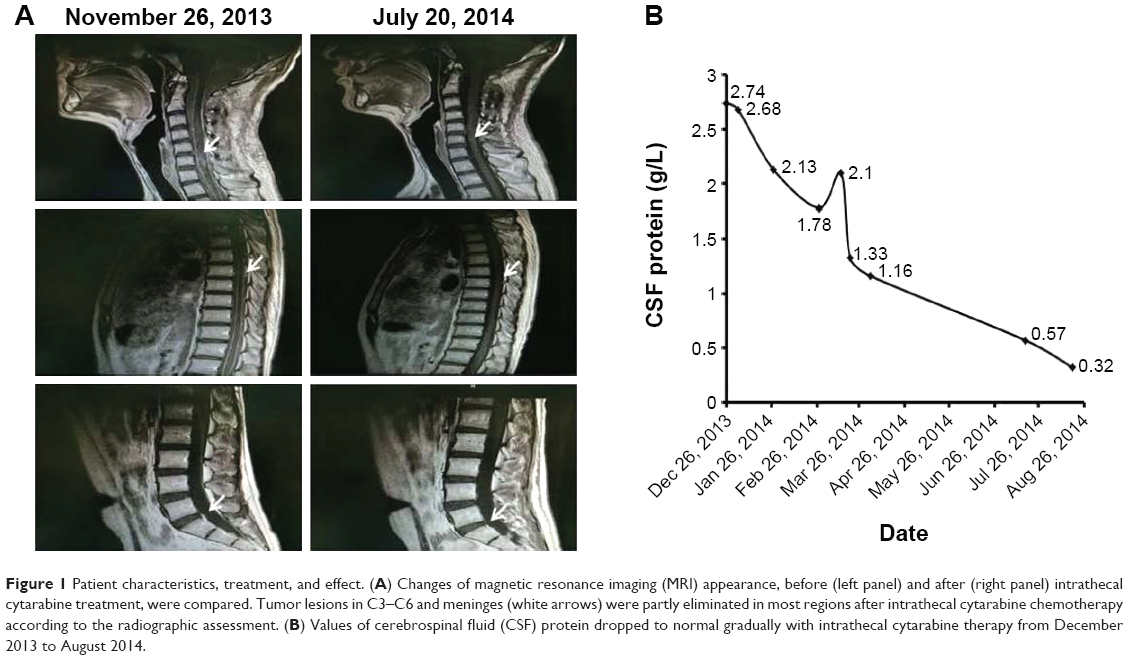 | Figure 1 Patient characteristics, treatment, and effect. (A) Changes of magnetic resonance imaging (MRI) appearance, before (left panel) and after (right panel) intrathecal cytarabine treatment, were compared. Tumor lesions in C3–C6 and meninges (white arrows) were partly eliminated in most regions after intrathecal cytarabine chemotherapy according to the radiographic assessment. (B) Values of cerebrospinal fluid (CSF) protein dropped to normal gradually with intrathecal cytarabine therapy from December 2013 to August 2014. |
Cytarabine and PI3K/Akt/mTOR pathway inhibitors could inhibit human malignant glioma cell growth
U87 cells were selected to represent human glioma for studies. Cell viability was measured by MTT assay, plate colony formation assay, and trypan-blue dye exclusion test. Apoptosis was assessed by flow cytometry. We discovered that cytarabine treatment resulted in decreased viability in U87 cells in a dose- and time-dependent manner, with IC50 values of 554.34±70.3 and 530.7±100.7 nM after 72 and 96 h of treatment, respectively (Figure 2A). Trypan-blue dye viability test showed that both cytarabine and LY294002 and rapamycin inhibited cell growth and decreased cell viability (Figure 2B). Plate colony formation assay indicated that cytarabine significantly decreased plating efficiency of U87 cells (Figure 2C and D). Flow cytometry showed that cytarabine induced cell apoptosis in a dose-dependent manner (Figure 2E and F).
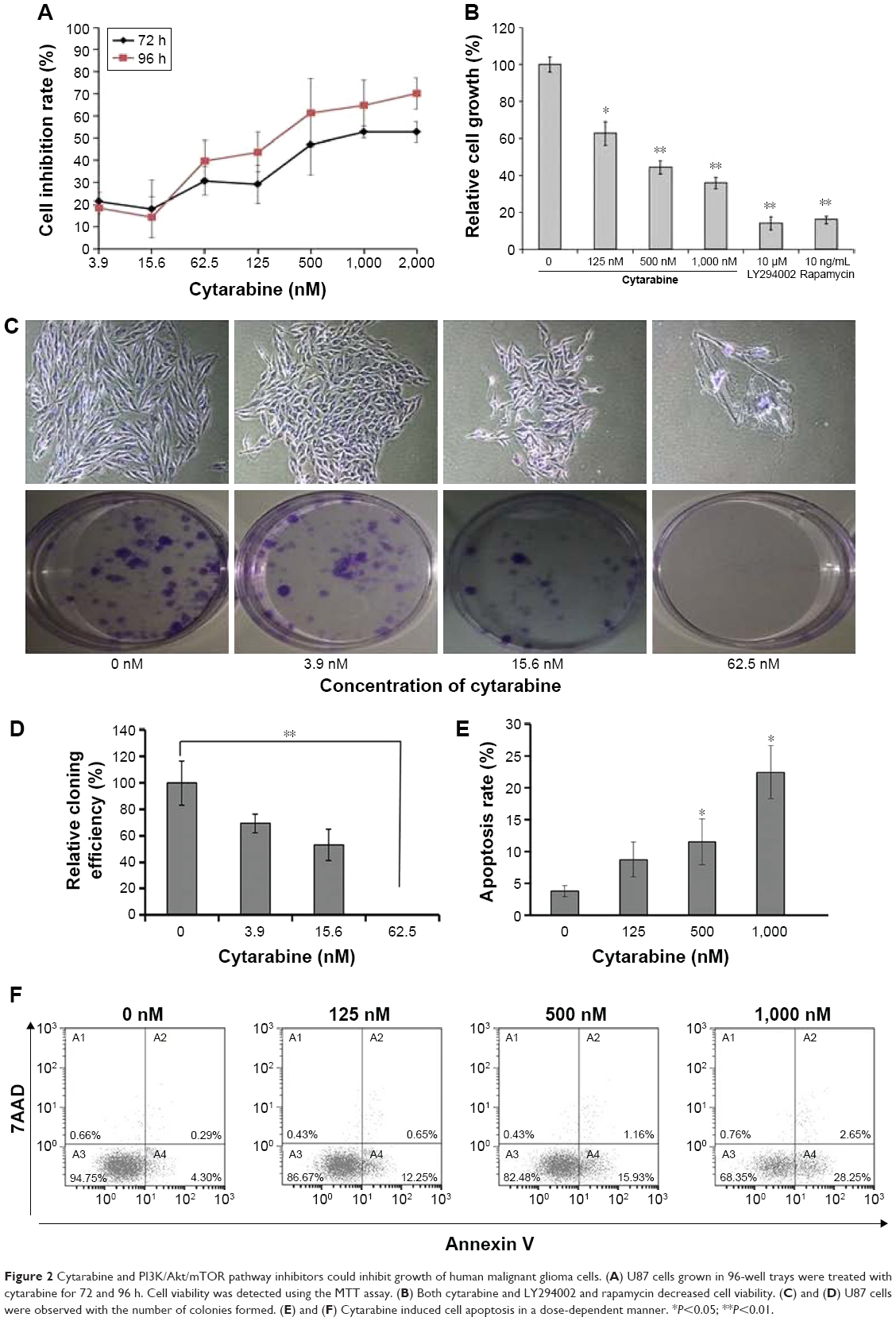 | Figure 2 Cytarabine and PI3K/Akt/mTOR pathway inhibitors could inhibit growth of human malignant glioma cells. (A) U87 cells grown in 96-well trays were treated with cytarabine for 72 and 96 h. Cell viability was detected using the MTT assay. (B) Both cytarabine and LY294002 and rapamycin decreased cell viability. (C) and (D) U87 cells were observed with the number of colonies formed. (E) and (F) Cytarabine induced cell apoptosis in a dose-dependent manner. *P<0.05; **P<0.01. |
Cytarabine could downregulate PI3K/Akt/mTOR pathway in human malignant glioma cells
Subsequent studies aimed at elucidating the underlying molecular mechanisms of cytarabine. To further delineate mechanisms by which cytarabine decreased cell survival and proliferation in U87 cells, WB assay was performed and revealed that cytarabine significantly decreased phosphorylation of Akt, mTOR, and p70S6K in a dose-dependent manner (Figure 3A and B). These results revealed that cytarabine inhibited cell proliferation and survival in human malignant glioma cells through downregulation of the PI3K/Akt/mTOR pathway.
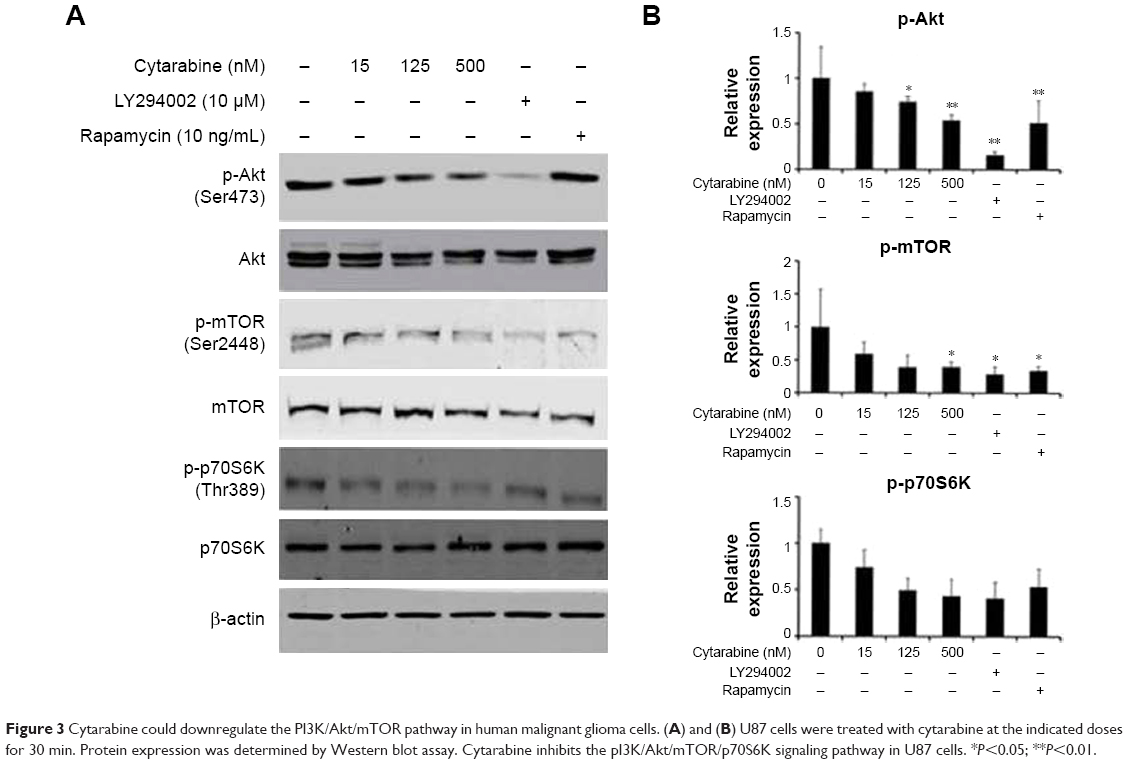 | Figure 3 Cytarabine could downregulate the PI3K/Akt/mTOR pathway in human malignant glioma cells. (A) and (B) U87 cells were treated with cytarabine at the indicated doses for 30 min. Protein expression was determined by Western blot assay. Cytarabine inhibits the pI3K/Akt/mTOR/p70S6K signaling pathway in U87 cells. *P<0.05; **P<0.01. |
Cytarabine could downregulate antiapoptotic molecules BCL2 and 4-1BB and upregulate proapoptotic molecule Bax
To explore mechanisms by which cytarabine induced apoptosis in U87 cells, we examined expression of antiapoptotic molecules BCL2 and 4-1BB and proapoptotic molecule Bax by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR) in cytarabine-treated cells. As shown in Figure 4A, after cytarabine treatment, BCL2 and 4-1BB mRNA levels decreased and Bax levels increased in U87 cells as compared with those in controls. Significant differences were observed in 4-1BB and BCL2. These findings defined a novel signaling cascade controlling survival of glioma cells and helped delineate mechanisms underlying antitumoral properties of cytarabine (Figure 4B).
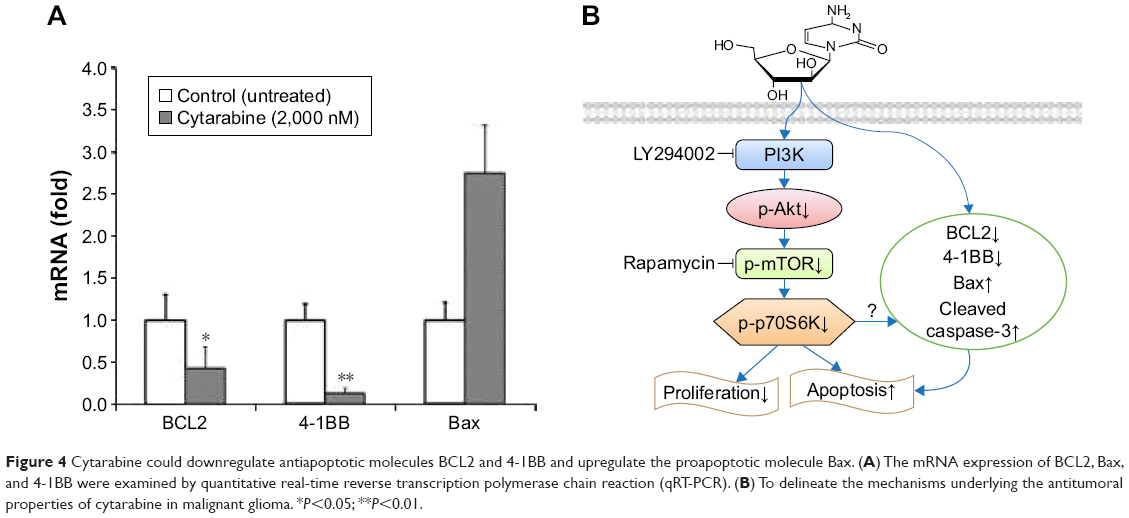 | Figure 4 Cytarabine could downregulate antiapoptotic molecules BCL2 and 4-1BB and upregulate the proapoptotic molecule Bax. (A) The mRNA expression of BCL2, Bax, and 4-1BB were examined by quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR). (B) To delineate the mechanisms underlying the antitumoral properties of cytarabine in malignant glioma. *P<0.05; **P<0.01. |
Cytarabine reduced tumor volume and growth of U87 cell xenografts in vivo
Cytarabine inhibited cell proliferation and induced apoptosis in glioma cells in vitro. Tests were conducted to determine whether cytarabine can inhibit progression of glioma cells in vivo. To test the effects of cytarabine on progression of human glioma cells, NOD-SCID mice inoculated with U87 cells were continuously treated with cytarabine (60 mg/kg/d) in two cycles until termination of the study. A total of 60 mg/kg cytarabine was intraperitoneally and peritumorally injected into mice with U87 tumor xenografts for two cycles; each cycle included a 10-day treatment and 7-day rest. Tumor growth in treated groups was significantly inhibited (P<0.05) compared with controls (Figure 5A–C). No significant differences occurred in tumor volume between intraperitoneal and peritumoral injections and mean body weight between control and treatment groups. However, two mice died after one cycle of treatment in the intraperitoneally injected group, indicating that intraperitoneal treatment resulted in severe adverse effects. Then, IHC staining of Ki-67, p-Akt, and cleaved caspase-3 was performed in xenograft tumor sections. We observed downregulation of the proliferation marker Ki-67 and p-Akt and upregulation of cleaved caspase-3 in cytarabine-treated groups (Figure 5D and E). Collectively, these results suggested that cytarabine inhibited glioma growth in vivo.
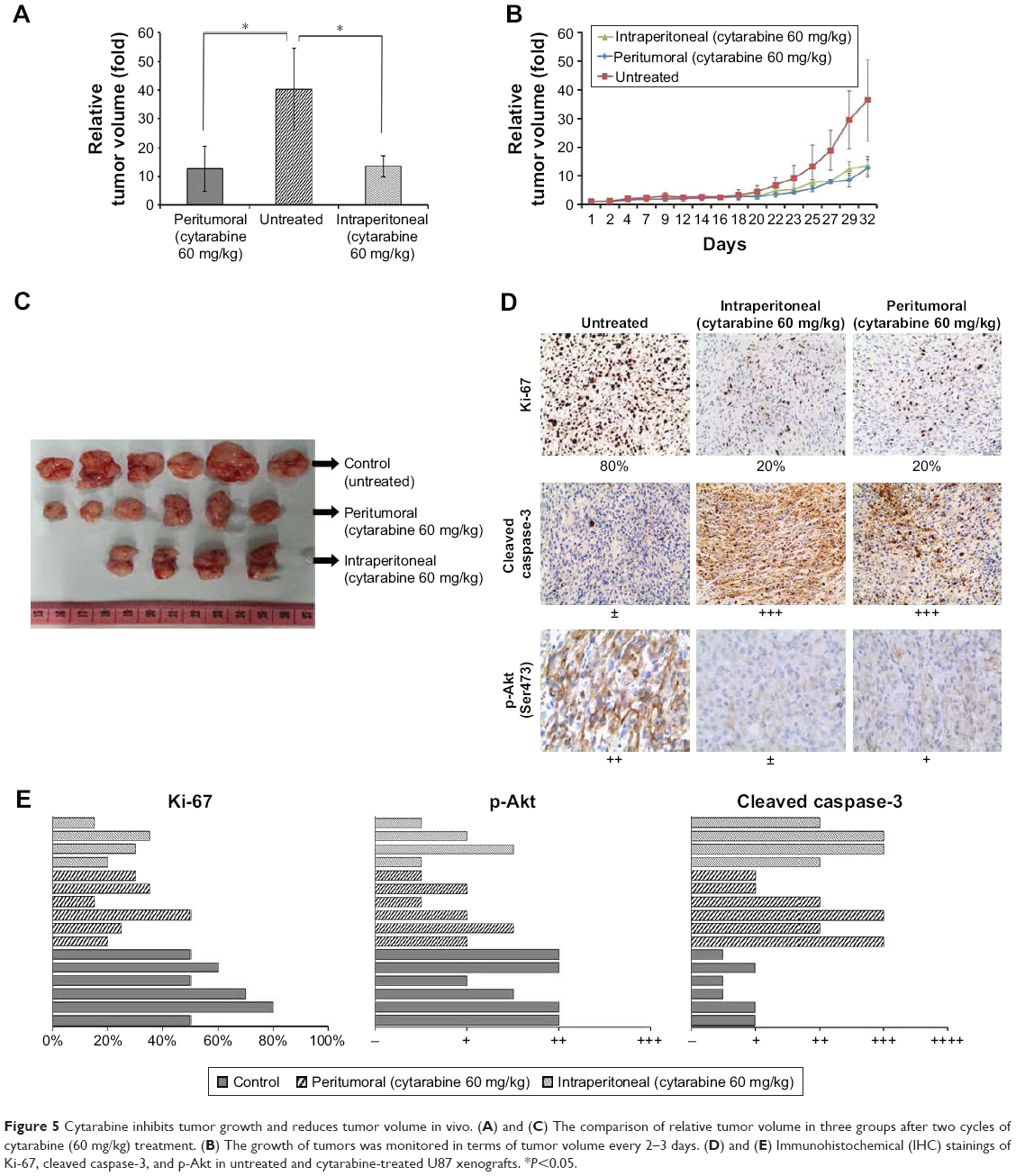 | Figure 5 Cytarabine inhibits tumor growth and reduces tumor volume in vivo. (A) and (C) The comparison of relative tumor volume in three groups after two cycles of cytarabine (60 mg/kg) treatment. (B) The growth of tumors was monitored in terms of tumor volume every 2–3 days. (D) and (E) Immunohistochemical (IHC) stainings of Ki-67, cleaved caspase-3, and p-Akt in untreated and cytarabine-treated U87 xenografts. *P<0.05. |
Discussion
Cytarabine is a deoxycytidine nucleoside analog. This antimetabolite drug incorporates into human DNA and consequently kills cells by interfering with DNA and RNA synthesis.21 This drug is most commonly used for ICV administration and possesses a prominent position in treatment of hematological malignancies (eg, leukemia and lymphomas).22 Liposomal cytarabine is a slow-release formulation of cytarabine. This compound is manufactured through encapsulating aqueous cytarabine in spherical multi-vesicular particles known as DepoFoam (DepoCyte; SkyePharma, San Diego, CA, USA).23 Exposure to cytarabine in the CSF is prolonged in this formulation. Liposomal formulation exerts its effect in CSF for a week in children and almost 2 weeks in adults, whereas conventional cytarabine exerts its effect in CSF for less than 24 h.
Few published reports discussed the role of liposomal cytarabine in LM from malignant glioma. Feasibility and safety of intrathecal liposomal cytarabine administration to neoplastic meningitis were demonstrated by Bomgaars et al24 in a Phase I trial and reported by Peyrl et al25 in central nervous system (CNS) tumors. Glantz et al26 conducted a randomized controlled trial comparing intrathecal DepoCyte to MTX in patients (n=14) with LM in malignant glioma and confirmed that DepoCyte produced a response rate comparable to that of MTX and significantly increased the time before neurological progression while offering the benefit of a less demanding dose schedule. Slavc et al27 reported intrathecal therapy consisting of alternating cycles of liposomal cytarabine and etoposide in 57 patients with various malignant brain tumors and concluded the feasibility of alternating intraventricular liposomal cytarabine and etoposide. In the present study, we reported a clinical case showing evidence of advantage that can be attributed to the use of cytarabine, which is generally well tolerated and results in longer remission. In our clinical case, PFS was 11 months, which was longer than the published survival time of 191 or 24 weeks following LM.5
The present study elucidated cytotoxic antitumoral effects of cytarabine in both in vitro cell research and in vivo mice tests and demonstrated, for the first time, the ability of cytarabine to inhibit growth of the glioma cell line through the PI3K/Akt/mTOR pathway. Akt belongs to a family of serine/threonine protein kinases that participate in cell proliferation and survival, as demonstrated by several downstream cellular targets that it regulates; these cellular targets include Bad, caspase-9, glycogen synthase kinase 3, Forkhead transcription factors, and nuclear factor-kappa B.28 Aberrant activation of Akt is frequently observed in malignant gliomas29 and was reported in approximately 70% of gliomas30,31 and 80% of GBM,30 in correlation with PI3K/Akt signaling being altered in 88% of GBM.32 The Akt pathway was reported to inhibit cell growth, induce radiosensitizing effects on GBM,33 and stimulate apoptosis in a range of mammalian cells.34,35 In our study, cytarabine significantly decreased phosphorylation of Akt in a dose-dependent manner in WB assay and IHC staining. mTOR regulated multiple cell growth and cellular functions by controlling mRNA translation, ribosome biogenesis, autophagy, and metabolism.36 mTOR is activated by Akt. Furthermore, mTOR is required for maintenance of astrocytic character in mice glioma cells, and inhibition of mTOR results in regional apoptosis in these tumors.37 Rapamycin and its synthetic analogs, Torisel and Afinitor, were assessed in clinical trials of recurrent malignant gliomas, proving their modest efficacy.38 mTOR is the primary autophagy repressor.39 mTORC1 is a negative regulator of autophagy and a downstream target of the PI3K/Akt pathway.40 Anticancer agents that target this pathway induce autophagy and inhibit growth. Our research confirmed that cytarabine can repress phosphorylation of mTOR, showing similarity to actions of rapamycin. Activated mTOR phosphorylated p70S6K, which induces GBM formation in mice.30 S6K1 correlated with Ki67 labeling index, indicating that p-p70S6K is correlated with proliferation of tumor cells.9 After WB assay, we confirmed that cytarabine significantly decreased phosphorylation of p70S6K in a dose-dependent manner.
BCL2, Bax, and caspase-3 are apoptosis-related genes. In addition, 4-1BB is a known regulator of cell apoptosis and growth.41 Caspase-3 has been shown to induce apoptosis in microglia and astrocytes of developing brains.42,43 It exists in either an active cleaved caspase form, or an inactive pro-caspase form. Results of our study showed that cytarabine could downregulate expression of BCL2 and 4-1BB, and upregulate Bax and cleaved caspase-3, suggesting that this compound could cause apoptosis of glioma cells.
Results of our in vitro and in vivo experiments suggest a novel mechanism underlying cytarabine antiglioma function through downregulation of the PI3K/Akt/mTOR pathway and antiapoptotic molecules BCL2 and 4-1BB, and upregulation of cleaved caspase-3 and proapoptotic molecule Bax. However, we lack information on how cytarabine regulates expression of BCL-2, Bax, 4-1BB, and caspase-3 through PI3K/Akt/mTOR. Further studies are required to assess the relationship between these factors.
Incorporation of intraventricular chemotherapy into initial treatment of medulloblastoma yielded promising results. In the German HIT-SKK’92 trial, 10-year PFS rate was 82% for patients who had no postoperative residual tumor and 50% for patients with residual tumors.44 These findings were favorable compared with 29%–41% 5-year PFS rates observed in previous studies that excluded CSF-directed chemotherapy.45 Based on encouraging results of CSF-directed chemotherapy at the initial treatment of medulloblastoma, similar benefits may be observed by incorporating CSF-directed therapy into the initial treatment of high-grade glioma for LM prevention and therapy. Our research confirmed that intrathecal cytarabine can be used as standard prophylaxis and treatment for LM.
Conclusion
Inhibition of the PI3K/Akt/mTOR pathway and regulation of apoptosis-related genes such as BCL2, Bax, cleaved caspase-3, and 4-1BB involved cytarabine in glioma. Our research suggests that using intrathecal cytarabine may be a valid strategy to control LM occurrence and progression in malignant gliomas.
Acknowledgments
The authors gratefully acknowledge all the staff in the laboratory at the Beijing Shijitan Hospital for their enthusiastic assistance. The authors thank the enrolled patient and his family for cooperating with follow-up requirements.
Disclosure
The authors report no conflicts of interest in this work.
References
Arita N, Taneda M, Hayakawa T. Leptomeningeal dissemination of malignant gliomas. Incidence, diagnosis and outcome. Acta Neurochir (Wien). 1994;126(2–4):84–92. | ||
Onda K, Tanaka R, Takahashi H, Takeda N, Ikuta F. Cerebral glioblastoma with cerebrospinal fluid dissemination: a clinicopathological study of 14 cases examined by complete autopsy. Neurosurgery. 1989;25(4):533–540. | ||
Erlich SS, Davis RL. Spinal subarachnoid metastasis from primary intracranial glioblastoma multiforme. Cancer. 1978;42(6):2854–2864. | ||
Noh JH, Lee MH, Kim WS, et al. Optimal treatment of leptomeningeal spread in glioblastoma: analysis of risk factors and outcome. Acta Neurochir (Wien). 2015;157(4):569–576. | ||
Arita N, Hayakawa T, Mogami H, Ushio Y. [Meningeal gliomatosis – clinical features and results of treatment]. Gan No Rinsho. 1989;35(11):1308–1312. Japanese [with English abstract]. | ||
Grabb PA, Albright AL, Pang D. Dissemination of supratentorial malignant gliomas via the cerebrospinal fluid in children. Neurosurgery. 1992;30(1):64–71. | ||
Lawton CD, Nagasawa DT, Yang I, Fessler RG, Smith ZA. Leptomeningeal spinal metastases from glioblastoma multiforme: treatment and management of an uncommon manifestation of disease. J Neurosurg Spine. 2012;17(5):438–448. | ||
Lim DA, Cha S, Mayo MC, et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol. 2007;9(4):424–429. | ||
Annovazzi L, Mellai M, Caldera V, et al. mTOR, S6 and AKT expression in relation to proliferation and apoptosis/autophagy in glioma. Anticancer Res. 2009;29(8):3087–3094. | ||
Li XY, Zhang LQ, Zhang XG, et al. Association between AKT/mTOR signalling pathway and malignancy grade of human gliomas. J Neurooncol. 2011;103(3):453–458. | ||
Yoshioka K, Yoshida K, Cui H, et al. Endothelial PI3K-C2alpha, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med. 2012;18(10):1560–1569. | ||
Hu L, Hofmann J, Jaffe RB. Phosphatidylinositol 3-kinase mediates angiogenesis and vascular permeability associated with ovarian carcinoma. Clin Cancer Res. 2005;11(22):8208–8212. | ||
Galetic I, Andjelkovic M, Meier R, Brodbeck D, Park J, Hemmings BA. Mechanism of protein kinase B activation by insulin/insulin-like growth factor-1 revealed by specific inhibitors of phosphoinositide 3-kinase – signifcance for diabetes and cancer. Pharmacol Ther. 1999;82(2–3):409–425. | ||
Chakravarti A, Zhai G, Suzuki Y, et al. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. 2004;22(10):1926–1933. | ||
Benesch M, Siegler N, Hoff KV, et al. Safety and toxicity of intrathecal liposomal cytarabine (depocyte) in children and adolescents with recurrent or refractory brain tumors: a multi-institutional retrospective study. Anticancer Drugs. 2009;20(9):794–799. | ||
Bosnjak M, Ristic B, Arsikin K, et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS One. 2014;9(4):e94374. | ||
Kang MK, Kang SK. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev. 2007;16(5):837–847. | ||
Liu G, Yuan X, Zeng Z, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer. 2006;5:67. | ||
Folkins C, Man S, Xu P, Shaked Y, Hicklin DJ, Kerbel RS. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67(8):3560–3564. | ||
Doetsch F, García-Verdugo JM, Alvarez-Buylla A. Regeneration of a germinal layer in the adult mammalian brain. Proc Natl Acad Sci U S A. 1999;96(20):11619–11624. | ||
Lamba JK. Genetic factors influencing cytarabine therapy. Pharmacogenomics. 2009;10(10):1657–1674. | ||
Galmarini CM, Jordheim L, Dumontet C. Pyrimidine nucleoside analogs in cancer treatment. Expert Rev Anticancer Ther. 2003;3(5):717–728. | ||
Murry DJ, Blaney SM. Clinical pharmacology of encapsulated sustained-release cytarabine. Ann Pharmacother. 2000;34(10):1173–1178. | ||
Bomgaars L, Geyer JR, Franklin J, et al. Phase I of intrathecal liposomal cytarabine in children with neoplastic meningitis. J Clin Oncol. 2004;22(19):3916–3921. | ||
Peyrl A, Sauermann R, Traunmueller F, et al. Pharmacokinetics and safety of intrathecal liposomal cytarabine in children aged <3 years. Clin Pharmacokinet. 2009;48(4):265–271. | ||
Glantz MJ, Jaeckle KA, Chamberlain MC, et al. A randomized controlled trial comparing intrathecal sustained-release cytarabine (DepoCyt) to intrathecal methotrexate in patients with neoplastic meningitis from solid tumors. Clin Cancer Res. 1999;5(11):3394–3402. | ||
Slavc I, Peyrl A, Azizi AA, et al. Long-term intraventricular therapy alternating etoposide and liposomal cytarabine is feasible and safe: experience in 57 children and adolescents with malignant brain tumors. Neuro Oncol. 2016;18 (Suppl 6):vi27–vi28. | ||
Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(20):10983–10985. | ||
Kita D, Yonekawa Y, Weller M, Ohgaki H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007;113(3):295–302. | ||
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25(1):55–57. | ||
Rajasekhar VK, Viale A, Socci ND, Wiedmann M, Hu X, Holland EC. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol Cell. 2003;12(4):889–901. | ||
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. | ||
Koul D, Shen R, Bergh S, et al. Inhibition of Akt survival pathway by a small-molecule inhibitor in human glioblastoma. Mol Cancer Ther. 2006;5(3):637–644. | ||
Ahn JY, Hu Y, Kroll TG, Allard P, Ye K. PIKE-A is amplified in human cancers and prevents apoptosis by up-regulating Akt. Proc Natl Acad Sci U S A. 2004;101(18):6993–6998. | ||
Cheng JQ, Altomare DA, Klein MA, et al. Transforming activity and cell cycle-dependent expression of the AKT2 oncogene: evidence for a link between cell cycle regulation and oncogenesis. Oncogene. 1997;14(23):2793–2801. | ||
Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. | ||
Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7(4):356–368. | ||
Galanis E, Buckner JC, Maurer MJ, et al; North Central Cancer Treatment Group. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. J Clin Oncol. 2005;23(23):5294–5304. | ||
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(22):124–131. | ||
Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129(7):1261–1274. | ||
Cheuk AT, Mufti GJ, Guinn BA. Role of 4-1BB:4-1BB ligand in cancer immunotherapy. Cancer Gene Ther. 2004;11(3):215–226. | ||
Zhang Y, Goodyer C, LeBlanc A. Selective and protracted apoptosis in human primary neurons microinjected with active caspase-3, -6, -7 and -8. J Neurosci. 2000;20(22):8384–8389. | ||
Dalmau I, Vela JM, González B, Finsen B, Castellano B. Dynamics of microglia in the developing rat brain. J Comp Neurol. 2003;458(2):144–157. | ||
Rutkowski S, Bode U, Deinlein F, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med. 2005;352(10):978–986. | ||
Rutkowski S. Current treatment approaches to early childhood medulloblastoma. Expert Rev Neurother. 2006;6(8):1211–1221. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.