")
Back to Journals » Drug Design, Development and Therapy » Volume 10
Anticonvulsant effects of isomeric nonimidazole histamine H3 receptor antagonists
Authors Sadek B, Saad A, Schwed JS, Weizel L, Walter M, Stark H
Received 2 June 2016
Accepted for publication 20 July 2016
Published 7 November 2016 Volume 2016:10 Pages 3633—3651
DOI https://doi.org/10.2147/DDDT.S114147
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Wei Duan
Bassem Sadek,1 Ali Saad,1 Johannes Stephan Schwed,2,3 Lilia Weizel,2 Miriam Walter,2 Holger Stark2,3
1Department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, United Arab Emirates University, Al Ain, United Arab Emirates; 2Biocenter, Institute of Pharmaceutical Chemistry, Goethe University, Frankfurt, Germany; 3Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmaceutical and Medicinal Chemistry, Heinrich Heine University, Düsseldorf, Germany
Abstract: Phenytoin (PHT), valproic acid, and modern antiepileptic drugs (AEDs), eg, remacemide, loreclezole, and safinamide, are only effective within a maximum of 70%–80% of epileptic patients, and in many cases the clinical use of AEDs is restricted by their side effects. Therefore, a continuous need remains to discover innovative chemical entities for the development of active and safer AEDs. Ligands targeting central histamine H3 receptors (H3Rs) for epilepsy might be a promising therapeutic approach. To determine the potential of H3Rs ligands as new AEDs, we recently reported that no anticonvulsant effects were observed for the (S)-2-(4-(3-(piperidin-1-yl)propoxy)benzylamino)propanamide (1). In continuation of our research, we asked whether anticonvulsant differences in activities will be observed for its R-enantiomer, namely, (R)-2-(4-(3-(piperidin-1-yl)propoxy)benzylamino)propaneamide (2) and analogs thereof, in maximum electroshock (MES)-, pentylenetetrazole (PTZ)-, and strychnine (STR)-induced convulsion models in rats having PHT and valproic acid (VPA) as reference AEDs. Unlike the S-enantiomer (1), the results show that animals pretreated intraperitoneally (ip) with the R-enantiomer 2 (10 mg/kg) were moderately protected in MES and STR induced models, whereas proconvulsant effect was observed for the same ligand in PTZ-induced convulsion models. However, animals pretreated with intraperitoneal doses of 5, 10, or 15 mg/kg of structurally bulkier (R)-enantiomer (3), in which 3-piperidinopropan-1-ol in ligand 2 was replaced by (4-(3-(piperidin-1-yl)propoxy)phenyl)methanol, and its (S)-enantiomer (4) significantly and in a dose-dependent manner reduced convulsions or exhibited full protection in MES and PTZ convulsions model, respectively. Interestingly, the protective effects observed for the (R)-enantiomer (3) in MES model were significantly greater than those of the standard H3R inverse agonist/antagonist pitolisant, comparable with those observed for PHT, and reversed when rats were pretreated with the selective H3R agonist R-(α)-methyl-histamine. Comparisons of the observed antagonistic in vitro affinities among the ligands 1–6 revealed profound stereoselectivity at human H3Rs with varying preferences for this receptor subtype. Moreover, the in vivo anticonvulsant effects observed in this study for ligands 1–6 showed stereoselectivity in different convulsion models in male adult rats.
Keywords: histamine, H3 receptor, isomeric antagonists, anticonvulsant activity, stereoselectivity
Introduction
Epilepsy is the propensity to have frequent seizures unprovoked by systemic or acute neurologic insults and it affects millions of people worldwide.1 The recent drug therapy causes adverse side effects including ataxia, attention deficit, and cognitive problems, and lifelong medication is required.2–4 Therefore, pharmacological intervention with antiepileptic drugs (AEDs) represents a major managing strategy for this disease. Cognitive impairment accompanies some types of epilepsy at onset and worsens with chronic, poorly controlled seizures particularly in the developing brain.3 Consequently, appropriate early management of seizures removes the insult on the brain cognitive function, though many AEDs are not so harmless from the same perspective.5 Histamine was discovered to be a neurotransmitter that exerts its biological activities via interaction with four distinct G-protein-coupled histamine receptors (H1R–H4R). H1R, H2R, and H4R are found in the brain and periphery; H3Rs, on the other hand, are abundant in central nervous system (CNS). While activation of H1R and H2R mediates slow excitatory postsynaptic potentials, H3Rs act as autoreceptors that control the synthesis and release of histamine, and as heteroreceptors modulating the release of other neurotransmitters such as acetylcholine, glutamate, gamma-aminobutyric acid (GABA), norepinephrine, serotonin, and dopamine in variable brain regions.6–11 Evidence from experimental and clinical studies supported the notion that histamine plays an important role in the pathogenesis of seizure disorders. For example, drugs that deplete brain histamine levels increased the duration of clonic convulsions induced by maximal electroshock (MES) in mice.12 Furthermore, histidine was found to increase the threshold of seizures induced by pentylenetetrazol (PTZ) in different animal models.7 In addition, H1R antagonist diphenhydramine has been demonstrated to increase focal spikes or spike and wave complexes in patients with grand mal or petit mal epilepsy, suggesting that histamine acts as an endogenous anticonvulsant.13–20 Recently, H3R antagonists/inverse agonists earned a growing interest in the treatment of neuropsychiatric disorders.11,21,22 Supported by positive outcomes in preclinical studies (chemically induced or electrically induced convulsions in rodents), H3R antagonists/inverse agonists paved their way to clinical Phase III with the recent success of pitolisant (PIT) in treatment of epilepsy.23 Dose dependently (20–60 mg), PIT, alone or in combination with other AEDs, showed a favorable EEG profile in human photosensitivity model.24 PIT is one of the first nonimidazole-based H3R antagonists/inverse agonists that, compared to imidazole-based agents such as thioperamide, have lower drug interaction and increased brain penetration.25 Furthermore and in comparison to other AEDs, H3R antagonists/inverse agonists might have a unique feature by their cognition-enhancing potential as indicated by several lines of evidence from preclinical studies.26 Supported by the abovementioned results, we used the multiple-target approach involving the designed structural overlap of H3R pharmacophore with main structural elements of different AEDs on the market. A related approach has previously been realized by linking the known antagonist H3R pharmacophore (3-piperidinopropoxy)aryl to known neuroleptics.27,28 Moreover, recent studies have suggested that this multitargeting approach can successfully be applied to afford dual serotonin transporter/H3R ligands.29,30 Furthermore, we recently reported that no anticonvulsant effects were observed for the (S)-2-(4-(3-(piperidin-1-yl)propoxy)benzylamino)propanamide (1).31 Therefore and in continuation of our research, we asked whether anticonvulsant differences in activities will be observed based on isomeric differences in the present series of compounds. Consequently, a series of H3R ligands (1–6) incorporating aminopropanamide and hydantoin present in lacosamide, safinamide, and phenytoin (PHT), correspondingly, has been synthesized and examined on its in vitro antagonistic binding affinities at human H3Rs (hH3Rs) (Figure 1). Since antagonism of H1Rs has been linked to convulsion, and to support the receptor-subtype selectivity, the in vitro binding affinities of new H3R ligands 1–6 at human H1Rs (hH1Rs) have further been tested. Moreover, the in vitro affinities of 1–6 at hH4Rs have been investigated as hH4Rs were found, among H1Rs–H4Rs, to display the highest receptor homology to hH3R. In addition, the in vivo anticonvulsant effects of the novel series in MES-, PTZ-, and strychnine (STR)-induced convulsion models in Wistar rats were tested.
 | Figure 1 Structures of considered AEDs and of newly designed final compounds 1–6. |
Experimental procedures
Chemistry
General
Educts, reactants, and solvents were commercially obtained from Merck (Whitehouse Station, NJ, USA), Sigma-Aldrich Co. (St Louis, MO, USA), Alfa Aesar (Ward Hill, MA, USA), ABCR (Karlsruhe, Germany), and Acros Organics (Thermo Fisher Scientific, Waltham, MA, USA). Melting points were determined on a Büchi 510 melting point apparatus (Büchi, Flawil, Switzerland) and are uncorrected. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker AMX 300 (300 MHz) spectrometer (BrukerOptik GmbH, Ettlingen, Germany). 1H NMR data are reported in the following order: chemical shift (δ) in ppm downfield from tetramethylsilane as internal reference; multiplicity (br, broad; s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet); approximate coupling constants (J) in Hertz (Hz); and number and assignment of protons (imi, imidazolidine; meth, methyl; pip, piperidine; pyr, pyridine; ph, phenyl; prop, propyl). Carbon nuclear magnetic resonance (13C NMR) spectra were recorded on a Bruker AC 200 (50 MHz) spectrometer (Bruker). Electrospray ionization (ESI)-mass spectrometry (MS) was performed on a Fisons Instruments VG Platform II (Manchester, Great Britain) in positive polarity. Data are listed as mass number [M+H]+ and relative intensity (%). Elemental analyses (C, H, N) were measured on CHN-Rapid (Heraeus, Hanau, Germany) and were within ±0.4% of the theoretical values for all final compounds. Preparative column chromatography was performed on silica gel (particle size, 63–200 μm; Merck). IntelliFlash TM310 (Varian) (Agilent, Böblingen, Germany). For flash chromatography, prepacked columns were used. The adsorbent agent was silica gel of particle size 50 μm (Super Flash, Varian), and column size used was 8–40 g. The mobile phase used was a mixture of dichloromethane (DCM) and ammonia-saturated methanol (MeOH), and detection was carried out using ultraviolet detector (detection wave lengths: λ=256 and 280 nm). Rotation angle α [°] was determined with the Polarimeter 341 (Perkin Elmer, Rodgau, Germany). Specific rotation [α]D20 depends on layer thickness “l” of used cuvette (10 cm), the concentration “c” of the example, the used solvents (water for 1 and 2 and MeOH for 3 and 4), and the measured rotation angle α. Measurement temperature (20°C) and wavelength of the used light were standardized (λ=589 nm). For the calculation of specific rotation, the following formula was used: [α]D20: (α×100)/(c×l).
Hydrogenations were carried out on an autoclave model IV, 500 mL (Carl Roth GmbH & Co. KG, Karlsruhe, Germany). The microwave oven used was a Biotage Initiator 2.0 (Biotage, Sweden). High-pressure amidation was carried out in a high-pressure Haberautoklav Modell IV Typ 50S (Carl Roth GmbH & Co. KG; 2–199 bar).
The precursors containing the 1-(3-phenoxypropyl)piperidine precursors were prepared as described previously.31–34 Briefly, piperidine was alkylated with 3-chloropropan-1-ol under basic conditions (Scheme 1). The resulting alcohol precursor P1 was chlorinated (P2) and subsequently transferred to ether synthesis coupling to phenol derivative to afford precursors P3, P4, and P7. The intermediate precursor aldehyde compound P6 was achieved through Williamson ether synthesis of chloride derivative (P5) and 4-hydroxyaldehyde as described by Sadek et al31 (Scheme 1). Precursor P9 could be synthesized in two different synthetic ways as described previously. The first way involved the oxidation reaction of precursor P7 and Cu(II)Br2, whereas the second way involved the application of Williamson ether synthesis of P2 and 1-(4-hydroxyphenyl)-2-phenylethane-1,2-dion (P8) as previously described.35 Accordingly, the final compounds 1 and 2 were prepared through reaction of P3 with the respective 2-aminopropaneamide, whereas compounds 3 and 4 were achieved through reaction of P6 with corresponding 2-aminopropaneamide. Final product 5 was achieved as previously described, whereas final compound 6 was prepared through reaction of P9 with urea applying hydantoin ring closure reaction.31 All structures and purities of newly synthesized compounds were confirmed by combined analytical techniques (1H NMR, 13C NMR, MS, elemental analysis, and specific rotation, if applicable).
 | Scheme 1 Synthesis of precursors P1–P9 and final H3R ligands 1–6. |
Synthesis of precursors P1–P9
3-(Piperidin-1-yl)propan-1-ol hydrochloride (P1)
Piperidine (23.3 mL, 0.235 mol), 3-chloropropane-1-ol (13.1 mL, 0.157 mol), K2CO3 (32.5 g, 0.235 mol), and KI (26 g, 0.157 mol) were refluxed in absolute acetone (200 mL) for 72 hours. This mixture cooled down to room temperature (RT), inorganic components were removed by filtration, and the filtrate was concentrated to dryness. Purification of the crude orange oil was achieved by means of distillation under reduced pressure (95°C). The oily product was crystallized as hydrochloride from 2-propanol in isopropylic HCl 5–6 N (white solid, 26.5 g, 95%; Scheme 1).31–34
1-(3-Chloropropyl)piperidine hydrochloride (P2)
Alcohol P1 (13.4 g, 0.08 mol) was suspended in toluene (100 mL). Under inert atmosphere and at 0°C, an excess of thionyl chloride (11.6 mL, 0.16 mol) was added dropwise. Once the exothermic reaction had decayed, the mixture was stirred for 3 hours at 60°C. Upon completion of the reaction, thionyl chloride and toluene were distilled off. The crude product was recrystallized from ethanol (beige solid, 14.8 g, quantitative conversion; Scheme 1).31–34
4-(3-(Piperidin-1-yl)propoxy)benzaldehyde (P3)
4-Hydroxybenzaldehyde (1.12 g, 9.18 mmol) was dissolved in 50 mL absolute acetonitrile. K2CO3 (1.4 g, 10.09 mmol) was added at RT. P2 (2 g, 10.09 mmol) was added dropwise and the mixture was refluxed to boiling for 16 hours under inert argon atmosphere. After cooling down, the inorganic components were separated by filtration and the filtrate was dried under reduced pressure. The residue was dissolved in DCM and was extracted with 2 N NaOH. All the combined organic phases were washed with saturated NaCl solution and water. They were dried with Na2SO4, filtrated, and dried under reduced pressure. The product was a yellow-beige solid (2.2 g, 95%; Scheme 1).31–34 Chemical formula: C15H21NO2. 1H NMR (DMSO-d6): δ 9.87 (s, 1H, CHO), 7.85 (d, J=7.5, 2H, ph-2,6H), 7.14 (d, J=7.5, 2H, ph-3,5H), 4.13 (t, J=6.3, 2H, prop-1H2), 2.42–2.30 (m, 6H, pip-2,6H, prop-1H2), 1.95–1.83 (m, 2H, prop-2H2), 1.54–1.45 (m, 4H, pip-3,5H2), 1.43–1.35 (m, 2H, pip-4H2). 13C NMR (DMSO-d6) δ 191.24 (CO), δ 163.65 (ph-4C), δ 131.79 (ph-2,6C), δ 129.49 (ph-1C), δ 114.87 (ph-3,5C), δ 66.52 (prop-1C), δ 54.94 (prop-3C), δ 54.85 (pip-2,6C), δ 26.06 (prop-2C), δ25.54 (pip-3,5C), δ 20.28 (pip-4C). ESI-MS (m/z): 248.1 [M + H]+ (100).
(4-(3-(Piperidin-1-yl)propoxy)phenyl)methanole hydrochloride (P4)
For the synthesis of P4, precursor P2 (2.1 g, 10.5 mmol), 4-hydroxybenzyl alcohol (2.6 g, 21.0 mmol), K2CO3 (8.7 g, 63.0 mmol), and KI (1.7 g, 10.5 mmol) were refluxed in absolute acetone (100 mL) for 2 days. After purification (as described for compound P3), the resulting light solid was crystallized as hydrochloride from 2-propanol (1.5 g, 50%; Scheme 1).31,36,37 Chemical formula: C15H23NO2 × HCl. 1H NMR (DMSO-d6): δ 10.74 (br s, 1H, NH), 7.23 (d, J=8.6, 2H, ph-2,5H), 6.90 (d, J=8.6, 2H, ph-3,5H), 4.43 (s, 2H, CH2–OH), 4.04 (t, J=6.0, 2H, prop-1H2), 3.43 (m, 2H, pip-2,6Heq), 3.16–3.13 (m, 2H, prop-3H2), 2.92–2.76 (m, 2H, pip-2,6Hax), 2.23–2.19 (m, 2H, prop-2H2), 1.85–1.78 (m, 5H, pip-3,5H2, pip-4Heq), 1.45–1.34 (m, 1H, pip-4Hax). 13C NMR (DMSO-d6) δ 157.09 (ph-4C), δ 134.86 (ph-1C), δ 128.17 (ph-2,6C), δ 113.55 (ph-3,5C), δ 65.02 (prop-1C), δ 62.43 (CH2–OH), δ 53.38 (prop-1C), δ 51.65 (pip-2,6C), δ 23.23 (prop-2C), δ 22.15 (pip-3,5C), δ 9.76 (pip-4C). ESI-MS (m/z): 250.0 [M+H]+ (100).
(1-(3-(4-Chlormethyl)phenoxy)propyl)piperidine hydrochloride (P5)
Preparation of P5 was performed as described by Sadek et al31 (Scheme 1). In short, the alcohol precursor P4 (1.62 g, 5.7 mmol) was suspended in 50 mL absolute toluene. Under inert argon atmosphere and at 0°C, an excess of thionyl chloride (0.82 mL, 11.4 mmol) was added dropwise. Once the exothermic reaction decayed, the mixture was refluxed for 3 hours at 60°C. Upon completion of the reaction, toluene and thionyl chloride were distilled off under reduced pressure. The residue was suspended in toluene and was dried for three times. The crude product was recrystallized from ethanol/diethyl ether (white-beige solid, quantitative conversion). Chemical formula: C15H22ClNO × HCl 1H NMR (DMSO-d6): δ 10.44 (br s, 1H, NH+), 7.34 (d, J=8.6, 2H, ph-3,5H), 6.94 (d, J=8.6, 2H, ph-2,6H), 4.71 (s, 2H, CH2-Cl), 4.05 (t, J=6.0, 2H, prop-3H2), 3.49–3.44 (m, 2H, pip-2,6Heq), 3.17–3.10 (m, 2H, prop-1H2), 2.92–2.79 (m, 2H, pip-2,6Hax), 2.21–2.12 (m, 2H, prop-2H2), 1.79–1.66 (m, 5H, pip-3,5H2, pip-4Heq), 1.38–1.34 (m, 1H, pip-4Hax). 13C NMR (DMSO-d6): δ 158.22 (ph-1C), 130.40 (ph-3,5C), 129.93 (ph-4C), 114.51 (ph-3,5C), 65.09 (prop-3C), 53.27 (prop-1C), 51.87 (pip-2,6C), 46.21 (CH2-Cl), 23.14 (prop-2C), 22.24 (pip-3,5C), 21.38 (pip-4C). ESI-MS (m/z): 267.9 [M+H]+ (100).
4-(4-(3-(Piperidin-1-yl)propoxy)benzyloxy)benzaldehyde (P6)
As a general synthesis route, a Williamson ether synthesis with alkylhalogenids was performed as described previously by Sadek et al31 (Scheme 1). Precursor P5 (1.55 g, 5.09 mmol), 4-hydroxybenzaldehyde (566 mg, 4.63 mmol), and K2CO3 (960 mg, 6.95 mmol) were dissolved in 40 mL DMF and the mixture refluxed for 3 days at 100°C. The crude product was purified by column chromatography (eluent: DCM: MeOH + NH3, 99:1 to 98:2) with a yield of 77% of theoretical (1.3 g). Chemical formula: C22H27NO3. 1H NMR (DMSO-d6): δ 9.88 (s, 1H, CHO), 7.86 (d, J=7.5, 2H, ph′-2,6H), 7.38 (d, J=7.5, 2H, ph′-3,5H), 7.22 (d, J=7.5, 2H, ph-2,6H), 6.97 (d, J=7.5, 2H, ph-3,5H), 5.15 (s, 2H, meth-CH2), 4.00 (t, J=6.25, 2H, prop-1H2), 2.42–2.32 (m, 6H, pip-2,6H, prop-3H2), 1.86 (m, 2H, prop-2H2), 1.51–1.48 (m, 4H, pip-3,5H2), 1.40 (m, 2H, pip-4H). 13C NMR (DMSO-d6): δ 191.22 (CO), 163.36 (ph′-4C), 158.58 (ph-4C), 131.73 (ph′-2,6C), 131.66 (ph′-1C), 129.66 (ph-2,6C), 128.05 (ph-1C), 115.27 (ph′-3,5C), 114.42 (ph-3,5C), 69.48 (prop-1C), 65.96 (meth-C), 55.08 (prop-3C), 54.07 (pip-2,6C), 26.25 (prop-2C), 25.55 (pip-3,5C), 24.09 (pip-4C). ESI-MS (m/z): 354.1 [M+H]+ (100).
2-Phenyl-1-(4-(3-(piperidin-1-yl)propoxy)phenyl)ethanone (P7)
As a general synthesis route, a Williamson ether synthesis using alkylhalogenids was performed as described also by Sadek et al31 (Scheme 1). Precursor P2 (513 mg, 2.59 mmol), benzyl-4-hydroxyphenylketone (500 mg, 2.36 mmol), and K2CO3 (326 mg, 2.36 mmol) were dissolved in 20 mL abs acetone. The yield is 773 mg (97% of theoretical). Chemical formula: C22H27NO2 1H NMR (DMSO-d6): δ 8.05 (d, J=10.0, 2H, ph-2,6H), 7.33 (m, 5H, ph′-2,3,4,5,6H), 7.11 (d, J=10.0, 2H, ph-3,5H), 4.36 (s, 2H, eth-2H2), 4.15 (t, J=6.25, 2H, prop-1H2), 2.46–2.36 (m, 6H, pip-2,6H, prop-3H2), 1.98–1.87 (m, 2H, prop-2H2), 1.59–1.50 (m, 4H, pip-3,5H2), 1.47–1.40 (m, 2H, pip-4H2). 13C NMR (DMSO-d6): δ 196.19 (CO), 162.67 (ph-4C), 135.32 (ph′-1C), 130.79 (ph-3,5C), 129.56 (ph′-2,6C), 129.04 (ph′-4C), 128.33 (ph′-3,5C), 126.48 (ph-1C), 114.14 (ph-2,6C), 66.63 (prop-1C), 54.91 (prop-3C), 54.09 (pip-2,6C), 44.42 (eth-2C), 26.32 (prop-2C), 25.59 (pip-3,5C), 23.86 (pip-4C). ESI-MS (m/z): 338.0 [M+H]+ (100).
1-(4-Hydroxyphenyl)-2-phenylethane-1,2-dion (P8)
Benzyl-4-hydroxyphenylketone (1.66 g, 7.82 mmol) and Cu(II)Br2 (3.49 g, 15.64 mmol) were suspended in a mixture of DMSO and ethylacetate 1:1 (16 mL total) and refluxed with stirring under inert argon gas atmosphere for 16 hours at 60°C according to previously described procedures (Scheme 1).35 After cooling down to RT, ice water and DCM were added to the preparation. The product was extracted by using only the organic phase. The organic phase was washed several times with water until the green color of the water phase disappeared. Afterward, the organic phase was filtrated and the solvent was evaporated. Further purification was performed by column chromatography (eluent: DCM: MeOH + NH3, 95:5) and a yellowish oil was obtained as product (yield 1.01 g, 57% of theoretical). Chemical formula: C14H10O3. 1H NMR (DMSO-d6): δ 10.96 (s, 1H, OH), 7.88 (d, J=7.5, 2H, ph-2,6H), 7.82–7.75 (m, 3H, ph′-2,4,6H), 7.62 (t, J=8.75, 2H, ph′-3,5H), 6.96 (d, J=7.5, 2H, ph-3,5H). 13C NMR (DMSO-d6): δ 195.33 (eth-1C), 193.03 (eth-2C), 164.19 (ph-4C), 135.19 (ph′-4C), 132.60 (ph′-1C), 132.39 (ph-2,6C), 129.39 (ph′-2,3,5,6C), 123.86 (ph-1C), 116.22 (ph-3,5C). ESI-MS (m/z): 225.09 [M−H]+ (100) Measurement was performed in negative mode.
1-Phenyl-2-(4-(3-(piperidin-1-yl)propoxy)phenyl)ethane-1,2-dione (P9)
Precursor P9 could be synthesized in two different synthetic ways (Scheme 1). The first way involved the precursor P7 (770 mg, 2.28 mmol) and Cu(II)Br2 (1.02 g, 4.56 mmol), which were suspended in a mixture of DMSO and absolute ethylacetate 1:1 (6 mL total) and were refluxed with stirring under argon atmosphere for 2 days at 60°C.35 After cooling down, ice water and DCM were added to the preparation. The crude product was extracted within the organic phase. The organic phase was washed several times with water until the green color vanished. The organic phase was filtrated and the solvent was evaporated. Further purification was performed by column chromatography (eluent: DCM: MeOH + NH3, 95:5). The product (P9) was obtained as yellow-orange oil (yield 296 mg, 37% of theoretical). The second way involved the application of Williamson ether synthesis as previously described. Precursor P2 (723 mg, 3.65 mmol), precursor P8 (750 mg, 3.32 mmol), and K2CO3 (458 mg, 3.32 mmol) were suspended in 30 mL absolute acetone and refluxed for 3 days at 60°C. Purification was performed by column chromatography (eluent: DCM: MeOH + NH3, 98:2 to 95:5). The yield was 677 mg (58% of theoretical). Chemical formula: C22H25NO3. 1H NMR (DMSO-d6): δ 7.90–7.87 (m, 4H, ph-2,6H, ph′-2,6H), 7.79 (t, J=7.5, 1H, ph′-4H), 7.62 (t, J=9.0, 2H, ph′-3,5H), 7.15 (d, J=9.0, 2H, ph-3,5H), 4.15 (t, J=6.0, 2H, prop-1H2), 2.85–2.62 (m, 6H, pip-2,6H, prop-3H2), 2.01 (m, 2H, prop-2H2), 1.59 (m, 4H, pip-3,5H2), 1.45 (m, 2H, pip-4H2). 13C NMR (DMSO-d6): δ 195.06 (eth-1C), 193.19 (eth-2C), 164.03 (ph-4C), 135.37 (ph′-4C), 132.39 (ph′-1C), 132.14 (ph-2,6C), 129.46 (ph′-3,5C), 129.44 (ph′-2,6C), 125.19 (ph-1C), 115.29 (ph-3,5C), 65.97 (prop-1C), 53.80 (prop-3C), 52.89 (pip-2,6C), 24.29 (prop-2C), 23.69 (pip-3,5C), 22.27 (pip-4C). ESI-MS (m/z): 352.3 [M+H]+ (100).
(S)-2-(4-(3-(Piperidin-1-yl)propoxy)benzylamino)propaneamide dihydrogenoxalate (1)
Preparation of final product 1 was achieved by suspension of aldehyde P3 (1 g, 4.04 mmol) and commercially available (S)-2-aminopropaneamide hydrochloride (530 mg, 4.26 mmol) in 30 mL of absolute MeOH, and stirring the mixture for 16 hours under inert argon atmosphere at RT. Subsequently, NaBH4 (242 mg, 6.38 mmol) was added at 0°C, and the mixture was stirred for 10–15 minutes at RT. Once reaction saturation was achieved, a solution of NaHCO3 was added to quench the reaction mixture. Then, DCM was used to extract the organic product. All combined organic phases were washed with saturated NaCl solution and water, dried with Na2SO4, filtrated, and finally dried under reduced pressure. The crude product was purified with flash chromatography (DCM: MeOH + NH3, 95:5). The product (1) was precipitated with oxalic acid and recrystallized in a solvent mixture of absolute ethanol and acetonitrile (Scheme 1). White solid in 51% yield, melting point (MP): 155°C, [α]D20: +2.52. 1H NMR (DMSO-d6): δ 7.28 (s, 1H, NH2), 7.18 (d, J=7.5, 2H, ph-2,6H), 6.94 (s, 1H, NH2), 6.85 (d, J=7.5, 2H, ph-3,5H), 3.94 (t, J=6.25, 2H, prop-1H2), 3.60–3.41 (q, J=15.8, 2H, meth-H2), 3.01–2.93 (q, J=7.5, 1H, prop′-2H), 2.37–2.28 (m, 6H, pip-2,6H, prop-1H2), 2.21 (s, 1H, NH), 1.87–1.76 (m, 2H, prop-2H2), 1.51–1.43 (m, 4H, pip-3,5H2), 1.37 (m, 2H, pip-4H2), 1.11 (d, J=7.5, 3H, CH3). 13C NMR (DMSO-d6) δ 171.30 (prop′-1C), δ 164.28 (ox-C), d 158.46 (ph-4C), δ 131.22 (ph-2,6C), δ 124.91 (ph-1C), δ 114.47 (ph-3,5C), δ 65.09 (prop-1C), δ 54.33 (prop′-2C), δ 53.34 (prop-3C), δ 52.10 (pip-2,6C), δ 48.15 (meth-C), δ 23.44 (prop-2C), δ 22.57 (pip-3,5C), δ 21.45 (pip-4C), δ 16.27 (CH3). ESI-MS (m/z): 320.1 [M+H]+ (100). Elemental analysis: calculated for C18H29N3O2 ×2 (COOH)2; calculated: C =52.90, H =6.66, N =8.41; found: C =52.63, H =6.59, N =8.35.
(R)-2-(4-(3-(Piperidin-1-yl)propoxy)benzylamino)propaneamide dihydrogenoxalate (2)
Preparation of final product 2 was as described for 1, with the only modification being the way in which (R)-aminopropaneamide hydrochloride was used (Scheme 1). The crude product was purified with flash chromatography (DCM: MeOH + NH3, 95:5). The product 2 was precipitated with oxalic acid and recrystallized in a solvent mixture of absolute ethanol and acetonitrile. White solid in 52% yield, MP: 131°C, [α]D20: −2.61. Chemical formula: C18H29N3O2 ×2 (COOH)2. 1H NMR (DMSO-d6; 300 Hz): δ 7.29 (s, 1H, NH2), 7.19 (d, J=9.0, 2H, ph-2,6H), 6.95 (s, 1H, NH2), 6.85 (d, J=9.0, 2H, ph-3,5H), 3.95 (t, J=7.5, 2H, prop-1H2), 3.61–3.43 (q, J=18.0, 2H, meth-H2), 3.01–2.94 (q, J=6.9, 1H, prop′-2H), 2.38–2.29 (m, 6H, pip-2,6H, prop-1H2), 1.87–1.78 (m, 2H, prop-2H2), 1.51–1.44 (m, 4H, pip-3,5H2), 1.40–1.34 (m, 2H, pip-4H2), 1.11 (d, J=6.0, 3H, CH3). 13C NMR (DMSO-d6): δ 171.23 (prop′-1C), 164.52 (ox-C), 158.47 (ph-4C), 131.29 (ph-2,6C), 124.79 (ph-1C), 114.45 (ph-3,5C), 65.06 (prop-1C), 54.27 (prop′-2C), 53.34 (prop-3C), 52.06 (pip-2,6C), 48.08 (meth-C), 23.40 (prop-2C), 22.56 (pip-3,5C), 21.45 (pip-4C), 16.25 (CH3). ESI-MS (m/z): 320.1 [M+H]+ (100). Elemental analysis calculated for C18H29N3O2 ×2 (COOH)2; calculated: C =52.90; H =6.66; N =8.41; found: C =52.66; H =6.71; N =8.41.
(R)-2-(4-(4-(3-(Piperidin-1-yl)propoxy)benzyloxy)benzylamino)propanamide dihydrogenoxalate (3)
Preparation of final product 3 was achieved by suspension of aldehyde P6 (700 mg, 1.98 mmol) and commercially available (R)-2-aminopropaneamide hydrochloride (260 mg, 2.085 mmol) in 20 mL of absolute MeOH, and stirring the mixture for 16 hours under inert argon atmosphere at RT. Afterward, NaBH4 (118 mg, 3.13 mmol) was added at 0°C and the mixture was stirred for 10–15 minutes at RT. Once a reaction stop was achieved, a saturated NaHCO3 solution was added to quench the reaction mixture. DCM was used to extract the organic product. All combined organic phases were washed with saturated NaCl solution and water, dried with Na2SO4, filtrated, and finally dried under reduced pressure. The crude product was purified with flash-chromatography (eluent: DCM: MeOH + NH3, 95:5). The final product 3 was precipitated with oxalic acid and recrystallized in a solvent mixture of absolute ethanol and acetonitrile (yellow solid, yield 992 mg, 77% of theoretical; Scheme 1). Chemical formula: C25H35N3O3 ×2.5 (COOH)2, MP: 93°C, [α]D20: +3.44. NMR analytics refer to the free base! 1H NMR (DMSO-d6): δ 7.39 (d, J=7.5, 2H, ph′-2,6H), 7.36 (s, 1H, NH2), 7.26 (d, J=7.5, 2H, ph-2,6H), 7.02 (s, 1H, NH2), 7.00 (d, J=10.0, 4H, ph-3,5H, ph′-3,5H), 5.05 (s, 2H, methoxy-CH2), 4.05 (t, J=6.25, 2H, prop-1H2), 3.68–3.49 (q, J=15.0, 2H, meth-H2), 3.08–3.00 (q, J=6.7, 1H, prop′-2H), 2.45–2.32 (m, 6H, pip-2,6H, prop-1H2), 1.96–1.85 (m, 2H, prop-2H2), 1.59–1.52 (m, 4H, pip-3,5H2), 1.45 (m, 2H, pip-4H2), 1.19 (d, J=7.5, 3H, CH3). 13C NMR (DMSO-d6): δ 158.30 (prop′-1C), 157.17 (ph-4C), 132.61 (ph′-4C), 129.30 (ph′-2,6C), 128.98 (ph-2,6C), 128.95 (ph-1C, ph′-1C), 114.42 (ph-3,5C), 114.29 (ph′-3,5C), 68.91 (prop-1C), 65.92 (methoxy-C), 56.35 (prop′-2C), 55.09 (prop-3C), 54.07 (pip-2,6C), 50.44 (meth-C), 26.28 (prop-2C), 25.57 (pip-3,5C), 24.11 (pip-4C), 19.29 (CH3). ESI MS (m/z): 426.4 [M+H]+ (100). Elemental analysis calculated: C =55.38; H =6.20; N =6.46; found: C =55.68; H =6.41; N =6.50.
(S)-2-(4-(4-(3-(Piperidin-1-yl)propoxy)benzyloxy)benzylamino)propanamide dihydrogenoxalate (4)
Preparation of final product 4 was achieved by suspension of aldehyde P6 (530 mg, 1.499 mmol) and commercially available (S)-2-aminopropaneamide hydrochloride (197 mg, 1.578 mmol) in 15 mL of absolute MeOH, and stirring the mixture for 16 hours under inert argon atmosphere at RT. Afterward, NaBH4 (90 mg, 2.368 mmol) was added at 0°C and the mixture was stirred for 10–15 minutes at RT. Once a reaction stop was achieved, a saturated NaHCO3 solution was added to quench the reaction mixture. DCM was used to extract the organic product. All combined organic phases were washed with saturated NaCl solution and water, dried with Na2SO4, filtrated, and finally dried under reduced pressure. The crude product was purified with flash chromatography (DCM: MeOH + NH3, 95:5). The final product 4 was precipitated with oxalic acid and recrystallized in a solvent mixture of absolute ethanol and acetonitrile (yellow solid, yield 946 mg, 97% of theoretical; Scheme 1). Chemical formula: C25H35N3O3 ×2.5 (COOH)2, MP: 101°C, [α]D20: −3.61. NMR analytics refer to the free base! 1H NMR (DMSO-d6): δ 7.91 (s, 1H, NH2), 7.54 (s, 1H, NH2), 7.37 (d, J=7.5, 4H, ph′, ph-2,6H), 7.05 (d, J=7.5, 2H, ph-3,5H), 6.98 (d, J=10.0, 2H, ph′-3,5H), 5.05 (s, 2H, methoxy-CH2), 4.04 (t, J=6.25, 2H, prop-1H2), 3.98 (s, 2H, meth-H2), 3.73–3.64 (q, J=7.5, 1H, prop′-2H), 3.14 (m, 6H, pip-2,6H, prop-1H2), 2.16–2.10 (m, 2H, prop-2H2), 1.75 (m, 4H, pip-3,5H2), 1.55 (m, 2H, pip-4H2), 1.38 (d, J=10.0, 3H, CH3). 13C NMR (DMSO-d6): δ 164.17 (ox-C), 158.56 (prop′-1C), 157.98 (ph-4C), 131-15 (ph′-4C), 129.41 (ph, ph′-2,6C), 129.04 (ph-1C, ph′-1C), 114.81 (ph-3,5C), 114.38 (ph′-3,5C), 70.94 (prop-1C), 68.92 (methoxy-C), 54.35 (prop′-2C), 53.42 (prop-3C), 52.13 (pip-2,6C), 48.14 (meth-C), 23.51 (prop-2C), 22.62 (pip-3,5C), 21.45 (pip-4C), 16.28 (CH3). ESI-MS (m/z): 426.6 [M+H]+ (100). Elemental analysis calculated: C =55.38; H =6.20; N =6.46; found: C =55.70; H =6.53; N =6.61.
5,5-Diphenyl-3-(3-(piperidin-1-yl)propyl)imidazolidin-2,4-dione (5)
P2 (450 mg, 2.265 mmol), PHT (800 mg, 3.17 mmol), and K2CO3 (940 mg, 6.8 mmol) were suspended in 50 mL absolute acetone as previously described (Scheme 1).31 This mixture was refluxed under inert argon atmosphere for 48 hours. After cooling to RT, inorganic components were removed by filtration and the residue concentrated under reduced pressure. Further purification of final product 5 was performed with flash chromatography (DCM: MeOH + NH3, 95:5). The colorless oil was precipitated with water. White solid in 28% yield, MP: 164°C. 1H NMR (DMSO-d6): δ 9.66 (s, 1H, NH), 7.43 (m, 10H, ph-2,3,4,5,6H, ph′-2,3,4,5,6H), 3.51 (t, J=7.5, 2H, prop-1H2), 2.28–2.19 (m, 6H, pip-2,6H, prop-3H2), 1.78–1.66 (m, 2H, prop-2H2), 1.49–1.46 (m, 4H, pip-3,5H2), 1.39 (m, 2H, pip-4H2). 13C NMR (DMSO-d6, δ): δ 173.11 (imi-4CO), δ 155.33 (imi-2CO), δ 139.65 (ph-1C, ph′-1C), δ 128.47 (ph-3,5C, ph′-3,5C), δ 128.06 (ph-4C, ph′-4C), δ 126.55 (ph-2,6C, ph′-2,6C), δ 68.90 (imi-5C), δ 55.56 (prop-3C), δ 53.88 (pip-2,6C), δ 36.58 (prop-1C), δ 25.45 (pip-3,5C), δ 24.89 (prop-2C), δ 24.04 (pip-4C). ESI-MS (m/z): 378.4 [M+H]+ (100). Elemental analysis calculated for C23H27N3O2; calculated: C =73.18, H =7.21, N =11.13, found: C =72.94, H =7.27, N =10.98.
5-Phenyl-5-(4-(3-(piperidin-1-yl)propoxy)phenyl)imidazolidine-2,4-dione (6)
Synthesis of product 6 was performed by using a hydantoin ring closure reaction (Scheme 1). Precursor molecule P9 (400 mg, 1.14 mmol), urea (137 mg, 2.28 mmol), 1 mL NaOH 30%, and as solvent 1 mL EtOH were dissolved in a microwave reaction vial. Microwave conditions were 10 minutes at 120°C. After the reaction finished, the mixture was diluted with water and side products were separated by filtration. Approximately 5 N HCL was added to the filtrate until the crude product precipitated. A flash chromatography was performed afterward (eluent: DCM: MeOH + NH3, 95:5). A yellowish-white solid product was obtained with a 40% yield, MP: 200°C, 1H NMR (DMSO-d6): δ 11.01 (s, 1H, imi-3NH), 9.22 (s, 1H, imi-1NH), 7.35 (m, 5H, ph′-2,3,4,5,6H), 7.20 (d, J=7.5, 2H, ph-2,6H), 6.94 (d, J=7.5, 2H, ph-3,5H), 4.00 (t, J=6.25, 2H, prop-1H2) 2.39–2.32 (m, 6H, pip-2,6H, prop-3H2), 1.88–1.78 (m, 2H, prop-2H2), 1.49 (m, 4H, pip-3,5H2), 1.37 (m, 2H, pip-4H2). 13C NMR (DMSO-d6): δ 175.06 (imi-4CO), 158.23 (ph-4C), 155.92 (imi-2CO), 140.12 (ph′-1C), 131.74 (ph-1C), 128.39 (ph′-3,5C), 127.89 (ph′-4C), 127.81 (ph-2,6C), 126.51 (ph′-2,6C), 114.25 (ph-3,5C), 69.75 (imi-5C), 65.90 (prop-1C), 55.01 (prop-3C), 54.00 (pip-2,6C), 26.11 (prop-2C), 25.45 (pip-3,5C), 24.01 (pip-4C). ESI-MS (m/z): 394.6 [M+H]+ (100). Elemental analysis calculated: C =69.41; H =6.96; N =10.56; found C =69.36; H =6.86; N =10.51.
In vitro pharmacology
Antagonist binding to human histamine H3 receptor (hH3R)
As described in previous publications,31,37–41 the antagonist affinity for human histamine H3Rs was tested utilizing radioligand binding assays and HEK-293 cell membrane preparations. The competition binding experiments were conducted on membranes (20–25 μg/well in a final volume of 0.2 mL binding buffer), which were incubated with [3H]Nα-MeHA (2 nM; 85 Ci/mmol) and a variety of concentrations range of the respective test ligand. For each compound, assays were conducted at least in 3–5 experiments in triplicates with seven appropriate concentrations in the range of 0.01 nM–10 μM of the respective test compound, and the produced nonspecific binding was measured in the presence of unlabelled PIT (10 μM). Accordingly, the maximal binding concentration (Bmax) was determined to be 0.89 pmol/mg and the Kd value of [3H]Nα-MeHA resulted to be 2.98 nmol/L (pKd =8.53±0.01) in saturation binding experiments. Moreover, filtration through GF/B filters pretreated with 0.3% m/v polyethyleneimine using an Inotech cell harvester was utilized to separate the bound radioligand from free radioligand. Accordingly, unbound radioligand was removed from the mixture by three washing cycles with 0.3 mL/well of ice-cold 50 mM Tris HCl buffer, pH 7.4, containing 120 mM NaCl. The filters were saturated in a sample bag with 9 mL scintillator liquid (Betaplate Scint, PerkinElmer Inc., Waltham, MA, USA) and counted using a PerkinElmer MicroBeta Trilux scintillation counter. Consequently, the analysis of competition binding data was conducted by the software GraphPad Prism 3.02 (GraphPad Software, Inc., La Jolla, CA, USA) using nonlinear least squares fit. Ki values were calculated from the half maximal inhibitory concentration (IC50) values according to the Cheng–Prusoff equation.42
Antagonist binding to human histamine H1 receptor (hH1R)
As described previously, binding assays conducted on CHO-K1 cells stably expressing the hH1R were used to determine the antagonist affinity of the respective test compound for hH1Rs.31,37–41 The experimental assays were conducted in triplicates with at least four appropriate concentrations in the range of 100 nM–100 μM of the respective test compound. In addition, the resulting nonspecific binding was measured in the presence of the standard H1R antagonist chlorpheniramine hydrogenmaleate at a concentration of 10 mM. Consequently, the concentrations of bound and free radioligand were separated in similar way described earlier. In addition, the unbound radioligand was removed with four washes of 5 mL of ice-cold HEPES buffer. Liquid scintillation counting using a PerkinElmer MicroBeta Trilux scintillation counter was used to determine the amount of radioactivity collected on the filter used in the current experiment. Accordingly, competition binding data were analyzed using the software GraphPad Prism 3.02 (GraphPad Software, Inc.) using nonlinear least squares fit, and Ki values were calculated from the IC50 values according to the Cheng–Prusoff equation.31,43–45
Antagonist affinity to human histamine H4 receptor (hH4R)
As described previously, binding assay experiments were conducted to determine the antagonist affinity of the respective test compound to hH4Rs.31,37–41 Briefly, the competition binding experiments were run on incubating membranes, 35 μg/well (prepared from Sf9 cells expressing hH4R, coexpressed with G protein Gγi2 and Gβ1γ2 subunits) in a final volume of 0.2 mL containing binding buffer and [3H]histamine (10 nM, 15.3 Ci/mmol) in a 96-well microtiterplate. Assays were conducted in triplicates with seven appropriate concentrations in the range of 0.1 nM–100 μM of the respective test compound. Nonspecific binding, bound radioligand, and free radioligand were determined as described previously, and competition binding data were analyzed using the software GraphPad Prism 3.02 (GraphPad Software, Inc.) using nonlinear least squares fit. Ki values were calculated from the IC50 values according to the Cheng–Prusoff equation.31,43–45
In vivo pharmacology
General remarks
Animals
Bred male Wistar rats (Central Animal Facility of the UAE University) of body weight 180–200 g were used for this study. All animals were kept in an air-conditioned animal facility room with controlled temperature (24°C±2°C) and humidity (55%±15%) under a 12-hour light/dark cycle. The animals were allowed free access to food and water. The experiments of this study were carried out between 900 and 1200 hours, and all procedures were performed according to the guidelines of the European Communities Council Directive of November 24, 1986 (86/609/EEC) and was approved for the epilepsy study by the College of Medicine and Health Sciences/United Arab Emirates University (Institutional Animal Ethics Committee, approval number: A9-14). All efforts were made to minimize animal suffering and to reduce the number of animals used. In addition, all animal procedures with regard to convulsion studies were carried out by the same experimenter and in a blind manner.
Drugs
PHT, valproic acid (VPA), R-(α)-methyl-histamine (RAMH), PTZ, and STR were purchased from Sigma-Aldrich Co. The H3R standard antagonist/inverse agonist PIT was synthesized by the Department of Technology and Biotechnology of Drugs (Kraków, Poland) as described previously (Figure 1).46 PHT, pyrilamine, and the H3R standard ligand PIT, as well as test compounds 1–6 were dissolved in isotonic saline solution and administered intraperitoneally (IP) at a volume of 1 mL/kg. Doses of test compounds (5, 10, and 15 mg/kg) were expressed in terms of the free base.
MES-induced seizure
As previously described,31,37–41 the convulsions were induced in rats with a 50 Hz alternating current of 120 mA intensity. The current was applied through ear electrodes for 1 second. Protection against the spread of MES-induced convulsion was defined as the abolition of the tonic hind limb extension (THLE) component of the seizure.31,38,41,47 Animals were divided into nine treatment groups (eight rats each) as the following: group 1: saline, group 2: PHT, group 3: PIT, and groups 4–9: test compounds 1–6 at 10 mg/kg, IP. All compounds were injected 30 minutes prior to the test. For the most promising compound, further doses (5 and 15 mg/kg, IP) were investigated in additional animal groups. To explore the mechanism of how most promising compounds influences MES-induced convulsions, we have selected the most effective dose of 3 for further analysis. In one group of animals of eight rats, the selected dose of 3 coinjected together with RAMH 10 mg/kg 5 minutes apart and as described previously.31,38,39,41,48–50 All doses were selected according to previously described methods.31,38,41,47 For the reference drug PHT, the dose (10 mg/kg, IP) that provided a full protection in the MES model in rats without mortality was used.
Chemically induced seizures
In our study and according to previously used protocols, two chemical agents have been used to induce convulsions: PTZ and STR. Initially, different doses of these agents have been tested (in six to eight rats per dose) to determine the minimum dose that cause convulsions in all treated animals.51–55 This minimum dose was then used to screen potential AEDs. PTZ 60 mg/kg or STR 3.5 mg/kg was administered IP to all groups (six to eight rats per group), ie, saline and treated rats. Vehicle or tested compounds (VPA 100 or 300 mg/kg in PTZ and STR, respectively,39 PIT 10 mg/kg, or test compounds 1–6 at 10 mg/kg, all IP) were administered 30 minutes before PTZ or STR injection. All doses of test compounds were selected according to previously described methods.51–55 For the reference drug VPA, the dose (100 or 300 mg/kg, IP) that provided a full protection in the respective seizure model in rats without mortality was used. Immediately, animals were observed for 30 minutes (experiment period) for any convulsion signs, and graded scores have been used to evaluate the severity of seizures as following: score 0= no seizures, score 1= eye or facial twitches, score 2= convulsive waves across the body, score 3= myo-clonic jerks or rearing, score 4= turn over onto one side position, and score 5= turn over onto back position, generalized tonic–clonic seizures, or die during the experiment period. For the most promising compound, further doses (5 and 15 mg/kg, IP) were investigated in additional animal groups. The most effective dose of promising test compound 4 was selected for further analysis. In one group of animals of eight rats, the selected dose of 4 was coinjected together with RAMH 10 mg/kg 5 minutes apart.
Statistics
For statistical comparisons, the software package SPSS 20.0 (IBM Corporation, Armonk, NY, USA) was used. All in vivo data were expressed as the mean ± SEM. Data were analyzed by one-way analysis of variance, followed by the Bonferroni post hoc test for multiple comparisons. The criterion for statistical significance was set at a P-value of less than 0.05.
Results
Pharmacology
In vitro antagonist affinities at hH1R, hH3R, and hH4Rs
The new compounds were first tested for their H3R activity obtained by [3H]Nα-MeHA binding assay on HEK-293 cell membrane preparation stably expressing hH3R. To expand their pharmacological profile, their binding affinities in competition-binding experiments with seven-point measurements in at least duplicates (n≥2) were also tested. Displacement assays were carried out using membrane suspension of cell lines stably expressing the hH1R (CHO) with [3H]pyrilamine. Moreover, binding assay on Sf9 cells transiently expressing hH4R was used to examine their affinities at hH4R (Table 1).
 | Table 1 In vitro affinities at hH1Rs, hH2Rs, and hH4Rs and in vivo anticonvulsant effects of final compounds 1–6 |
In vivo seizure models
The current used in the MES test and the dose of PTZ and STR used in the study produced convulsions in 100% of animals without mortality.
Protective effect of H3R ligands 1–6 on MES-induced convulsion
The protective effects of acute systemic administration of 1–6 against MES-induced seizures in rats are shown in Figure 2. One-way analysis of variance showed that treatment with the reference drug PHT, standard H3R antagonist PIT, and novel H3R ligands 1–6 (10 mg/kg, IP) exhibited a significant protection against MES-induced seizures (F(8,54) =57.39; P<0.001). Importantly and amid the compounds tested, H3R ligand 3 significantly showed the most promising protection against MES-induced convulsion in rats pretreated with a dose of 10 mg/kg, IP as compared with the saline- or PIT (10 mg/kg)-treated group with (F(1,12)=229.53 and F(1,12)=38.06, respectively, all P<0.001). More importantly, ligand 3 showed comparable protective effects when compared with the group pretreated with reference drug PHT (10 mg/kg, IP) with (F(1,12) =1.44, P=0.254).
 | Figure 2 Protective effect of acute systemic injection of H3R ligands 1–6 on MES-induced convulsions in rats. |
Dose-dependent protective effects of H3R ligand 3 on MES-induced convulsions
As shown in Figure 3, rats pretreated with 5 mg/kg of H3R ligand 3 were significantly protected against convulsions when compared with the saline-treated group (F(1,12) =31.36, P<0.001). Moreover, significant increases in protective effects were observed after pretreatment with 10 and 15 mg/kg of H3R ligand 3 when compared with 5 mg/kg of the same compound with (F(1,12) =74.34, P<0.001) and (F(1,12) =92.86, P<0.001), respectively.
 | Figure 3 Dose-dependent protective effect of H3R ligand 3 against MES-induced convulsions. |
Effect of RAMH pretreatment on the protection provided by H3R ligand 3 on MES-induced convulsions
Figure 4 shows the reversal of protective effect of H3R ligand 3 by pretreatment with 10 mg/kg of the CNS penetrant histamine H3R agonist RAMH 15 minutes before MES challenge with (F(1,12) =0.004; P=0.948, for the comparison between 3 + RAMH and 3 alone). More importantly, RAMH alone did not affect MES-induced seizures (P=0.517 saline-saline vs saline-RAMH).
 | Figure 4 Effect of RAMH (10 mg/kg, IP) pretreatment on the protection by H3R ligand 3 (10 mg/kg, IP) on MES-induced convulsions. |
Protective effect of H3R ligands 1–6 pretreatment on PTZ-induced convulsions
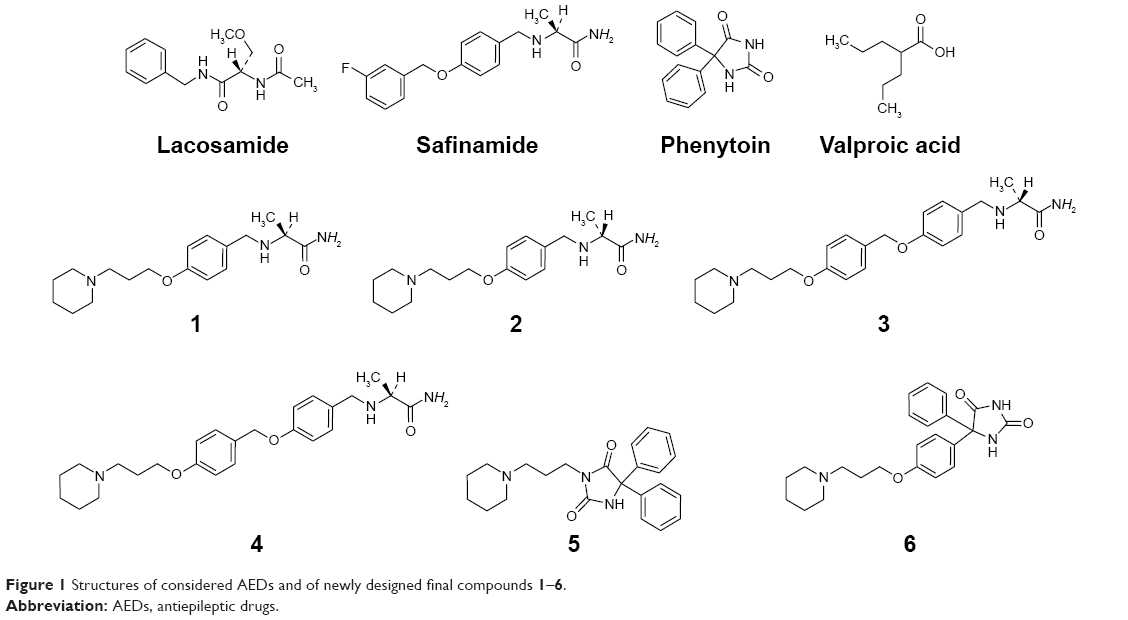
Figure 5 shows the effects of H3R ligands 1–6 (10 mg/kg, IP) and the standard H3R antagonist PIT in comparison to the protective effects of the reference AED VPA on PTZ-induced seizures in rats. In PTZ-induced seizure, H3R ligands 4 and 6 provided significant protective effects relative to the saline-treated group after 10, 20, and 30 minutes of observation (P<0.050). More importantly, H3R ligand 4 provided full protection when compared with saline-treated group after 10, 20, and 30 with (F(1,12) =101.4; P<0.001), (F(1,12) =44.74; P<0.001), and (F(1,12) =10.5; P<0.05), respectively. Similarly, H3R ligand 6 exhibited full protection when compared with saline-treated group after 10, 20, and 30 with (F(1,12) =101.4; P<0.05), (F(1,12) =44.74; P<0.05), and (F(1,12) =16.8; P<0.05), respectively. Importantly, H3R ligand 3 (10 mg/kg, IP) did not provide significant protective effect relative to the saline-treated after 10, 20, and 30 minutes (F(1,12) =0.10, 0.005, and 0.021, respectively, all NS). Similarly, PIT failed to provide protection in the PTZ-induced seizure model after 10, 20, and 30 minutes (F(1,12) =0.03, 0.01, and 0.50, respectively, all NS). However, VPA (100 mg/kg) provided significant protection when compared with saline-treated group after 10, 20, and 30 minutes with (F(1,12) =101.4; P<0.005), (F(1,12) =44.74; P<0.05), and (F(1,12) =16.8; P<0.05), respectively.
 | Figure 5 Protective effect of H3R ligands 1–6 pretreatment on PTZ-induced convulsions in rats. |
Dose-dependent protective effect of H3R ligands 4 and 6 on PTZ-induced convulsions
Figure 6 shows the protective and dose-dependent effects of acute administration of H3R ligands 4 and 6 (5, 10, or 15 mg/kg, IP) on PTZ-induced seizures in rats. One-way analysis of variance showed that pretreatment with reference AED VPA and H3R ligand 6 (5, 10, and 15 m/kg, IP) exerted a significant protective effect against PTZ-induced convulsions with (F(3,24) =101.40; P<0.001), (F(3,24) =44.74; P<0.001), and (F(3,24) =10.50; P<0.001) for 10, 20, and 30 minutes observation, respectively. Moreover, the results showed that significant and full protective effect was provided at 10, 20, and 30 minutes after pretreatment with 10 and 15 mg/kg of H3R ligands 4 and 6 (P<0.001).
 | Figure 6 Dose-dependent protective effect of H3R ligands 2, 4, and 5 against PTZ-induced convulsions. |
Effect of RAMH pretreatment on the protection provided by H3R ligand 4 on PTZ-induced convulsions
Figure 7 shows the protective effect of H3R ligand 4 when coadministered with 10 mg/kg of the CNS penetrant histamine H3R agonist RAMH 15 minutes before PTZ challenge. For the comparison between H3R ligand 4 + RAMH and H3R ligand 4 alone for 10 and 20 minutes of observation, the observed results showed that RAMH failed to reverse the protection provided by H3R ligand 4 with (F(1,12) =101.4; P<0.001), (F(1,12) =44.74; P<0.001), and (F(1,12) =10.5; P<0.05), respectively. Importantly, RAMH alone did not affect convulsion score for 10, 20, and 30 minutes of observation with P=0.813, 0.725, and 0.754, respectively.
 | Figure 7 Effect of RAMH (10 mg/kg, IP) pretreatment on the protection by H3R ligand 4 (10 mg/kg, IP) on PTZ-induced convulsions in rats. |
Protective effect of H3R ligands 1–6 pretreatment on STR-induced convulsions
Figure 8 shows the effects of H3R ligands 1–6 (10 mg/kg, IP) and the standard H3R antagonist PIT in comparison to the protective effects of reference AED VPA in STR-induced seizures in rats. In STR-induced seizure, only H3R ligand 2 provided significant protective effects relative to the saline-treated group after 10, 20, and 30 minutes of observation (P<0.05). The results showed that H3R ligand 2 provided significant protection when compared with saline-treated group after 10, 20, and 30 with (F(1,12) =24.71; P<0.001), (F(1,12) =8.68; P<0.05), and (F(1,12) =8.33; P<0.05), respectively. Moreover, the standard H3R ligand PIT (10 mg/kg, IP) failed to provide protection in STR-induced seizure model after 10, 20, and 30 minutes (F(1,12) =0, 3.32, and 3.50; all NS), respectively. However, VPA (300 mg/kg, IP) provided significant protection when compared with saline-treated group after 10, 20, and 30 with (F(1,12) =41.82; P<0.05), (F(1,12) =144.15; P<0.05), and (F(1,12) =81.82; P<0.05), respectively.
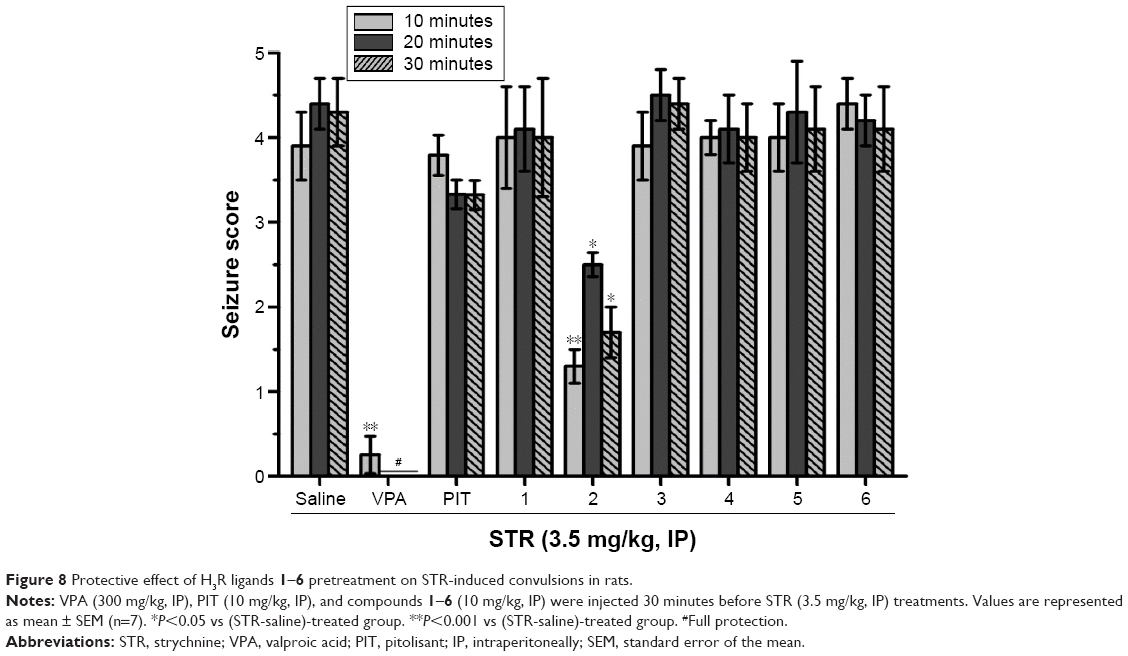 | Figure 8 Protective effect of H3R ligands 1–6 pretreatment on STR-induced convulsions in rats. |
Discussion
In vitro pharmacology
Examined H3R ligands 1–6 differed in their chemical structure either in the lipophilic part being benzylaminopropanamide or benzyloxyphenylethylaminoacetamide of 3-(piperidin-1-yl)propan-1-ol as well as in their stereochemistry (Table 1). When tested for their H3R activity in [3H]Nα-MeHA binding assay on HEK-293 cell membrane preparations stably expressing hH3R, compounds 1–6 exhibited moderate-to-high in vitro affinities in the nanomolar as well as subnanomolar concentration range with Ki values in the range of 5–43 nM. In addition, compounds 1–6 showed a high subtype selectivity for hH3R when tested on their antagonist affinities in displacement assays using cell membrane preparations of CHO cell lines stably expressing the hH1R with [3H]pyrilamine, and in binding assays on Sf9 cell membrane preparations transiently expressing hH4R (Table 1). Our observed antagonist in vitro results revealed modest-to-profound stereoselectivity for in vitro affinities at hH3Rs with varying preferences for this receptor subtype, as the (R)-enantiomer (2) showed five-times increased affinity at hH3Rs when compared to its (S)-enantiomer (1). Moreover, replacement of benzylaminopropanamide in 2 with the bulkier benzyloxyphenylethylaminoacetamide resulted in the design of the (R)-enantiomer (3), with an increase in antagonist affinity at hH3Rs with more than 1.5 times. Interestingly, the observed influence of chirality on the H3R antagonist affinity of the respective enantiomer has evidently been comprehended with the design of the S-enantiomer (4) of compound 3, as the affinity of 4 was decreased by more than four times when compared to 3 (Table 1). Noticeably, replacement of 3-(piperidin-1-yl)propan-1-ol in H3R ligand 5 with the bulkier 1-(3-phenoxypropyl)piperidine in 6 together with being 5-hydantoin-substituted derivative intensely increased the antagonist affinity for H3R by more than 89 times.
In vivo pharmacology
Encouraged by these in vitro outcomes, H3R ligands 1–6 were further investigated on their anticonvulsant effect against MES-, PTZ-, and STR-induced convulsion models in rats. The MES test is linked with the electrical induction of the convulsion, whereas the PTZ and STR tests involve chemical induction to generate the convulsion.39,51 The H3R ligands (1–6) were administered IP to the rats at doses of 10 mg/kg in the MES, PTZ, and STR screens. PHT was selected as the reference AED and PIT as a standard H3R antagonist in MES model, whereas VPA was chosen as the reference AED in PTZ and STR model. When investigated on their anticonvulsant effect against MES-induced convulsion in rats, no protection was observed for (S)-enantiomer (1, 10 mg/kg, IP). However, PHT (10 mg/kg, IP, P<0.001) fully and PIT (10 mg/kg, IP, P<0.05) moderately protected animals in the MES model (Figure 2). Contrary, the observed results showed that the protection provided in animals pretreated with 10 mg/kg, IP of (R)-enantiomer (2) was significantly higher when compared to the (S)-enantiomer-(1)-treated group (saline)-treated group (P<0.005 and P<0.05, respectively; Figure 2). Interestingly, animals pretreated with the bulkier (R)-enantiomer (3, 10 mg/kg, IP) were significantly and potently protected against MES-induced convulsion with observed effect comparable to that of the reference AED PHT (P=0.2540). Moreover, the anticonvulsant effect observed for 3 (R-enantiomer) was significantly higher than that exhibited by (S)-enantiomer (2) and standard H3R antagonist PIT (P<0.001). Importantly, the influence of chirality among the current series of ligands has been further comprehended, as the (S)-enantiomer (4), unlike the (R)-enantiomer (3), failed to protect animals against MES-induced convulsions (P=0.1039 vs saline-treated group) (Figure 2). Interestingly, the observed results for the isomeric H3R ligands 1–4 are in agreement with previous studies that demonstrated the chiral dependency of several AEDs, eg, lacosamide and safinamide.56,57 Accordingly, (R)-enantiomer of lacosamide exhibited potent anticonvulsant effect, whereas (S)-enantiomer was virtually inactive in MES model. Likewise, additional studies have clearly shown that anticonvulsant effect of several functionalized amino acids and derivatives thereof resided in the (R)-enantiomers whereas (S)-enantiomers were almost inactive.56,58,59 Notably, our major findings for in vivo anticonvulsant activity of the novel ligands 1–6 investigated in MES model correlated directly with their observed in vitro hH3R antagonist affinity. Accordingly, the obtained anticonvulsant properties for compound 3 were the highest, and in vitro hH3R antagonist affinity measured for 3 with 5.35±2.35 nM was the highest among investigated compounds 1–6. Surprisingly, the development of 5-(piperidin-1-yl)propoxyphenyl-substituted hydantoin (6) from the 3-piperidin-1-ylpropyl-substituted derivative (5) failed to provide significant protection in animals challenged with MES convulsions (P=0.9359 vs [6, 10 mg/kg, IP]-treated group; P=0.7054 vs saline-treated group), despite the significant increase observed for in vitro hH3R antagonist affinity by more than 89 times (Figure 2; Table 1). In a second experiment, H3R ligand 3 being the most promising H3R ligand in MES model was chosen to further establish a dose–response relationship of protective effect observed. Interestingly, our findings indicated that the protective effect of 3 was dependent on the dose administered; hence, rats pretreated with 3 in a dose of 10 and 15 mg/kg, IP were significantly higher protected against MES-induced convulsions when compared to the (3, 5 mg/kg, IP)-treated group (P<0.001; Figure 3). In addition, the results showed that the 3-provided protection was abrogated when the rats were pretreated with 10 mg/kg, IP of the histamine H3R agonist RAMH 15 minutes before MES challenge (P=0.9478 vs [saline]-treated group; Figure 4). Notably, RAMH administered alone (10 mg/kg, IP) did not offer either a protective or proconvulsant effect in MES-challenged rats (P=0.5173 vs [saline]-treated group; Figure 4). These findings suggest that H3R antagonism plays a critical role in the protective effect observed in animals pretreated with H3R ligand 3 in MES model. Therefore, blockade of these receptors by selective H3R ligand, for example, 3, would lead to an enhanced neuronal histamine release in the brain, resulting in anticonvulsant effect. Previous reports suggested that H3R ligands protected animals in PTZ convulsion models.23,60 In this study, the effects of H3R ligands 1–6 and PIT were also monitored in PTZ-induced convulsion model in rats. Surprisingly, for the most promising H3R ligand (3, (R)-enantiomer) in MES model the results obtained showed no protection in PTZ model. However, H3R ligands with virtually no protection in MES model, namely, 4 and 6, showed full protection against PTZ-induced convulsions during 30 minutes time of observation (Figure 5). In addition, the results observed for H3R ligand 4 were dependent on the dose administered, as animals pretreated with 10 and 15 mg/kg, IP of 4 were fully protected whereas (5 mg/kg)-treated group showed convulsions comparable to that of the saline-treated group (P<0.005; Figure 6). Moreover, the (R)-enantiomer (2) with moderate protection in MES model was found to be proconvulsant in a dose-dependent manner in the PTZ model when administered in a dose of 10 mg/kg, IP (P<0.005; Figures 5 and 6). Furthermore, the results observed among H3R ligands 1–6 showed no protection in STR-induced convulsion model with the exception of 2 in a dose of 10 mg/kg, IP (P<0.005; Figure 8). The discrepancy in the effects of ligands 3, 4, and 6 could be explained by the different levels of histamine release as a result of the convulsion in different models. In this respect, a significant increase of histamine was found in brain following MES convulsions, whereas a tendency toward decrease in histamine levels was found following PTZ-induced clonic convulsions, and no influence of histamine level was found in STR-induced convulsion model.38,41,61 The later finding was also comprehended in the present study, as the protective effect of H3R ligand 4 was not abrogated when animals were pretreated with RAMH (10 mg/kg, IP) 15 minutes before PTZ challenge, demonstrating that other mechanisms might explain the anticonvulsant effect observed for 4 in the PTZ model (Figure 7). In addition, RAMH administered alone (10 mg/kg, IP) did not offer either a protective or proconvulsant effect in rats challenged by PTZ model (Figure 7). Moreover, the discrepancy in the effects observed could also be explained by the different constructs and predictive validities of the MES, PTZ, and STR models. The endpoint in the MES test is THLE, which is considered to be a predictive model for generalized tonic-clonic seizures, whereas the PTZ and STR tests are used as predictors of anticonvulsant drug activity against nonconvulsive (absence or myoclonic) seizures. However, several available AEDs (eg, pregabalin and gabapentin) that are protective against nonconvulsive seizures in epilepsy patients failed in the PTZ or STR test, and others (eg, levetiracetam, tiagabine, and vigabatrin) without efficacy against partial seizures in the MES test were shown to suppress partial seizures in epilepsy patients.51
Conclusion
It can be concluded that by using different synthetic approaches we were able to design isomeric H3R ligands with very high in vitro affinity. We were also able to explore preliminary structure–affinity relationships on histamine hH1R, hH3R, and hH4R receptor subtypes with this small number of derivatives. Our obtained results showed that the anticonvulsant activity in MES model resided primarily in the structurally bulkier (R)-enantiomer (3) with observed protection comparable to that of PHT in rats, whereas the anticonvulsant activity in PTZ model resided primarily in its (S)-enantiomer (4) and was similar to that of VPA. Therefore, our in vivo results highlight the importance of chirality influence on the H3R ligand ability to exhibit anticonvulsant effects in three different convulsion models. In light of the controversial findings and the chirality dependencies, we conclude that the novel compounds 3, 4, and 6 may have potent anticonvulsant activity and can be used as potential templates for further drug design. This is promising because additional trials in epilepsy as well as convulsion models in other species are necessary to clarify the pharmacological profile of the current class to develop proper, clinically useful AEDs. Also, further investigations addressing in vitro anticonvulsant effects, rat pharmacokinetics/pharmacodynamics analysis, and ion channel affinity for ligands 3, 4, and 6 are warranted to comprehend their in vitro anticonvulsant effect and to exclude possibilities of off-target effects.
Acknowledgments
Support to Bassem Sadek was provided in form of grants received from UAE University. We greatly thank Prof JC Schwartz (Bioprojet, Saint-Grégoire, France) for providing HEK-293 cells stably expressing the recombinant hH3R in full length. The CHO-K1 cells stably expressing the human H1R receptor were generously gifted by Prof H Timmermann and Prof R Leurs (Amsterdam, the Netherlands). We gratefully thank Prof R Seifert (Hannover, Germany) for providing Sf9 cells and baculovirus stock solutions encoding for hH4R and G-protein Gαi2 and Gβ1γ2 subunits. The authors would also like to acknowledge Mr Dhanasekaran Subramanian and Mr Mohamed Shafiullah for their technical assistance. Support for this work has been kindly given to HS by the EU COST Actions BM0806, BM1007, CM1103, and CM1207 as well as the Hesse LOEWE Schwerpunkte Fh-TMP, OSF and NEFF, the Else KrönerStiftung, TRIP, and the Deutsches Konsortium für Translationale Krebsforschung, DKTK.
Disclosure
The authors report no conflicts of interest in this work.
References
Brigo F, Ausserer H, Tezzon F, Nardone R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy Behav. 2013;27(3):439–442. | ||
Schmidt D. The clinical impact of new antiepileptic drugs after a decade of use in epilepsy. Epilepsy Res. 2002;50(1–2):21–32. | ||
Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. Lancet Neurol. 2004;3(11):663–672. | ||
Hermann B, Meador KJ, Gaillard WD, Cramer JA. Cognition across the lifespan: antiepileptic drugs, epilepsy, or both? Epilepsy Behav. 2010;17(1):1–5. | ||
Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88(3):1183–1241. | ||
Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302(5911):832–837. | ||
Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63(6):637–672. | ||
Arrang JM, Garbarg M, Schwartz JC. Autoinhibition of histamine synthesis mediated by presynaptic H3-receptors. Neuroscience. 1987;23(1):149–157. | ||
Schlicker E, Fink K, Detzner M, Gothert M. Histamine inhibits dopamine release in the mouse striatum via presynaptic H3 receptors. J Neural Transm Gen Sect. 1993;93(1):1–10. | ||
Schlicker E, Betz R, Gothert M. Histamine H3 receptor-mediated inhibition of serotonin release in the rat brain cortex. Naunyn Schmiedebergs Arch Pharmacol. 1988;337(5):588–590. | ||
Sadek B, Stark H. Cherry-picked ligands at histamine receptor subtypes. Neuropharmacology. 2016;106:56–73. | ||
Yokoyama H. The role of central histaminergic neuron system as an anticonvulsive mechanism in developing brain. Brain Dev. 2001;23(7):542–547. | ||
Scherkl R, Hashem A, Frey HH. Histamine in brain – its role in regulation of seizure susceptibility. Epilepsy Res. 1991;10(2–3):111–118. | ||
Kamei C, Ishizawa K, Kakinoki H, Fukunaga M. Histaminergic mechanisms in amygdaloid-kindled seizures in rats. Epilepsy Res. 1998;30(3):187–194. | ||
Chen Z, Li WD, Zhu LJ, Shen YJ, Wei EQ. Effects of histidine, a precursor of histamine, on pentylenetetrazole-induced seizures in rats. Acta Pharmacol Sin. 2002;23(4):361–366. | ||
Yawata I, Tanaka K, Nakagawa Y, Watanabe Y, Murashima YL, Nakano K. Role of histaminergic neurons in development of epileptic seizures in EL mice. Brain Res Mol Brain Res. 2004;132(1):13–17. | ||
Takano T, Sakaue Y, Sokoda T, et al. Seizure susceptibility due to antihistamines in febrile seizures. Pediatr Neurol. 2010;42(4):277–279. | ||
Miyata I, Saegusa H, Sakurai M. Seizure-modifying potential of histamine H1 antagonists: a clinical observation. Pediatr Int. 2011;53(5):706–708. | ||
Haruyama W, Fuchigami T, Noguchi Y, et al. The relationship between drug treatment and the clinical characteristics of febrile seizures. World J Pediatr. 2008;4(3):202–205. | ||
Zolaly MA. Histamine H1 antagonists and clinical characteristics of febrile seizures. Int J Gen Med. 2012;5:277–281. | ||
Sadek B, Saad A, Sadeq A, Jalal F, Stark H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav Brain Res. 2016;312:415–430. | ||
Gemkow MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today. 2009;14(9–10):509–515. | ||
Bhowmik M, Khanam R, Vohora D. Histamine H3 receptor antagonists in relation to epilepsy and neurodegeneration: a systemic consideration of recent progress and perspectives. Br J Pharmacol. 2012;167(7):1398–1414. | ||
Kasteleijn-Nolst Trenite D, Parain D, Genton P, Masnou P, Schwartz JC, Hirsch E. Efficacy of the histamine 3 receptor (H3R) antagonist pitolisant (formerly known as tiprolisant; BF2.649) in epilepsy: dose-dependent effects in the human photosensitivity model. Epilepsy Behav. 2013;28(1):66–70. | ||
Sander K, Kottke T, Stark H. Histamine H3 receptor antagonists go to clinics. Biol Pharm Bull. 2008;31(12):2163–2181. | ||
Witkin JM, Nelson DL. Selective histamine H3 receptor antagonists for treatment of cognitive deficiencies and other disorders of the central nervous system. Pharmacol Ther. 2004;103(1):1–20. | ||
Van der Schyf CJ, Geldenhuys WJ, Youdim MB. Multifunctional drugs with different CNS targets for neuropsychiatric disorders. J Neurochem. 2006;99(4):1033–1048. | ||
von Coburg Y, Kottke T, Weizel L, Ligneau X, Stark H. Potential utility of histamine H3 receptor antagonist pharmacophore in antipsychotics. Bioorg Med Chem Lett. 2009;19(2):538–542. | ||
Keith JM, Barbier AJ, Wilson SJ, et al. Dual serotonin transporter inhibitor/histamine H3 antagonists: development of rigidified H3 pharmacophores. Bioorg Med Chem Lett. 2007;17(19):5325–5329. | ||
Altenbach RJ, Black LA, Strakhova MI, et al. Diaryldiamines with dual inhibition of the histamine H(3) receptor and the norepinephrine transporter and the efficacy of 4-(3-(methylamino)-1-phenylpropyl)-6-(2-(pyrrolidin-1-yl)ethoxy)naphthalen-1-ol in pain. J Med Chem. 2010;53(21):7869–7873. | ||
Sadek B, Schwed JS, Subramanian D, et al. Non-imidazole histamine H3 receptor ligands incorporating antiepileptic moieties. Eur J Med Chem. 2014;77:269–279. | ||
Isensee K, Amon M, Garlapati A, et al. Fluorinated non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem Lett. 2009;19(8):2172–2175. | ||
Stocks MJ, Cheshire DR, Reynolds R. Efficient and regiospecific one-pot synthesis of substituted 1,2,4-triazoles. Org Lett. 2004;6(17):2969–2971. | ||
Meier G, Apelt J, Reichert U, et al. Influence of imidazole replacement in different structural classes of histamine H(3)-receptor antagonists. Eur J Pharm Sci. 2001;13(3):249–259. | ||
Baek JB, Harris FW. Development of an improved synthetic route to an AB phenylquinoxaline monomer. J Polym Sci A Polym Chem. 2005;43(4):801–814. | ||
Sander K, Kottke T, Hoffend C, et al. First metal-containing histamine H3 receptor ligands. Org Lett. 2010;12(11):2578–2581. | ||
Sander K, Kottke T, Weizel L, Stark H. Kojic acid derivatives as histamine H(3) receptor ligands. Chem Pharm Bull (Tokyo). 2010;58(10):1353–1361. | ||
Sadek B, Kuder K, Subramanian D, et al. Anticonvulsive effect of nonimidazole histamine H3 receptor antagonists. Behav Pharmacol. 2014;25(3):245–252. | ||
Sadek B, Saad A, Subramanian D, Shafiullah M, Lazewska D, Kiec-Kononowiczc K. Anticonvulsant and procognitive properties of the non-imidazole histamine H3 receptor antagonist DL77 in male adult rats. Neuropharmacology. 2016;106:46–55. | ||
Sadek B, Schreeb A, Schwed JS, Weizel L, Stark H. Drug-likeness approach of 2-aminopyrimidines as histamine H3 receptor ligands. Drug Des Devel Ther. 2014;8:1499–1513. | ||
Sadek B, Shehab S, Wiecek M, et al. Anticonvulsant properties of histamine H3 receptor ligands belonging to N-substituted carbamates of imidazopropanol. Bioorg Med Chem Lett. 2013;23(17):4886–4891. | ||
Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22(23):3099–3108. | ||
Schlotter K, Boeckler F, Hubner H, Gmeiner P. Fancy bioisosteres: metallocene-derived G-protein-coupled receptor ligands with subnanomolar binding affinity and novel selectivity profiles. J Med Chem. 2005;48(11):3696–3699. | ||
van Staveren DR, Metzler-Nolte N. Bioorganometallic chemistry of ferrocene. Chem Rev. 2004;104(12):5931–5985. | ||
Schibli R, Schubiger PA. Current use and future potential of organometallic radiopharmaceuticals. Eur J Nucl Med Mol Imaging. 2002;29(11):1529–1542. | ||
Lazewska D, Ligneau X, Schwartz JC, Schunack W, Stark H, Kiec-Kononowicz K. Ether derivatives of 3-piperidinopropan-1-ol as non-imidazole histamine H3 receptor antagonists. Bioorg Med Chem. 2006;14(10):3522–3529. | ||
Sadek B, Khanian SS, Ashoor A, et al. Effects of antihistamines on the function of human alpha7-nicotinic acetylcholine receptors. Eur J Pharmacol. 2014;746:308–316. | ||
Bahi A, Sadek B, Nurulain SM, Lazewska D, Kiec-Kononowicz K. The novel non-imidazole histamine H receptor antagonist DL77 reduces voluntary alcohol intake and ethanol-induced conditioned place preference in mice. Physiol Behav. 2015;151:189–197. | ||
Bahi A, Sadek B, Schwed SJ, Walter M, Stark H. Influence of the novel histamine H(3) receptor antagonist ST1283 on voluntary alcohol consumption and ethanol-induced place preference in mice. Psychopharmacology (Berl). 2013;228(1):85–95. | ||
Bahi A, Schwed JS, Walter M, Stark H, Sadek B. Anxiolytic and antidepressant-like activities of the novel and potent non-imidazole histamine H(3) receptor antagonist ST-1283. Drug Des Devel Ther. 2014;8:627–637. | ||
Loscher W. Critical review of current animal models of seizures and epilepsy used in the discovery and development of new antiepileptic drugs. Seizure. 2011;20(5):359–368. | ||
Branco Cdos S, Scola G, Rodrigues AD, et al. Anticonvulsant, neuroprotective and behavioral effects of organic and conventional yerba mate (Ilex paraguariensis St. Hil.) on pentylenetetrazol-induced seizures in Wistar rats. Brain Res Bull. 2012;92:60–68. | ||
Sowemimo AA, Adio O, Fageyinbo S. Anticonvulsant activity of the methanolic extract of Justicia extensa T. Anders. J Ethnopharmacol. 2011;138(3):697–699. | ||
Sancheti J, Shaikh MF, Chaudhari R, et al. Characterization of anticonvulsant and antiepileptogenic potential of thymol in various experimental models. Naunyn Schmiedebergs Arch Pharmacol. 2013;387(1):59–66. | ||
Coppola G, Arcieri S, D’Aniello A, et al. Levetiracetam in submaximal subcutaneous pentylentetrazol-induced seizures in rats. Seizure. 2010;19(5):296–299. | ||
Rogawski MA, Tofighy A, White HS, Matagne A, Wolff C. Current understanding of the mechanism of action of the antiepileptic drug lacosamide. Epilepsy Res. 2015;110:189–205. | ||
Zolkowska D, Dhir A, Krishnan K, Covey DF, Rogawski MA. Anticonvulsant potencies of the enantiomers of the neurosteroids androsterone and etiocholanolone exceed those of the natural forms. Psychopharmacology (Berl). 2014;231(17):3325–3332. | ||
Choi D, Stables JP, Kohn H. Synthesis and anticonvulsant activities of N-Benzyl-2-acetamidopropionamide derivatives. J Med Chem. 1996;39(9):1907–1916. | ||
Kohn H, Conley JD, Leander JD. Marked stereospecificity in a new class of anticonvulsants. Brain Res. 1988;457(2):371–375. | ||
Kakinoki H, Ishizawa K, Fukunaga M, Fujii Y, Kamei C. The effects of histamine H3-receptor antagonists on amygdaloid kindled seizures in rats. Brain Res Bull. 1998;46(5):461–465. | ||
Vohora D, Pal SN, Pillai KK. Thioperamide, a selective histamine H3 receptor antagonist, protects against PTZ-induced seizures in mice. Life Sci. 2000;66(22):PL297–PL301. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.