")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
Antiapoptotic and Anti-Inflammatory Effects of CPCGI in Rats with Traumatic Brain Injury
Authors Niu F, Qian K , Qi H, Zhao Y, Jiang Y , Sun M
Received 11 September 2020
Accepted for publication 16 November 2020
Published 7 December 2020 Volume 2020:16 Pages 2975—2987
DOI https://doi.org/10.2147/NDT.S281530
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yuping Ning
Fei Niu,1,* Ke Qian,2,* Hongyan Qi,3 Yumei Zhao,4 Yingying Jiang,4 Ming Sun4
1Department of Neurotrauma, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, Beijing 100070, People’s Republic of China; 2Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, People’s Republic of China; 3Department of Acupuncture, Lianyungang TCM Hospital Affiliated to Nanjing University of Chinese Medicine, Lianyungang City 222000, Jiangsu Province, People’s Republic of China; 4Department of Neuropharmacology, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, Beijing 100070, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ming Sun
Department of Neuropharmacology, Beijing Key Laboratory of Central Nervous System Injury, Beijing Neurosurgical Institute, Capital Medical University, 119 South Fourth Ring West Road, Fengtai District, Beijing 100070, People’s Republic of China
Tel/Fax + 86-10-59975497
Email [email protected]
Background: Compound porcine cerebroside and ganglioside injection (CPCGI) has been used for the treatment of certain brain disorders. Apoptosis and inflammation were reported to be involved in the pathogenesis of traumatic brain injury (TBI). Therefore, this study primarily investigated the effects of CPCGI on mitochondrial apoptotic signaling and PARP/NF-κB inflammatory signaling in a rat model of controlled cortical impact (CCI).
Materials and Methods: CPCGI (0.6 mL/kg) was administered intraperitoneally 30 min after the induction of CCI. Mitochondrial apoptotic signaling and PARP/NF-κB inflammatory signaling were evaluated 24 h after CCI, and apoptotic cell death, neutrophil infiltration, and astrocyte and microglial activation were determined by TUNEL and immunofluorescent staining 3 days after CCI.
Results: 1) CPCGI markedly enhanced cytosolic and mitochondrial Bcl-xL levels, the mitochondrial Bcl-xL/Bax ratio, and mitochondrial cytochrome (cyt) c levels and reduced cytosolic cyt c levels, caspase-3 activity, and nuclear AIF levels in brain tissues after traumatic injury; however, CPCGI had no significant effects on cytosolic or mitochondrial Bax levels, the cytosolic Bcl-xL/Bax ratio, or mitochondrial AIF levels. Moreover, CPCGI markedly reduced the TUNEL staining score in the contusion region. 2) CPCGI markedly reduced cytosolic and nuclear PARP levels and nuclear NF-κB p65 levels in brain tissues after traumatic injury but had no significant effect on cytosolic NF-κB p65 levels. In addition, CPCGI markedly reduced caspase-1 activity and the levels of caspase-1, ICAM-1, TNF-α, and IL-1β in brain tissues after traumatic injury and decreased the immunoreactivities of neutrophils, GFAP and Iba-1 in the region of CCI-induced contusion.
Conclusion: These data suggest that CPCGI can reduce brain injury due to trauma by suppressing both mitochondrial apoptotic signaling and PARP/NF-κB inflammatory signaling.
Keywords: compound porcine cerebroside and ganglioside injection, traumatic brain injury, mitochondrial apoptotic signaling, PARP/NF-κB inflammatory signaling
Introduction
Traumatic brain injury (TBI), which involves both primary and secondary injuries, is the leading cause of mortality and disability in adults under 45 years of age.1,2 To date, there is no effective drug for TBI, which places a heavy burden on society and the patients’ families.3,4 Primary injury due to initial impact is the main factor determining TBI prognosis, whereas secondary injury, which is triggered by primary injury, the results from neurochemical processes and intracellular signaling pathways, such as oxidative stress, inflammatory response, neuronal apoptosis and other detrimental factors, which aggravate brain primary injury after TBI.5 Therefore, alleviating secondary injury by intervening in neurochemical processes and intracellular signaling pathways is very important in the treatment of TBI.
Extensive data have confirmed the important roles of apoptosis and inflammation in the pathogenesis of TBI. During TBI, apoptotic signaling pathways, including the membrane death receptor pathway (ie, the external pathway) and the mitochondrial pathway (ie, the internal pathway), are activated, and caspase-3 is a primary effector of apoptosis. In addition, apoptosis-inducing factor (AIF), a mitochondrial flavoprotein, induces caspase-3-independent apoptosis.6,7 Inflammation due to TBI is characterized by the release of pro-inflammatory mediators, the infiltration of circulating inflammatory cells (including neutrophils, macrophages, lymphocytes, etc.), and the activation of astrocytes and microglia.6,8 Poly (ADP-ribose) polymerase (PARP), a nuclear enzyme, participates in inflammatory gene expression driven by nuclear factor-κB (NF-κB).9,10
Compound porcine cerebroside and ganglioside injection (CPCGI) is a neurotrophic drug that has been approved for use in the treatment of certain brain diseases in China. It reportedly plays a neuroprotective role in experimental cerebral ischemia and Alzheimer’s disease.11,12 A previous study revealed that CPCGI exerts protective effects against TBI by upregulating the Nrf2 signaling pathway, lessening oxidative stress and suppressing the overactivation of calpain.13 This study primarily investigated whether CPCGI protects the brain against TBI through repressing both mitochondrial apoptotic and PARP/NF-κB inflammatory signaling after TBI.
Materials and Methods
Induction of Brain Injury Due to Controlled Cortical Impact (CCI) in Rats
All animal experiments complied with the guidelines established by the National Institutes of Health for the care and use of laboratory animals and were approved by the animal care committee of the Beijing Neurosurgical Institute. Male adult Sprague Dawley rats (weighing 290–320 g, Beijing Vital River Experimental Animals Technology Ltd., Beijing, China) were housed under controlled conditions (22 °C, 12-h light/12-h dark cycle) and had free access to food and water. The rats were anesthetized by isoflurane inhalation and placed on a stereotaxic frame (RWD Life Science Co., Shenzhen, China). Brain injury was induced using a PCI3000 PinPoint Precision Cortical Impactor (Hatteras Instruments, Cary, NC, USA) as previously described.14,15 Briefly, a 5-mm-diameter craniotomy was created using a skull drill, while leaving the dura intact. The craniotomy was located at the middle of the right parietal bone between the coronal and lambdoid sutures and approximately 0.5 mm from the sagittal suture. A circular impact tip with a diameter of 4 mm was then accelerated and impacted the surface of the craniotomy (velocity, 2 m/s; compression time, 85 ms; and depth, 2.5 mm). After each impact, the impact tip was cleaned with sterile alcohol. The sham-operated rats underwent the same procedure without the impact. The incision was closed with 4–0 silk sutures after surgery. During the procedure, the temperature of the rats was maintained at 37 °C ± 0.5 °C with a thermal pad.
Experimental Protocol
CPCGI (provided by Buchang Pharmaceuticals Co., Ltd., Xian, China) was dissolved in 0.9% normal saline (NS). The rats were randomly divided into three groups as follows: 1) the CPCGI group: the rats were subjected to CCI and treated with 0.6 mL/kg CPCGI; 2) vehicle group: the rats were subjected to CCI and treated with a similar volume of NS (2 mL/kg); and 3) sham group: the rats were subjected to the sham operation and treated with 2 mL/kg NS. NS or CPCGI was administered intraperitoneally at a dose of 2 mL/kg 30 min after CCI. For the evaluation of mitochondrial-mediated apoptotic cell death and inflammatory signaling, traumatic brain tissues were collected 24 h post-CCI, and the levels of Bcl-xL, Bax, and cyt c in the cytosolic and mitochondrial fractions, the levels of AIF in the mitochondrial and nuclear fractions, the levels of PARP and NF-κB p65 in the cytosolic and nuclear fractions, the levels of intercellular adhesion molecule 1 (ICAM-1), tumor necrosis factor α (TNF-α), interleukin 1 β precursor (pro-IL-1β), IL-1β, caspase-1 precursor (pro-caspase-1) and caspase-1 p20, and the activities of caspase-1 and caspase-3 in the cytosolic fractions were assayed. In addition, the whole brain was dissected at 72 h post-CCI, and the levels of apoptotic cell death, neutrophil infiltration, and astrocyte and microglial activation were determined by terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL) and immunofluorescence staining, respectively. The animal number in each group was 6 in all experiments.
Sample Collection and Preparation
The tissues of the right injured hemisphere were dissected at 4 °C, and samples were prepared as described previously16,17 and modified as follows. Briefly, the rats were sacrificed under anesthesia. The whole brain was dissected, and the right (injured) hemisphere was cut into three sections beginning 2 mm from the anterior tip of the frontal lobe and 2 mm from the posterior tip of the occipital lobe. The tissue from the middle part of the brain was collected as brain tissue with traumatic injury. The brain tissues with traumatic injury were homogenized in 10 volumes of homogenization buffer A (20 mM N-2-hydroxyethylpiperazine-N’-2ʹ-ethanesulfonic acid (HEPES), 250 mM sucrose, 10% glycerol, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1% Triton-X 100, and 1% protease inhibitor cocktail, pH 7.5). The homogenates were centrifuged at 1000 g and 4 °C for 15 min to separate the homogenates into supernatant A and pellet A. Pellet A was washed three times with buffer A, suspended in an equal volume of buffer B (20 mM HEPES, 20% glycerol, 1.5 mM MgCl2, 420 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1% Triton-X 100, and 1% protease inhibitor cocktail, pH 7.5), and vortexed at maximum speed for 15 s. The suspension was placed on ice for 30 min and then centrifuged at 14,000 g and 4 °C for 30 min, and the supernatant containing the nuclear fraction was collected. Supernatant A, which contained the cytosolic and mitochondrial fractions, was further centrifuged at 16,000 g and 4 °C for 30 min to obtain supernatant B and pellet B. Supernatant B was used as the cytosolic fraction. Pellet B, which served as the mitochondrial fraction, was suspended in an equal volume of buffer A. The cytosolic, mitochondrial and nuclear fractions were stored at −70 °C. The protein concentrations in the cytosolic, mitochondrial and nuclear fractions were determined using the BCA method.
Caspase-1 and Caspase-3 Activity Assays
Caspase-1 and caspase-3 activities were measured using a previously described method with some modifications.18,19 Briefly, the cleavage of the fluorogenic substrates for caspase-1 (ac-YVAD-AFC, Santa Cruz Biotech, CA, USA) and caspase-3 (ac-DEVD-AFC, Santa Cruz Biotech, CA, USA) into their fluorescent products was used to measure caspase-1 and caspase-3 activity levels, respectively. The cytosolic fractions (10 µL) were incubated with caspase reaction buffer (250 μL; 50 mM HEPES, pH 7.4, 100 mM NaCl, 1 mM EDTA, and 10 mM DTT) containing the fluorogenic caspase-1 or caspase-3 substrate at a concentration of 20 μM for 2 h at 37 °C. The free AFC released by proteolysis was then assessed using a microplate reader (BioTek Instruments, Inc., Winooski, VT, USA; excitation, 400 nm; emission, 505 nm). The fluorescence arbitrary units were converted into micromoles of AFC released per hour and milligrams of protein using a standard curve of free AFC (Santa Cruz Biotech, CA, USA).
Western blot
The protein levels of Bcl-xL, Bax, cyt c, PARP, NF-κB p65, ICAM-1, TNF-α, pro-IL-1β, IL-1β, pro-caspase-1 and caspase-1 p20 in the cytosolic fraction, the protein levels of Bcl-xL, Bax, cyt c, and AIF in the mitochondrial fraction, and the protein levels of AIF, PARP and NF-κB p65 in the nuclear fraction were determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Briefly, equal amounts of cytoplasmic, mitochondrial or nuclear protein (30 μg) and molecular weight markers (Abcam Inc., MA, USA) were loaded and separated by SDS-PAGE. The proteins on the gel were transferred to a PVDF membrane (Millipore, MA, USA). The membranes were blocked with Tris-buffered saline containing 1% Tween 20. The membrane was then probed with antibodies that reacted with Bax (1:500), Bcl-xL (1:250), cyt c (1:400), AIF (1:1000), PARP (1:250), histone H3 (1:500), ICAM-1 (1:250, Abcam, Cambridge, United Kingdom), TNF-α (1:250), NF-κB p65 (1:500), pro-IL-1β/IL-1β (1:1000, R&D, MN, USA), pro-caspase-1/caspase-1 p20 (1:50, Novus Biologicals, CO, USA), β-actin (1:2000, Sigma-Aldrich Corp., MO, USA), and VDAC (1:5000, Millipore, MA, USA) overnight at 4 °C and subsequently with horseradish peroxidase-conjugated secondary antibodies. Antibody binding was visualized by chemiluminescence (Millipore, MA, USA). β-actin, VDAC and histone H3 were used as internal controls for the cytosolic, mitochondrial and nuclear fractions, respectively. The integrated optical density (IODs) of the protein bands in the cytosolic, mitochondrial and nuclear fractions were normalized to the β-actin, VDAC and histone H3 immunoreactivities, respectively, and the results are expressed as percentages of the levels in the sham-operated rats.
Immunofluorescent Staining of Neutrophils, Astrocytes and Microglia
Sham-operated or CCI rats were transcardially perfused with heparinized normal saline and 4% buffered paraformaldehyde under anesthesia 3 days after the application of CCI. The rats were decapitated, and the whole brains were dissected. The brains were fixed in 4% buffered paraformaldehyde for 24 h and embedded in paraffin, and 5-μm-thick sections through the dorsal striatum were collected. Anti-neutrophil (Novus Biologicals, CO, USA), anti-Iba-1 (Millipore, MA, USA) and anti-GFAP (Abcam, Cambridge, MA, USA) antibodies were used to label neutrophils, microglia and astrocytes, respectively. Briefly, the 5-μm coronal sections were deparaffinized and rinsed in distilled water for 5 min × 2. The sections were boiled in an 800 W microwave oven for 15 min in 10 mM citrate buffer (pH 6). Sections were allowed to come to room temperature and washed in phosphate-buffered saline (PBS) twice for 5 min each. The sections were treated with avidin/biotin blocking solution for 30 min, washed three times with PBS containing 0.1% Triton X-100 (PBST), and incubated with an anti-neutrophil antibody (1:100), anti-Iba-1 antibody (1:400) or anti-GFAP antibody (1:200) diluted in 10% goat serum-PBST overnight at 4 °C. The sections were washed 3 times with PBS and incubated with fluorochrome-conjugated secondary antibodies for 1 h at room temperature. After 3 washes with PBS, the sections were mounted with Shield Hardset mounting medium with DAPI (Vector Labs, Burlingame, CA, USA) and coverslips. Negative control samples were incubated without the primary antibody. The sections were viewed on a fluorescence microscope (Leica Inc., Germany), and images were captured using a digital camera by a blinded, trained observer. The neutrophil, Iba-1 or GFAP immunoreactivity was semiquantified according to the immunoreactive cells and/or intensity of immunoreactivity in traumatic brain tissue on a scale of 0–4: 0, no or few immunoreactive cells and/or no change in immunoreactivity from sham-operated animals; 1, random immunoreactive cells with a mild alteration in intensity of immunoreactivity; 2, a discrete region of immunoreactive cells with a moderate alteration in intensity of immunoreactivity; 3, many strong immunoreactive cells with marked alteration in intensity of immunoreactivity; and 4, the entire region is infiltrated with immunoreactive cells with serious alteration in intensity of immunoreactivity.20,21 The neutrophil, Iba-1 or GFAP immunoreactivity in four regions around the lesion and four regions in the lesion was evaluated, and the results were expressed as the average of immunoreactive cells and/or intensity of immunoreactivity in the eight regions.
TUNEL Staining
The degree of apoptotic cell death was analyzed by TUNEL staining.22 Briefly, the 5-μm coronal sections through the striatum were deparaffinized and rinsed in distilled water at room temperature. The samples were incubated with proteinase K (Roche Diagnostics GmbH, Germany) for 15 min at 37 °C, and endogenous biotin was blocked by covering the sections with avidin-blocking solution for 1 h at room temperature. The sections were washed with PBST and immersed in TdT buffer (30 mM Tris, pH 7.2, 140 mM sodium cacodylate, 1 mM cobalt chloride) for 30 min, followed by reaction with the TdT enzyme (Roche Diagnostics GmbH, Germany) and biotinylated 16-dUTP (Roche Diagnostics GmbH, Germany) at 37 °C for 2 h. The sections were immersed in reaction stopping buffer (150 mM NaCl, 15 mM sodium citrate, pH 7.4) for 15 min and then washed in PBST. The sections were incubated with streptavidin-dye 488 for 20 min. TUNEL-stained cells in four regions around lesion were observed using a fluorescence microscope by a blinded, trained observer, and the green fluorescence indicated the apoptotic cells. The positive TUNEL staining score was expressed as the intensity of green influence, which was semiquantified on a scale of 0–4 as follows: 0, damaged neurons comprised <5% of the sample; 1, 5–25%; 2, 25–50%; 3, 50–75%; and 4, >75%. The results were expressed as the average positive TUNEL staining score in the four regions.
Statistical Analysis
The data are expressed as the mean ± SEM. The protein levels of Bax, Bcl-xL, cyt c, AIF, PARP, ICAM-1, TNF-α, NF-κB p65, pro-IL-1β, IL-1β, pro-caspase-1 and caspase-1 p20 in the cytosolic, mitochondrial and nuclear fractions were evaluated by one-way ANOVA with a post hoc LSD test, and the immunoreactivities of neutrophil, Iba-1 and GFAP and the TUNEL staining index were evaluated by a nonparametric Kruskal–Wallis ANOVA with Dunn’s post hoc test. A P value of <0.05 was defined as significant.
Results
Antiapoptotic Effects of CPCGI After CCI
Effects of CPCGI on Apoptotic Cell Death
Five-micrometer-thick coronal sections of the dorsal striatum were used to evaluate apoptotic cell death in the brain area with traumatic injury. DNA fragmentation was determined by TUNEL staining, and the results are illustrated in Figure 1. The TUNEL-positive cells showed nuclear staining and exhibited morphological signs of apoptotic cell death. A large number of TUNEL-positive cells were detected in the injured area of the brain in the vehicle-treated rats, whereas no TUNEL-positive cells were observed in the brain of the sham-operated rats. Treatment with CPCGI significantly reduced the TUNEL-staining positive score in the brain region with traumatic injury (P < 0.05 vs vehicle-treated rats).
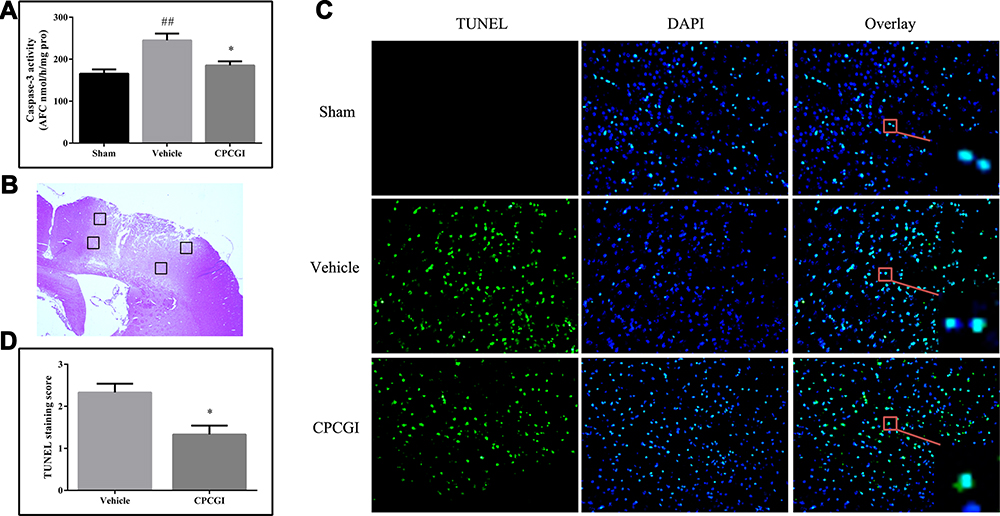 |
Figure 1 Effect of CPCGI on caspase-3 activity and apoptotic cell death after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A) Caspase-3 activity. (B) Four regions around the lesion were stained with TUNEL. (C) Representative images of TUNEL-stained sections from animals in all groups (original magnification, 200×). (D) CPCGI treatment significantly reduced the number of TUNEL-positive cells in CCI rats, as determined by the fluorescence intensity score. The data are expressed as the mean ± SEM. n = 6 per group. ##P < 0.01 vs sham-operated rats; *P < 0.05 vs vehicle-treated rats. |
Effects of CPCGI on Mitochondria-Mediated Caspase-3-Dependent Apoptotic Signaling
Compared with sham-operated rats, the cytosolic Bcl-xL levels and mitochondrial Bax levels showed no significant change in brain tissues with traumatic injury, but the levels of cytosolic Bax and mitochondrial Bcl-xL were decreased (Figure 2; both P < 0.05). Meanwhile, the cytosolic Bcl-xL/Bax ratio and cyt c levels were increased (both P < 0.05), while the mitochondrial Bcl-xL/Bax ratio and cyt c levels were decreased (both P < 0.01). Moreover, the cytosolic caspase-3 activity was enhanced in brain tissues with traumatic injury (Figure 1; P < 0.01 vs the sham-operated rats). CPCGI treatment markedly enhanced the cytosolic and mitochondrial Bcl-xL levels and the mitochondrial Bcl-xL/Bax ratio (P < 0.05, P < 0.01, and P < 0.01 vs the vehicle-treated rats, respectively), although it had no significant effects on the cytosolic and mitochondrial Bax levels or the cytosolic Bcl-xL/Bax ratio. Moreover, CPCGI significantly enhanced mitochondrial cyt c levels and reduced cytosolic cyt c levels and caspase-3 activity (P < 0.01, P < 0.05, and P < 0.05 vs vehicle-treated rats, respectively).
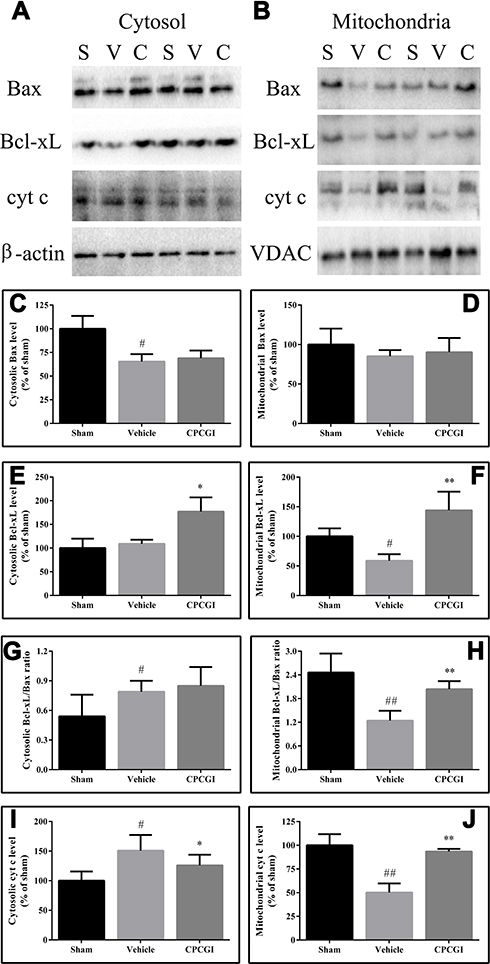 |
Figure 2 Effect of CPCGI on the levels of Bax, Bcl-xL and cyt c in the cytosolic and mitochondrial fractions of injured brain tissues collected 24 h post-CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A and B) Western blot analysis of the cytosolic and mitochondrial fractions using Bax, Bcl-xL or cyt c antibodies. (C–F), and (I and J) The bar graphs revealed the densitometric data from the Western blot analysis of the Bax, Bcl-xL and cyt c levels, respectively, in the cytosolic and mitochondrial fractions. (G and H) The bar graphs revealed the ratio of Bcl-xL/Bax in the cytosolic and mitochondrial fractions, respectively. All data are expressed as the mean ± SEM. n = 6 per group. #P < 0.05 and ##P < 0.01 vs sham-operated rats. *P < 0.05 and **P < 0.01 vs vehicle-treated rats. Abbreviations: S, sham group; V, vehicle group; C, CPCGI group. |
Effects of CPCGI on AIF-Mediated Caspase-3-Independent Apoptotic Signaling
To determine whether the caspase-independent apoptotic pathway contributed to the protective effect of CPCGI against CCI in rats, the AIF levels in the mitochondria and nucleus were analyzed by Western blot, and the results are presented in Figure 3. The mitochondrial AIF levels showed no marked changes in brain tissues with traumatic injury, while the nuclear AIF levels were increased significantly (P < 0.01 vs sham-operated rats). CPCGI treatment enhanced mitochondrial AIF levels without statistical significance, but it markedly reduced the nuclear AIF levels (P < 0.01 vs vehicle-treated rats).
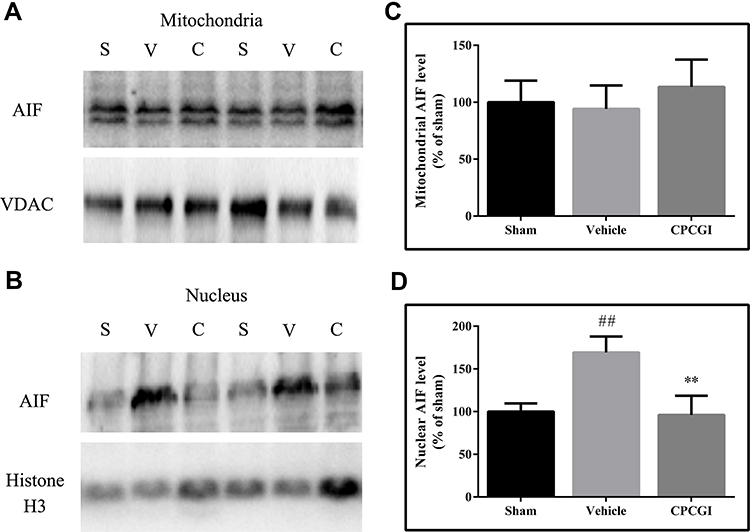 |
Figure 3 Effects of CPCGI on the AIF levels in mitochondrial and nuclear fractions from injured brain tissues collected 24 h post-CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A and B) Western blot analysis of the mitochondrial and nuclear fractions, respectively, using an AIF antibody. (C and D) The bar graphs reflect the densitometric data from the Western blot analysis of the AIF levels in the mitochondrial and nuclear fractions, respectively. All data are expressed as the mean ± SEM. n = 6 per group. ##P < 0.01 vs sham-operated rats. **P < 0.01 vs vehicle-treated rats. Abbreviations: S, sham group; V, vehicle group; C, CPCGI group. |
Anti-Inflammatory Effects of CPCGI After CCI
Effects of CPCGI on Neutrophil Infiltration in Brain Tissues with Traumatic Injury
Neutrophil infiltration was evaluated by neutrophil immunoreactivity. The brain sections in the sham-operated rats showed low neutrophil immunoreactivity, whereas the injured brain region in the vehicle-treated rats exhibited significant neutrophil immunoreactivity (Figure 4). CPCGI treatment markedly reduced the degree of neutrophil immunoreactivity in the brain region with traumatic injury (P < 0.05 vs vehicle-treated rats).
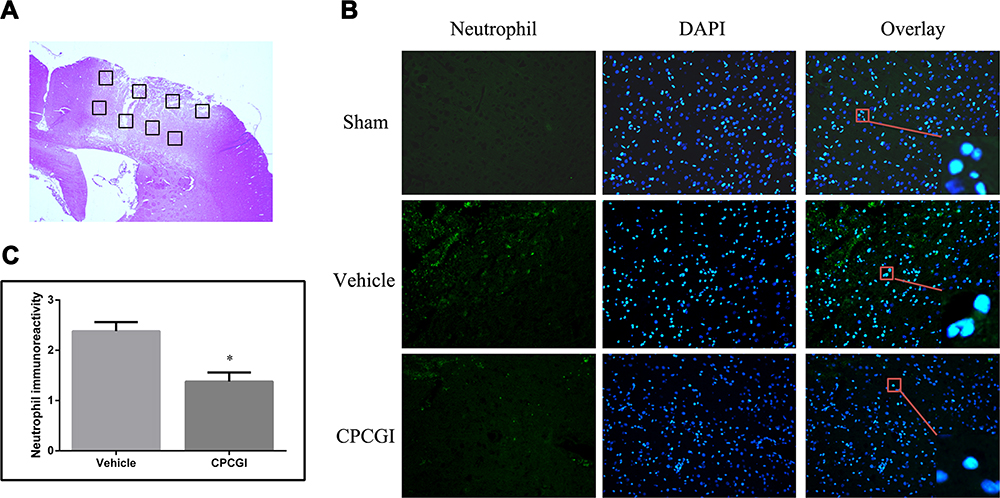 |
Figure 4 Effect of CPCGI on neutrophil infiltration in the injured brain region 72 h after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A) The area around the lesion was evaluated with immunofluorescent staining. (B) Representative neutrophil-stained sections from animals from all groups (original magnification, 200×). Green fluorescence represents the neutrophil immunoreactivity, and the blue fluorescence indicates DAPI staining. (C) The bar graphs show the neutrophil immunoreactivity in vehicle- and CPCGI-treated rats. The data are expressed as the mean ± SEM. n = 6 per group. *P < 0.05 vs the vehicle group. |
Effects of CPCGI on the Activation of Astrocytes and Microglia
The immunoreactivity of GFAP and Iba-1 represents the activation of astrocytes and microglia, respectively. The astrocytes and microglia in the brain sections were labeled by GFAP and Iba-1 immunofluorescence staining, respectively, and the results are shown in Figure 5. The brain sections showed weak GFAP and Iba-1 immunoreactivities in the sham-operated rats, and increased immunoreactivities of GFAP and Iba-1 in the brain region with traumatic injury in vehicle-treated rats (both P < 0.01). CPCGI treatment markedly reduced the immunoreactivities of GFAP and Iba-1 in the brain region with traumatic injury (*P < 0.01 and **P < 0.05 vs vehicle-treated rats, respectively).
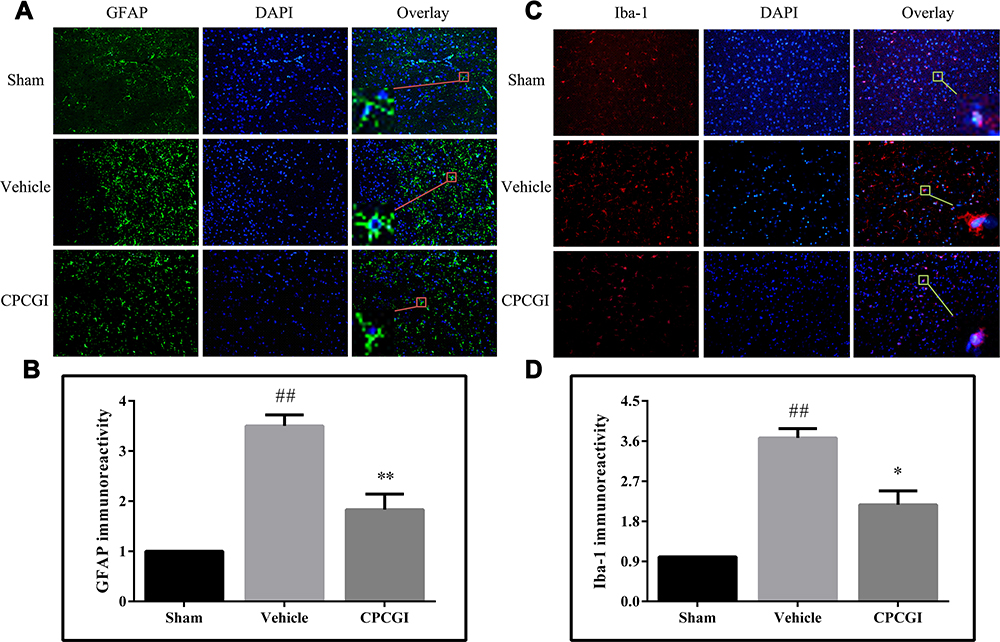 |
Figure 5 Effect of CPCGI on the activation of astrocyte and microglia in injured brain regions 72 h after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A and C) Representative GFAP and Iba-1 immunofluorescence images, respectively, of animals in all groups (original magnification, 200×). (B and D) The bar graphs revealed the GFAP and Iba-1 immunoreactivities in all groups. The data are expressed as the mean ± SEM. n = 6 per group. ##P < 0.01 vs sham-operated rats. *P < 0.05 and **P < 0.01 vs vehicle-treated rats. |
Effects of CPCGI on the Levels of PARP and NF-κB p65 in the Cytosol and Nucleus
PARP/NF-κB inflammatory signaling is reportedly involved in TBI. To evaluate the activation of PARP and NF-κB, the cytosolic and nuclear levels of PARP and NF-κB p65 were evaluated. In CCI-treated rats, the cytosolic PARP levels and the nuclear PARP and NF-κB p65 levels were increased significantly (Figure 6; all P < 0.01 vs sham-operated rats), although the cytosolic NF-κB p65 levels had no significant change. CPCGI treatment had no significant effects on the cytosolic NF-κB p65 levels. However, CPCGI markedly reduced the cytosolic and nuclear PARP levels and the nuclear NF-κB p65 levels after CCI (cytosolic and nuclear PARP: P < 0.05 and P < 0.01, respectively; nuclear NF-κB p65: P < 0.01). These data indicated the increased expression and nuclear translocation of PARP and NF-κB p65 after CCI and the reduced expression and nuclear translocation of PARP and NF-κB p65 by CPCGI treatment, suggesting the suppression of PARP/NF-κB inflammatory signaling after CCI.
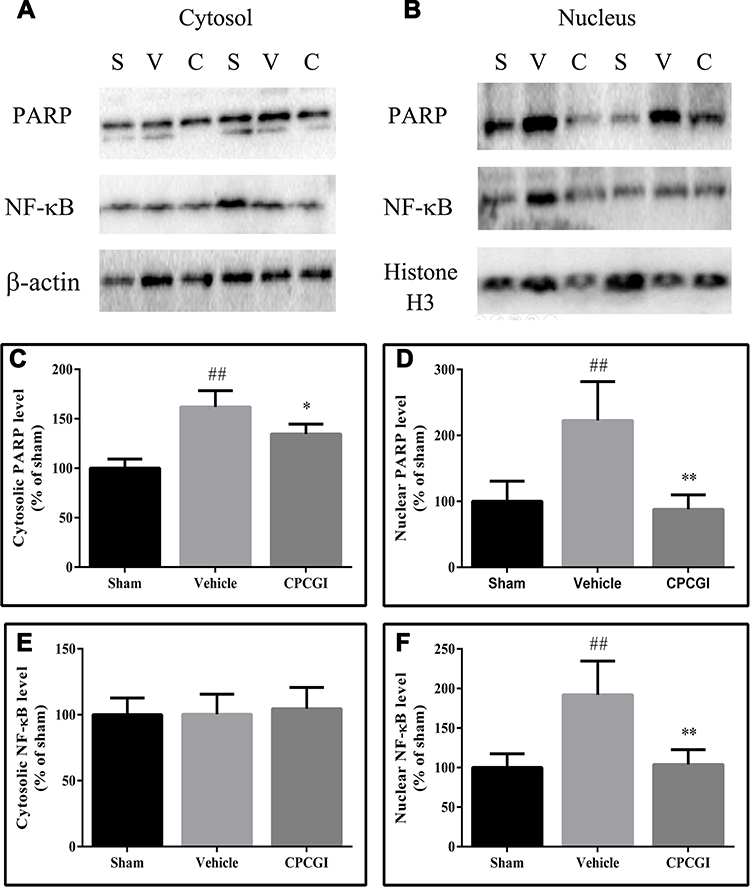 |
Figure 6 Effects of CPCGI on the cytosolic and nuclear levels of PARP and NF-κB p65 in injured brain tissues 24 h after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A and B) Western blot analysis of the cytosolic and nuclear fractions, respectively, using PARP and NF-κB p65 antibodies. (C–F) The bar graphs reflected the densitometric data from the Western blot experiments of PARP and NF-κB p65 in the cytosolic and nuclear fractions, respectively. All data are expressed as the mean ± SEM. n = 6 per group. ##P < 0.01 vs sham-operated rats. *P < 0.05 and **P < 0.01 vs vehicle-treated rats.Abbreviations: S, sham group; V, vehicle group; C, CPCGI group |
Effects of CPCGI on the Levels and Activities of Caspase-1
Caspase-1 is the effector enzyme of inflammasomes, and caspase-1 p20 is an active form of caspase-1. In this study, the cytosolic levels of pro-caspase-1 and caspase-1 p20 and the caspase-1 activity were determined, and the results are shown in Figure 7. The pro-caspase-1 and caspase-1 p20 levels and the caspase-1 activities in injured brain tissues were significantly increased 24 h after CCI (P < 0.05, P < 0.01 and P < 0.01 vs the sham-operated rats, respectively). CPCGI treatment markedly reduced the pro-caspase-1 and caspase-1 p20 levels and caspase-1 activities (P < 0.05, P < 0.05 and P < 0.01 vs vehicle-treated rats).
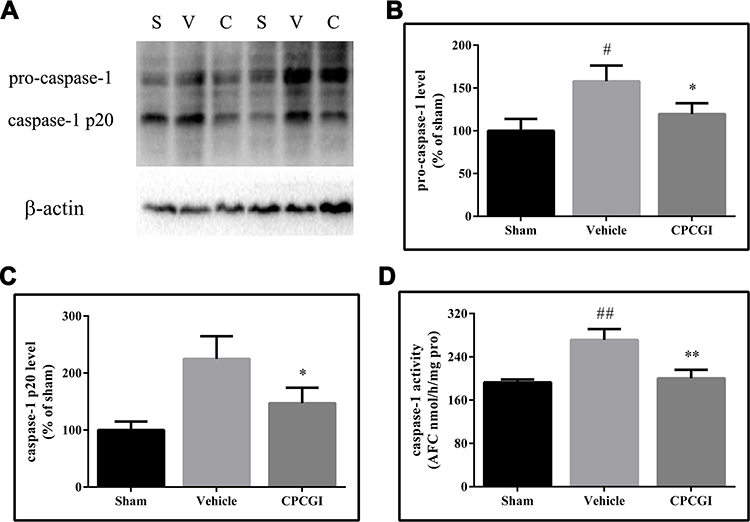 |
Figure 7 Effects of CPCGI on caspase-1 levels and caspase-1 activity in injured brain tissues 24 h after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A) Western blot analysis of the cytosolic fractions using caspase-1 antibody. (B and C) The bar graphs reflected the densitometric data from the Western blot analysis of caspase-1. (D) Caspase-1 activity. The data are presented as the mean ± SEM. #P < 0.05 and ##P < 0.01 vs sham-operated rats. n = 6 per group. *P < 0.05 and **P < 0.01 vs vehicle-treated rats. Abbreviations: S, sham group; V, vehicle group; C, CPCGI group. |
Effects of CPCGI on the Levels of Inflammatory Mediators
The protein levels of ICAM-1, TNF-α, pro-IL-1β and IL-1β in cytosol were determined by Western blot analysis, and the results are shown in Figure 8. The protein levels of ICAM-1, TNF-α, pro-IL-1β and IL-1β in the cytosol were significantly increased by CCI (ICAM-1: P < 0.01 vs the sham-operated rats; TNF-α, pro-IL-1β and IL-1β: all P < 0.05 vs the sham-operated rats). CPCGI treatment markedly reduced cytosolic ICAM-1, TNF-α, and IL-1β levels in brain tissues with traumatic injury (all P < 0.05) but had no significant effects on cytosolic pro-IL-1β levels.
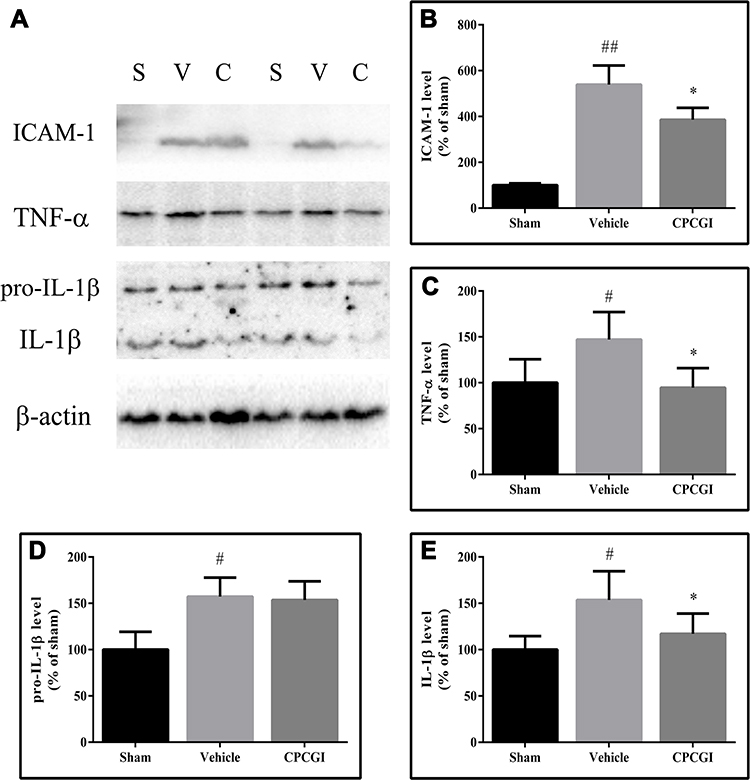 |
Figure 8 Effect of CPCGI on the levels of inflammatory factors in injured brain tissues 24 h after CCI. Vehicle or CPCGI was injected intraperitoneally 30 min post-CCI. (A) Western blot analysis of the cytosolic fractions using ICAM-1, TNF-α, pro-IL-1β and IL-1β antibodies. (B–E) The bar graphs reflected the densitometric data from the Western blot analysis of ICAM-1, TNF-α, pro-IL-1β and IL-1β in the cytosolic fractions. All the data are expressed as the mean ± SEM. n = 6 per group. #P < 0.05 and ##P < 0.01 vs sham-operated rats. *P < 0.05 vs vehicle-treated rats. Abbreviations: S, sham group; V, vehicle group; C, CPCGI group |
Discussion
In a previous study, we confirmed that CPCGI exerts protective effects against gray and white matter damage caused by CCI by stimulating Nrf2 signaling, alleviating oxidative stress and suppressing calpain overactivation. The present study revealed the following: 1) CPCGI treatment markedly enhanced the cytosolic and mitochondrial Bcl-xL levels and the mitochondrial Bcl-xL/Bax ratio, reduced the release of mitochondrial cyt c, suppressed caspase-3 activation, reduced the release of mitochondrial AIF and the nuclear translocation of AIF, and attenuated apoptotic cell death in brain regions with traumatic injury; and 2) CPCGI treatment markedly suppressed the nuclear translocation of PARP and NF-κB p65 levels, reduced pro-caspase-1 and caspase-1 p20 levels and caspase-1 activities, decreased ICAM-1, TNF-α, and IL-1β levels in brain tissues with traumatic injury, and suppressed the infiltration of neutrophils as well as the activation of astrocytes and microglia in brain regions with traumatic injury. These data suggest that CPCGI could reduce brain injury due to CCI by repressing mitochondria-mediated caspase-3-dependent and caspase-3-independent apoptotic signaling and PARP/NF-κB inflammatory signaling.
Mitochondria-mediated apoptotic signaling is reported to result in both caspase-dependent and caspase-independent apoptotic cell death.23 The Bcl-2 family of proteins, including antiapoptotic proteins (eg, Bcl-2 and Bcl-xL) and pro-apoptotic proteins (eg, Bax, Bad, and Bak), play an important role in the regulation of mitochondrial apoptotic signaling. Antiapoptotic Bcl-2 proteins can interact with pro-apoptotic Bcl-2 proteins in the mitochondrial outer membrane, and this interaction regulates the permeability of the mitochondrial outer membrane and the release of certain pro-apoptotic proteins, thus controlling apoptotic signaling. For example, Bcl-xL interacts with Bax to form Bcl-xL/Bax heterodimers, which results in the inhibition of Bax homodimerization and the suppression of apoptotic signaling.24,25 In response to apoptotic stimuli, cyt c is released from the mitochondria to the cytosol, where cyt c triggers the formation of apoptosomes composed of apoptotic protease-activating factor 1, caspase-9 and dATP, which leads to the activation of caspase-9 and caspase-3 and ultimately to caspase-3-dependent apoptotic cell death. In contrast, AIF released from the mitochondria translocates into the nucleus, where it induces internucleosomal DNA fragmentation, which leads to caspase-3-independent apoptotic cell death. It is well known that apoptotic cell death plays an important role in TBI,7 and the blockage of apoptotic signaling is one of the main therapeutic strategies for TBI.26 Mitochondrial-mediated caspase-3-dependent and caspase-3-independent apoptotic signaling is reportedly activated during TBI.27,28 In the brain tissues with traumatic injury, we found reduced Bcl-xL levels and a lower Bcl-xL/Bax ratio in the mitochondria, enhanced cyt c levels and caspase-3 activity in the cytosol and higher nuclear AIF levels. Moreover, DNA fragmentation was increased in the brain region with traumatic injury, as revealed by TUNEL staining. CPCGI treatment reversed these effects due to CCI. These data suggested that CCI activated mitochondria-mediated caspase-3-dependent and caspase-3-independent apoptotic signaling and that CPCGI suppressed mitochondrial apoptotic signaling after TBI.
The inflammatory response is another important detrimental event that occurs after TBI.29 Inflammation reportedly plays a critical role in secondary injury after TBI because it not only directly results in cell damage but also promotes other harmful events, such as oxidative stress, apoptosis, blood-brain barrier disruption and brain edema.30 The inflammatory response to TBI is characterized by the rapid activation of astrocytes and microglia, the mass production of pro-inflammatory mediators, and the infiltration of circulating inflammatory cells, such as neutrophils, macrophages, T cells, and other cells, into the traumatic parenchyma. PARP is involved in many vital cellular functions, such as DNA repair, cell proliferation and death, chromatin function and genomic stability.31 NF-κB is a critical transcription factor composed of the subunits p65 and p50. Under physiological conditions, NF-κB exists in the cytoplasm as a complex with its inhibitory protein, inhibitory κB (IκB). In the presence of inflammatory stimuli, IκB is degraded, which leads to the release of active NF-κB. Active NF-κB is translocated into the nucleus and stimulates the transcriptional activation of potentially deleterious pro-inflammatory genes.32 PARP acts as a transcriptional coregulator to facilitate the expression of NF-κB-driven inflammatory genes, such as ICAM-1, TNF-α, and IL-1β.33,34 During cerebral ischemia, PARP inhibition downregulates the expression of NF-κB-driven inflammatory genes and protects brain against ischemia.35,36 During TBI, PARP is activated by the increase in poly(ADP-ribose)-modified proteins during TBI.9,10,37 Furthermore, PARP inhibition suppresses the nuclear translocation of NF-κB p65, an active subunit of NF-κB, lessens the activation of microglia, and protects the brain against TBI.10,37,38 This study revealed the enhanced nuclear translocation of PARP and NF-κB p65 from the cytosol, upregulated the expression of NF-κB-driven inflammatory genes, increased neutrophil infiltration and the activation of astrocytes and microglia during TBI. In addition, the expression and activation of caspase-1, an effector of inflammasomes that processes pro-IL-1β to form bioactive IL-1β, were increased in brain tissues with traumatic injury. CPCGI treatment reversed these effects due to TBI. Our data combined with other evidence suggested that CPCGI could downregulate the expression of inflammatory mediators and reduce the inflammatory response due to TBI through suppressing PARP/NF-κB-mediated inflammatory signaling.
Conclusion
In summary, this study found that CPCGI treatment suppressed mitochondria-mediated caspase-3-dependent and caspase-3-independent apoptotic signaling and PARP/NF-κB inflammatory signaling and attenuated apoptotic cell death and the activation of inflammatory cells in brain tissues with traumatic injury in a rat model of CCI. A previous study revealed that CPCGI treatment reduced white matter and gray matter damage due to TBI through activating antioxidative signaling. These data suggest that the possible pharmacological mechanisms of CPCGI against TBI might involve antiapoptotic, anti-inflammatory and antioxidative actions. Our study mainly investigated the short-term protective effects and mechanisms of CPCGI on TBI, but data on the long-term protective effects and therapeutic window of CPCGI are currently lacking. Further studies are necessary to determine the long-term protective effects and therapeutic window of CPCGI on TBI and elucidate how CPCGI affects apoptotic signaling and PARP/NF-κB inflammatory signaling during TBI.
Data Sharing Statement
All the data are published in this article, and the materials can be obtained from the corresponding author upon reasonable request.
Acknowledgments
This study was supported by the Platform Construction of Basic Research and Clinical Translation of Nervous System Injury (PXM2020_026280_000002, Beijing Municipal Health Commission), Scientific Research and Cultivation Fund of Beijing Neurosurgical Institute (2020002) and grants from Buchang Pharmaceuticals Co., Ltd.
Disclosure
All authors have no conflicts of interest to declare.
References
1. McIntosh TK, Juhler M, Wieloch T. Novel pharmacologic strategies in the treatment of experimental traumatic brain injury: 1998. J Neurotrauma. 1998;15(10):731–769. doi:10.1089/neu.1998.15.731
2. McIntosh TK, Saatman KE, Raghupathi R, et al. The Dorothy Russell Memorial Lecture. The molecular and cellular sequelae of experimental traumatic brain injury: pathogenetic mechanisms. Neuropathol Appl Neurobiol. 1998;24(4):251–267. doi:10.1046/j.1365-2990.1998.00121.x
3. Stein DM, Feather CB, Napolitano LM. Traumatic brain injury advances. Crit Care Clin. 2017;33(1):1–13. doi:10.1016/j.ccc.2016.08.008
4. Aertker BM, Bedi S, Cox CS. Strategies for CNS repair following TBI. Exp Neurol. 2016;275(Pt 3):411–426. doi:10.1016/j.expneurol.2015.01.008
5. Rana A, Singh S, Sharma R, Kumar A. Traumatic brain injury altered normal brain signaling pathways: implications for novel therapeutics approaches. Curr Neuropharmacol. 2019;17(7):614–629. doi:10.2174/1570159X16666180911121847
6. Quillinan N, Herson PS, Traystman RJ. Neuropathophysiology of brain injury. Anesthesiol Clin. 2016;34(3):453–464. doi:10.1016/j.anclin.2016.04.011
7. Akamatsu Y, Hanafy KA. Cell death and recovery in traumatic brain injury. Neurotherapeutics. 2020;17(2):446–456. doi:10.1007/s13311-020-00840-7
8. Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72(3):355–362. doi:10.1001/jamaneurol.2014.3558
9. Besson VC. Drug targets for traumatic brain injury from poly(ADP-ribose)polymerase pathway modulation. Br J Pharmacol. 1992;119(3):695–704. doi:10.1111/j.1476-5381.2009.00229.x
10. Stoica BA, Loane DJ, Zhao Z, et al. PARP-1 inhibition attenuates neuronal loss, microglia activation and neurological deficits after traumatic brain injury. J Neurotrauma. 2014;31(8):758–772. doi:10.1089/neu.2013.3194
11. Wang M, Zhang Y, Feng L, et al. Compound porcine cerebroside and ganglioside injection attenuates cerebral ischemia-reperfusion injury in rats by targeting multiple cellular processes. Neuropsychiatr Dis Treat. 2017;13:927–935. doi:10.2147/NDT.S129522
12. Wang X, Zhao J. Neuroprotective effect of CPCGI on Alzheimer’s disease and its mechanism. Mol Med Rep. 2020;21(1):115–122.
13. Niu F, Qian K, Qi H, et al. CPCGI Reduces Gray and White Matter Injury by Upregulating Nrf2 Signaling and Suppressing Calpain Overactivation in a Rat Model of Controlled Cortical Impact. Neuropsychiatr Dis Treat. 2020;16:1929–1941. doi:10.2147/NDT.S266136
14. Bilgen M. A new device for experimental modeling of central nervous system injuries. Neurorehabil Neural Repair. 2005;19(3):219–226. doi:10.1177/1545968305278635
15. Onyszchuk G, Al-Hafez B, He YY, Bilgen M, Berman NE, Brooks WM. A mouse model of sensorimotor controlled cortical impact: characterization using longitudinal magnetic resonance imaging, behavioral assessments and histology. J Neurosci Methods. 2007;160(2):187–196. doi:10.1016/j.jneumeth.2006.09.007
16. Pei DS, Wang XT, Liu Y, et al. Neuroprotection against ischaemic brain injury by a GluR6-9c peptide containing the TAT protein transduction sequence. Brain. 2006;129(Pt 2):465–479. doi:10.1093/brain/awh700
17. Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. 2006;143(4):965–974. doi:10.1016/j.neuroscience.2006.09.014
18. O’Hare MJ, Hou ST, Morris EJ, et al. Induction and modulation of cerebellar granule neuron death by E2F-1. J Biol Chem. 2000;275(33):25358–25364. doi:10.1074/jbc.M001725200
19. Stefanis L, Park DS, Yan CY, et al. Induction of CPP32-like activity in PC12 cells by withdrawal of trophic support. Dissociation from apoptosis. J Biol Chem. 1996;271(48):30663–30671. doi:10.1074/jbc.271.48.30663
20. Yam PS, Dewar D, McCulloch J. Axonal injury caused by focal cerebral ischemia in the rat. J Neurotrauma. 1998;15(6):441–450. doi:10.1089/neu.1998.15.441
21. Derugin N, Wendland M, Muramatsu K, et al. Evolution of brain injury after transient middle cerebral artery occlusion in neonatal rats. Stroke. 2000;31(7):1752–1761. doi:10.1161/01.STR.31.7.1752
22. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119(3):493–501. doi:10.1083/jcb.119.3.493
23. Kim R, Emi M, Tanabe K. Caspase-dependent and -independent cell death pathways after DNA damage (Review). Oncol Rep. 2005;14(3):595–599.
24. Hardwick JM, Soane L. Multiple functions of BCL-2 family proteins. Cold Spring Harb Perspect Biol. 2013;5(2):a008722–a008722. doi:10.1101/cshperspect.a008722
25. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20(3):175–193.
26. Raghupathi R, Graham DI, McIntosh TK. Apoptosis after traumatic brain injury. J Neurotrauma. 2000;17(10):927–938. doi:10.1089/neu.2000.17.927
27. Slemmer JE, Zhu C, Landshamer S, et al. Causal role of apoptosis-inducing factor for neuronal cell death following traumatic brain injury. Am J Pathol. 2008;173(6):1795–1805. doi:10.2353/ajpath.2008.080168
28. Ng SY, Lee AYW. Traumatic brain injuries: pathophysiology and potential therapeutic targets. Front Cell Neurosci. 2019;13:528.
29. Simon DW, McGeachy MJ, Bayir H, Clark RS, Loane DJ, Kochanek PM. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat Rev Neurol. 2017;13(3):171–191.
30. Forstner P, Rehman R, Anastasiadou S, et al. Neuroinflammation after traumatic brain injury is enhanced in activating transcription factor 3 mutant mice. J Neurotrauma. 2018;35(19):2317–2329. doi:10.1089/neu.2017.5593
31. Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. doi:10.1124/pr.54.3.375
32. Ridder DA, Schwaninger M. NF-κB signaling in cerebral ischemia. Neuroscience. 2009;158(3):995–1006. doi:10.1016/j.neuroscience.2008.07.007
33. Hassa PO, Hottiger MO. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-κB in inflammatory disorders. Cell Mol Life Sci. 2002;59(9):1534–1553. doi:10.1007/s00018-002-8527-2
34. Chiarugi A, Moskowitz MA. Poly(ADP-ribose) polymerase-1 activity promotes NF-κB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem. 2003;85(2):306–317. doi:10.1046/j.1471-4159.2003.01684.x
35. Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8(1):96–103. doi:10.1016/j.coph.2007.10.005
36. Strosznajder RP, Czubowicz K, Jesko H, Strosznajder JB. Poly(ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol Neurobiol. 2010;41(2–3):187–196. doi:10.1007/s12035-010-8124-6
37. d’Avila JC, Lam TI, Bingham D, et al. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation. 2012;9:31. doi:10.1186/1742-2094-9-31
38. Tao X, Chen X, Hao S, et al. Protective actions of PJ34, a poly(ADP-ribose)polymerase inhibitor, on the blood-brain barrier after traumatic brain injury in mice. Neuroscience. 2015;291:26–36. doi:10.1016/j.neuroscience.2015.01.070
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.