")
Back to Journals » OncoTargets and Therapy » Volume 11
Antiangiogenic effects of AA-PMe on HUVECs in vitro and zebrafish in vivo
Authors Jing Y, Wang G, Xiao Q, Zhou Y, Wei Y, Gong Z
Received 22 November 2017
Accepted for publication 10 February 2018
Published 4 April 2018 Volume 2018:11 Pages 1871—1884
DOI https://doi.org/10.2147/OTT.S157747
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Yue Jing,1,2,* Gang Wang,1,* Qi Xiao,1 Yachun Zhou,1 Yingjie Wei,3 Zhunan Gong1
1Center for New Drug Research and Development, College of Life Science, Nanjing Normal University, Nanjing, China; 2Central Laboratory of Stomatology, Nanjing Stomatological Hospital, Medical School of Nanjing University, Nanjing, China; 3Key Laboratory of Oral Drug Delivery System of Chinese Materia Medica of State Administration of Traditional Chinese Medicine, Jiangsu Branch of China Academy of Chinese Medical Science, Nanjing, China
*These authors contributed equally to this work
Abstract: Angiogenesis plays a vital role in many physiological and pathological processes and several diseases are connected with its dysregulation. Asiatic acid (AA) has demonstrated anticancer properties and we suspect this might be attributable to an effect on angiogenesis. A modified derivative of AA, N-(2α,3β,23-acetoxyurs-12-en-28-oyl)-L-proline methyl ester (AA-PMe), has improved efficacy over its parent compound, but its effect on blood vessel development remains unclear.
Methods: In this study, we investigated the antiangiogenic activity of AA and AA-PMe in zebrafish embryos and human umbilical vein endothelial cells (HUVECs). First of all, we treated HUVECs with increasing concentrations of AA-PMe or AA, with or without vascular endothelial growth factor (VEGF) present, and assessed cell viability, tube formation, and cell migration and invasion. Quantitative real-time polymerase chain reaction and Western blot analysis were later used to determine the role of vascular endothelial growth factor receptor 2 (VEGFR2)-mediated signaling in AA-PMe inhibition of angiogenesis. We extended these studies to follow angiogenesis using Tg(fli:EGFP) transgenic zebrafish embryos. For these experiments, embryos were treated with varying concentrations of AA-PMe or AA from 24 to 72 hours postfertilization prior to morphological observation, angiogenesis assessment, and endogenous alkaline phosphatase assay. VEGFR2 expression in whole embryos following AA-PMe treatment was also determined.
Results: We found AA-PMe decreased cell viability and inhibited migration and tube formation in a dose-dependent manner in HUVECs. Similarly, AA-PMe disrupted the formation of intersegmental vessels, the dorsal aorta, and the posterior cardinal vein in zebrafish embryos. Both in vitro and in vivo AA-PMe surpassed AA in its ability to block angiogenesis by suppressing VEGF-induced phosphorylation of VEGFR2 and disrupting downstream extracellular regulated protein kinase and AKT signaling.
Conclusion: For the first time, this study reveals that AA-PMe acts as a potent VEGFR2 kinase inhibitor and exerts powerful antiangiogenic activity, suggesting it to be a promising therapeutic candidate for further research.
Keywords: AA-PMe, angiogenesis inhibitor, zebrafish, HUVEC
Introduction
Angiogenesis, the process by which new blood vessels sprout from preexisting endothelium, is essential for various biological processes including embryonic vascular development and differentiation, wound healing, and organ regeneration.1,2 Concurrently, angiogenesis regulates multiple pathological processes including tumor progression,1–6 infection,2 ischemic and inflammatory diseases,2 metastasis disorder,1 age-related macular degeneration,3,5 and diabetic retinopathy.1,5,7 Angiogenesis normally occurs under conditions in which pro- and antiangiogenic factors are in balance, maintaining homeostasis. In contrast, the tumor microenvironment promotes angiogenesis: hypoxic conditions within the tumor spur the expression and release of growth factors and cytokines that induce proliferation and migration of endothelial cells8,9 and stimulate blood vessel growth to increase the local blood supply.10
Vascular endothelial growth factor (VEGF) is a critical angiogenic factor well-documented for its mitogenic activity in endothelial cells and its role in promoting normal physiological as well as pathological neovascularization, such as that occurring in tumor angiogenesis.3,11 VEGF activates receptor tyrosine kinases in endothelial cells by binding one of two receptors: vascular endothelial growth factor receptor 1 (VEGFR1) or 2 (VEGFR2).12 VEGFR2 serves as the primary receptor and activates downstream signaling molecules including focal adhesion kinase, extracellular signal-regulated kinase (ERK), AKT/protein kinase B (PKB), mammalian target of rapamycin, and ribosomal protein S6 kinase (p70S6K).13 Under normal conditions, this results in the growth, migration, differentiation, and survival of endothelial cells in preexisting vasculature, but its activation can also be hijacked to drive pathologic angiogenesis.14 The VEGFR2 signaling pathway is thus a promising target for inhibiting angiogenesis in disease states.
At present, several VEGFR inhibitors have been used in Phase I and II clinical trials, including drugs aimed at VEGF or VEGFRs:15 bevacizumab, sunitinib malate, and sorafenib have all been approved by the US Food and Drug Administration for cancer treatment.16,17 However, clinical use of these treatments is restricted because they carry the risk of severe side effects including hypertension, bleeding, and gastrointestinal perforation.18,19 To identify safer and more efficient treatments for angiogenesis-related diseases attention is turning to small molecule therapeutic strategies targeting signal transduction pathways downstream of VEGFR2.
Consequently, there is increasing interest in natural products that contain a variety of antiangiogenic compounds and have a history of safe usage, including many Chinese herbs. Asiatic acid (2α,3β,23-trihydroxyurs-12-ene-28-oic acid; AA), a pentacyclic triterpenoid derived from the tropical medicinal plant Centella asiatica,20 is one such promising candidate. This study examines the antiangiogenic efficacy of N-(2α,3β,23-acetoxyurs-12-en-28-oyl)-L-proline methyl ester (AA-PMe), which was semi-synthesized in our laboratory with the goal of improving its antiangiogenic efficacy. Unmodified AA has been reported to aid in wound healing21,22 as well as inhibit UV-induced photoaging,23 glutamate- and β-amyloid-induced neurotoxicity,24–27 and hepatofibrosis.28 It has also demonstrated anticancer29–33 and antiangiogenic efficacy.34 Based on the antiangiogenic activity of AA,34 we researched the effects of AA-PMe on in vitro and in vivo angiogenesis and sought to determine the underlying mechanism.
In human umbilical vein endothelial cells (HUVECs) treated with AA-PMe we observed apoptosis induction as well as inhibition of cell migration and tube formation, so we decided to further explore these effects in an in vivo system. Zebrafish embryos are an established and efficient model for investigating vascular development and represent a practical and efficient system for screening angiogenesis drugs.35 The recent development of the transgenic zebrafish line Tg(fli1:EGFP), which carries a genetic modification that fluorescently labels blood vessels, represents a useful genetic model to accelerate angiogenesis research.36,37 We utilized this transgenic line to examine the impact of AA-PMe on angiogenesis and found a profound inhibitory effect. Our data further indicate that AA-PMe exerts its antiangiogenic activity through inhibition of VEGFR2-mediated signaling.
Materials and methods
Ethics statement
All animal experiments in this investigation were conducted in compliance with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication no 85-23, revised 1996) and approved by the Animal Care and Use Committee of Nanjing Normal University, China (permit number 2090658).
Chemicals and reagents
AA was extracted from C. asiatica (L.) Urban and structurally modified in our laboratory to produce AA-PMe.20 AA and AA-PMe were both dissolved in 1% dimethyl sulfoxide (DMSO; Sigma-Aldrich Co, St Louis, MO, USA) and their structures are presented in Figure 1. Endogenous alkaline phosphatase (EAP) staining was tested by a phosphatase substrate kit (Pierce; Thermo Fisher Scientific, Waltham, MA, USA). Recombinant human VEGF was purchased from Thermo Fisher Scientific. Primary antibodies for total Akt, pAkt-ser473, total ERK1/2, p-pERK1/2, total VEGFR2, and pVEGFR2-Tyr1175 were brought from Cell Signaling Technology (Danvers, MA, USA). GAPDH and tubulin antibody were from Abcam (Cambridge, UK).
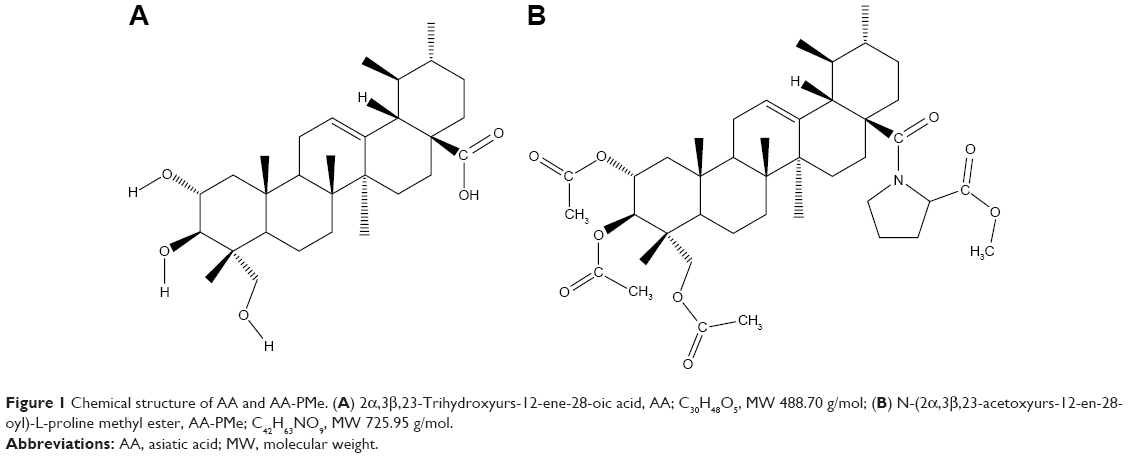 | Figure 1 Chemical structure of AA and AA-PMe. (A) 2α,3β,23-Trihydroxyurs-12-ene-28-oic acid, AA; C30H48O5, MW 488.70 g/mol; (B) N-(2α,3β,23-acetoxyurs-12-en-28-oyl)-L-proline methyl ester, AA-PMe; C42H63NO9, MW 725.95 g/mol. |
Cell culture
HUVECs were purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in complete growth medium (Dulbecco’s Modified Eagle’s Medium: F12) supplemented with 10% fetal bovine serum (FBS; Thermo Fisher Scientific), 0.1 mg/mL heparin, and 0.03%–0.05% mg/mL endothelial cell growth supplement.38 Cells were incubated at 37°C in a humidified atmosphere with 5% CO2.
Zebrafish
Transgenic zebrafish Tg(fli:EGFP) with enhanced green fluorescent protein (EGFP) expressing endothelial cells and wild-type zebrafish (Tuebingen line) were obtained from Model Animal Research Center of Nanjing University. Adult zebrafish were maintained at 28.5°C, pH 7±0.2 H2O in a 14:10 hour light/dark photoperiod and fed with live brine shrimp once a day and dry food twice a day.39 Embryos were produced by pairwise mating using a fish hatch box and were subsequently maintained at 28.5°C in E3 embryo medium: 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, and 0.33 mM MgSO4 in dH2O with added antifungal solvent (0.01% methylene blue).40 Embryos exhibiting normal development were dechorionated by forceps prior to drug treatment. All zebrafish studies were approved by the Institutional Animal Care and Use Committee at the Nanjing University of Technology.
In vitro viability assay
Proliferation assays were performed as previously described.41 Briefly, 8000 HUVECs/well were seeded in 96-well plates and allowed to attach for 12 hours. Cells were then treated for 24 hours with various concentrations of AA and AA-PMe or 0.1% DMSO vehicle control. Cell proliferation was measured by cell counting kit (CCK)-8 colorimetric assay according to the manufacturer’s protocol (Vazyme Biotech, Nanjing, China).
In vitro migration assay
HUVEC migration assay was performed using the wound healing method as previously described.42–44 Briefly, HUVECs were allowed to grow to full confluence in 6-well plates, manually disrupted by pipette tips, and washed twice with media to remove detached cells. Photomicrographs of initial wounds were taken at 100× magnification using a phase-contrast microscope and wound distance was measured using AxioVision Rel.4.7 software (Carl Zeiss Meditec AG, Jena, Germany). Cells were then treated with VEGF (10 ng/mL) and DMSO vehicle or the indicated concentrations of AA-PMe or AA and photomicrographs were taken at the indicated time points. After vehicle-treated HUVECs migrated to completely close the wound the experiment was terminated.
|

In vitro Transwell invasion assay
To determine cell invasion, Transwell plates (Costar; Corning Incorporated, Corning, NY, USA) pre-coated with Matrigel (BD Biosciences, San Jose, CA, USA) were placed on 24-well plates and the lower chamber was filled with 600 μL culture medium supplemented with VEGF (10 ng/mL) as a chemoattractant and vehicle or the indicated concentrations of AA-PMe or AA were added. Then, 1×105 HUVECs in 200 μL RPMI containing 1% FBS were added to the upper chamber. After 24 hours, non-migrated cells were removed with cotton swabs while cells that traversed the membrane and spread to the lower surface of the filters were fixed with 90% alcohol for 30 minutes at room temperature and stained with 0.1% crystal violet for visualization. Then, 100× images were captured on a phase-contrast microscope, imaged using AxioVision Rel.4.7 software, and invasive cells were quantified by manual counting. Thereafter, invasive cells were decolored with 33% acetic acid for 10 minutes. Samples were transferred to 96-well plates and the absorbance at 570 nm, indicative of the number of cells present, was measured with a microplate reader (Thermo Fisher Scientific). The percentage of invasion inhibition by AA-PMe or AA was calculated by dividing the difference between the absorbance of treated and untreated groups and multiplying by 100:
|

In vitro tube formation assay
To examine the effect of AA-PMe and AA on in vitro angiogenesis tube formation assays were performed as previously described.45 Briefly, 150 μL/well growth factor-reduced Matrigel was added to pre-chilled 24-well plates and polymerized for 45 minutes at 37°C. HUVECs (4×104) in complete media were then added to the Matrigel-coated plates. In the first part, varying concentrations of DMSO, AA-PMe, or AA were either added immediately or 6 hours after cells were seeded to allow initial capillary network formation. In the second part, varying concentrations of DMSO, AA-PMe, or AA were added 10 hours after HUVECs seeding. For further verifying the effect of AA-PMe on VEGF-stimulated tube formation, in the third experiment, HUVECs were washed twice with media containing 0.5% serum before seeding and plated with or without 10 ng/mL purified VEGF (Cell Signaling Technology) added in addition to AA-PMe or AA. For all experiments, tubular structures were photographed after 6 hours of treatment at 100× magnification using a phase-contrast microscope and were measured using AxioVision Rel.4.7 software.
Quantitative EAP assay
Quantitative EAP assay was performed as previously described using a Pierce phosphatase substrate kit (Thermo Fisher Scientific) according to the manufacturer’s protocol.46 Briefly, one 24-hour postfertilization (hpf) zebrafish embryo was added to each well of a 96-well plate, in 100 μL E3 embryo medium. DMSO vehicle or various concentrations of AA and AA-PMe were added and embryos were incubated at 28.5°C for 48 hours. Embryos at 72 hpf were treated with increasing concentrations of ethanol for dehydration purpose.
Embryos were washed three times with diethanolamine buffer and stained according to the manufacturer’s protocol; the reaction was stopped by the addition of 50 μL 2M NaOH and optical density was measured at 405 nm using a microplate reader. Percent vessel formation was calculated as the difference in optical density compared to the untreated control.
|
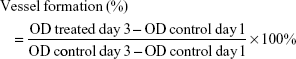
Assessment of blood vessel changes in whole zebrafish embryos
Following drug treatment, zebrafish embryos were anesthetized with 0.016% tricaine (Sigma-Aldrich Co). Embryo intersegmental blood vessel (ISV) and subintestinal vessel (SIV) plexus development were observed and imaged at 48 and 72 hpf, respectively (100× magnification), using a fluorescence microscope (IX71; Olympus Corporation, Tokyo, Japan).
Western blot analysis
The effects of AA-PMe and AA on intracellular signaling cascades were determined by Western blot. Zebrafish embryos were treated with DMSO control or the indicated concentrations of AA-PMe or AA from 24 to 72 hpf at 28.5°C; HUVEC cells were treated with AA-PMe or AA for 24 hours at 37°C. Cells and whole embryos were lysed for 30 minutes on ice in RIPA buffer (0.01% phenylmethane sulfonyl fluoride [Sigma-Aldrich Co], 150 mM NaCl, 50 mM Tris [pH =8], 0.1% sodium dodecyl sulfate, 0.2% ethylenediaminetetraacetic acid, 1% Triton X-100, 1% sodium deoxycholate), supplemented with protease and phosphatase inhibitors (Hoffman-La Roche Ltd, Basel, Switzerland). Lysates were cleared by centrifugation at 12,000 rpm at 4°C for 15 minutes and protein concentration was determined by bicinchoninic acid assay.
For each sample, 30–50 μg protein was run on a 10% agarose gel, then transferred to polyvinylidene fluoride membranes (Merck Millipore, Billerica, MA, USA) using a semidry transfer system (Bio-Rad Laboratories Inc, Hercules, CA, USA). Membranes were blocked with 5% nonfat milk in Tris buffered saline (TBST) containing 0.05% Tween-20 for 1 hour at room temperature, washed, and incubated overnight with primary antibodies (R&D) at recommended dilutions in TBST milk. After washing, blots were probed with horseradish peroxidase-conjugated secondary antibody (Abcam) for 1 hour, washed, and protein expression was detected by chemiluminescence using a Tanon-3500R image system (Tanon, Shanghai, China).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Transcriptional effects of AA and AA-PMe on VEGFR2 were determined by qRT-PCR. Embryos and HUVECs were treated as described for Western blots; then, total mRNA was extracted using Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Extracted RNA integrity was assessed by agarose-gel electrophoresis and quantified by OD260/OD280 absorbance. cDNA was reverse-transcribed from RNA using a PrimeScript™ RT reagent kit (TaKaRa, Kyoto, Japan) and qRT-PCR was performed with the SYBR PrimeScript RT-PCR kit (TaKaRa) on a StepOne Plus™ system (Thermo Fisher Scientific) according to the manufacturer’s instructions. Primer sequences are listed in Table 1. mRNA expression was calculated by the ΔΔCt method and quantified relative to GAPDH mRNA.
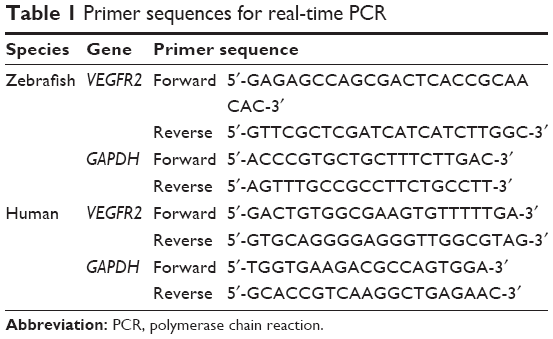 | Table 1 Primer sequences for real-time PCR |
Statistical analysis
All experiments were performed at least three times and values are given as mean ± SEM. Data were analyzed using GraphPad Prism 6.0 software (GraphPad Software, Inc, La Jolla, CA, USA) and statistical significance was assessed by T-test. P-values <0.05 were considered significant.
Results
AA-PMe decreases endothelial cell viability
To determine the effect of AA-PMe on endothelial cells HUVECs were treated with various doses of AA-PMe or AA (5, 10, and 25 μM), and the degree of cell death was determined at various time points (24, 48, and 72 hours) by CCK-8 assay. Both compounds demonstrated dose-dependent effects on cell viability over time, but the inhibitory effect of AA-PMe was much stronger (Figure 2A and B). It was observed that 10 and 25 μM AA-PMe reduced viability by as much as 20% after 24 hours, and after 72 hours viability decreased in response to all concentrations, falling by 23.0%–39.7% in a dose-dependent manner. AA significantly decreased viability by 20.8% and 24.1% after 48 and 72 hours only at its highest dose of 25 μM.
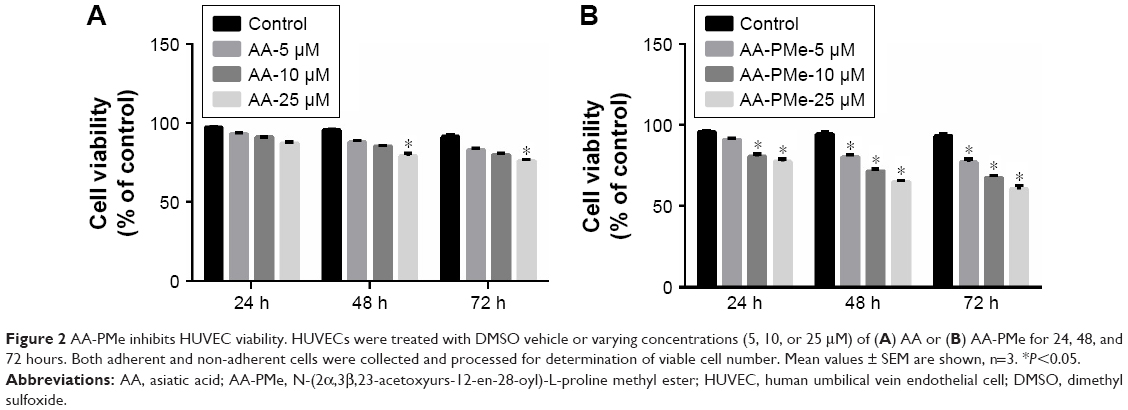 | Figure 2 AA-PMe inhibits HUVEC viability. HUVECs were treated with DMSO vehicle or varying concentrations (5, 10, or 25 μM) of (A) AA or (B) AA-PMe for 24, 48, and 72 hours. Both adherent and non-adherent cells were collected and processed for determination of viable cell number. Mean values ± SEM are shown, n=3. *P<0.05. |
AA-PMe inhibits VEGF-stimulated chemotactic motility and capillary structure formation in HUVECs
EC migration and invasion is essential for neo-angiogenesis and critical for tumor growth.47 VEGF is the most important pro-angiogenic factor and is known to enhance proliferation, survival, and capillary tube formation by endothelial cells.14 Accordingly, we investigated the effect of AA-PMe and AA treatment on VEGF-stimulated endothelial cell migration and invasion using wound healing and Transwell assays. Similar to our cell viability assays, both AA-PMe and AA inhibited wound healing and Matrigel invasion compared to VEGF-only controls, but AA-PMe showed a much stronger effect in both settings. After 6 hours treatment with 5, 10, and 25 μM doses, AA-PMe decreased migration distance by 44%, 62%, and 66% compared to 19%, 48%, and 57% migration inhibition by the same concentrations of AA (Figure 3A and B). The decrease in migration distance was even more pronounced after 12 hours, with AA-PMe treatment reducing the migrated distance by 34%, 56%, and 69%, compared to 25%, 38%, and 43% reductions by AA. Transwell experiments yielded even more striking results: after 24 hours, 5, 10, and 25 μM AA-PMe inhibited HUVEC invasion through Matrigel by 44%, 66%, and 83%, respectively, compared to 27%, 41%, and 54% inhibition by AA at the same concentrations (Figure 4A–C).
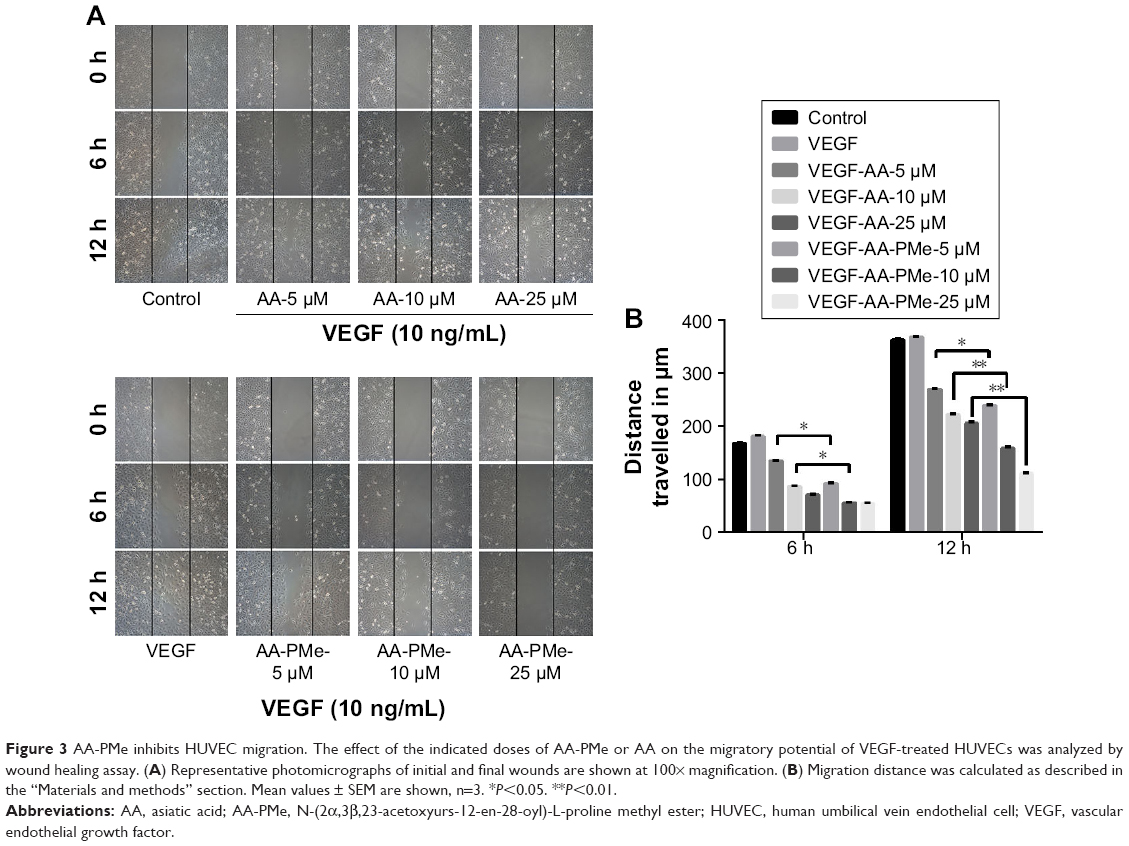 | Figure 3 AA-PMe inhibits HUVEC migration. The effect of the indicated doses of AA-PMe or AA on the migratory potential of VEGF-treated HUVECs was analyzed by wound healing assay. (A) Representative photomicrographs of initial and final wounds are shown at 100× magnification. (B) Migration distance was calculated as described in the “Materials and methods” section. Mean values ± SEM are shown, n=3. *P<0.05. **P<0.01. |
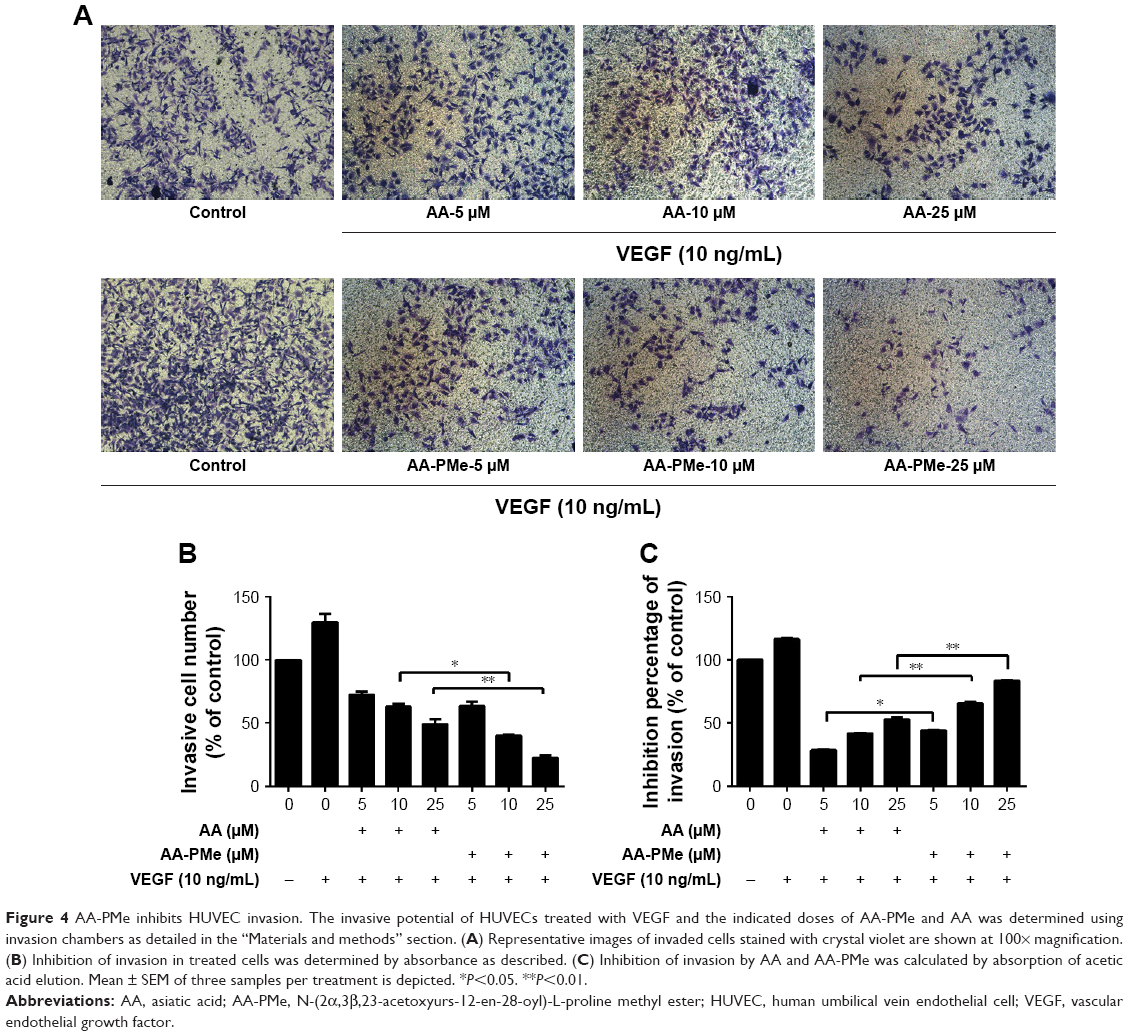 | Figure 4 AA-PMe inhibits HUVEC invasion. The invasive potential of HUVECs treated with VEGF and the indicated doses of AA-PMe and AA was determined using invasion chambers as detailed in the “Materials and methods” section. (A) Representative images of invaded cells stained with crystal violet are shown at 100× magnification. (B) Inhibition of invasion in treated cells was determined by absorbance as described. (C) Inhibition of invasion by AA and AA-PMe was calculated by absorption of acetic acid elution. Mean ± SEM of three samples per treatment is depicted. *P<0.05. **P<0.01. |
Another crucial step during neo-angiogenesis is the formation and merging of endothelial cell tubes to form a complex network of blood vessels. To understand the effects of AA-PMe and AA on early tube formation we cultured HUVECs in three-dimensional Matrigel and examined their structure after 6 hours. While untreated HUVECs formed a massive network of well-branched tubes after 6 hours, this was disrupted by treatment with 5–25 μM AA-PMe in a dose-dependent manner; however, AA had only minor effects on network formation even at 25 μM (Figure 5A). AA-PMe treatment also decreased the branch number by ~17%, 46%, and 65% at 5, 10, and 25 μM concentrations. Despite its weaker ability to disrupt tube formation, AA treatment did reduce branching by 10%, 30%, and 42% at the same doses (Figure 5D).
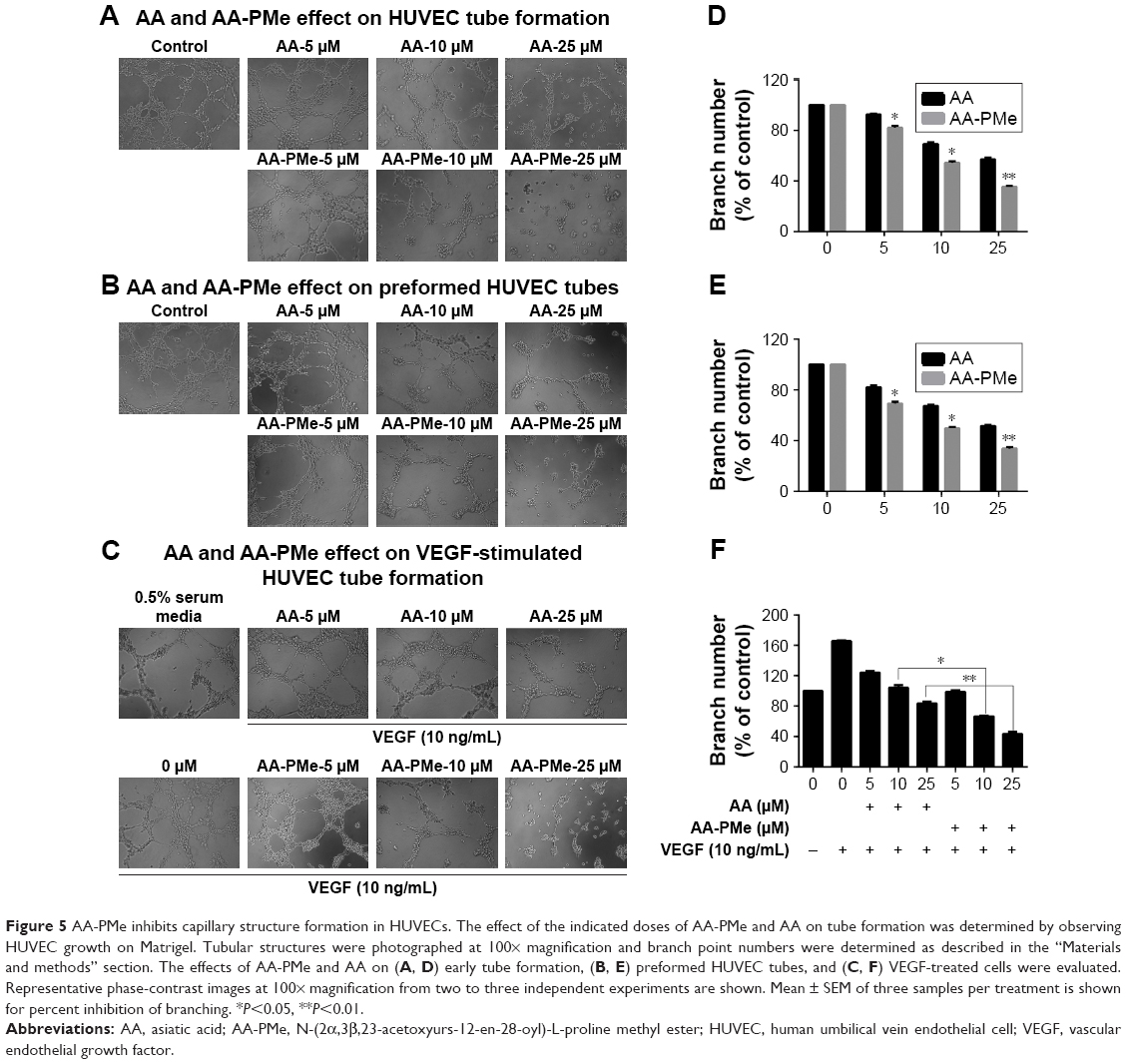 | Figure 5 AA-PMe inhibits capillary structure formation in HUVECs. The effect of the indicated doses of AA-PMe and AA on tube formation was determined by observing HUVEC growth on Matrigel. Tubular structures were photographed at 100× magnification and branch point numbers were determined as described in the “Materials and methods” section. The effects of AA-PMe and AA on (A, D) early tube formation, (B, E) preformed HUVEC tubes, and (C, F) VEGF-treated cells were evaluated. Representative phase-contrast images at 100× magnification from two to three independent experiments are shown. Mean ± SEM of three samples per treatment is shown for percent inhibition of branching. *P<0.05, **P<0.01. |
In a separate experiment, we examined the effect of AA-PMe and AA treatment on preformed tubes, beginning drug treatment after HUVECs had been allowed to form a tubular network for 6 hours. Again, the addition of AA-PMe significantly disrupted tube formation, reducing tube length by 30%, 52%, and 68% at 5, 10, and 25 μM, respectively, while AA reduced length by 17%, 32%, and 49% at the same doses (Figure 5B and E).
Finally, we tested whether AA-PMe and AA had similar effects on capillary development when HUVECs were treated with VEGF to stimulate growth. Although AA-PMe still decreased tube length and branch formation at 10 and 25 μM doses AA only had an effect at 25 μM (Figure 5C and F).
AA-PMe inhibits angiogenesis in transgenic zebrafish models
To extend these findings to the zebrafish model of whole organism angiogenesis Tg(fli:EGFP) embryos were treated with varying concentrations of AA and AA-PMe from 6 to 72 hpf, at which point all ISV had stretched to form dorsal longitudinal anastomotic vessels. In keeping with our in vitro findings, in vivo fluorescence imaging of EGFP expressing endothelial cells demonstrated potent inhibition of ISV, dorsal aorta (DA), and posterior cardinal vein (PCV) development.
At first, as shown in Figure 6, the ISVs were the most easily observed angiogenic vessels in the embryos at 72 hpf. AA-PMe showed potent inhibition on ISV formation at 10 μM, but AA showed no obvious effect at 10 μM (Figure 6A, left panel and central panel). We also found that the mean ISV length was 34.63±0.84 μm after treatment with 10 μM AA-PMe. The mean ISV length of the AA group was 44.96±0.45 μm at 10 μM, and it is obvious that treatment with AA-PMe resulted in shorter ISV than AA (Figure 6B).
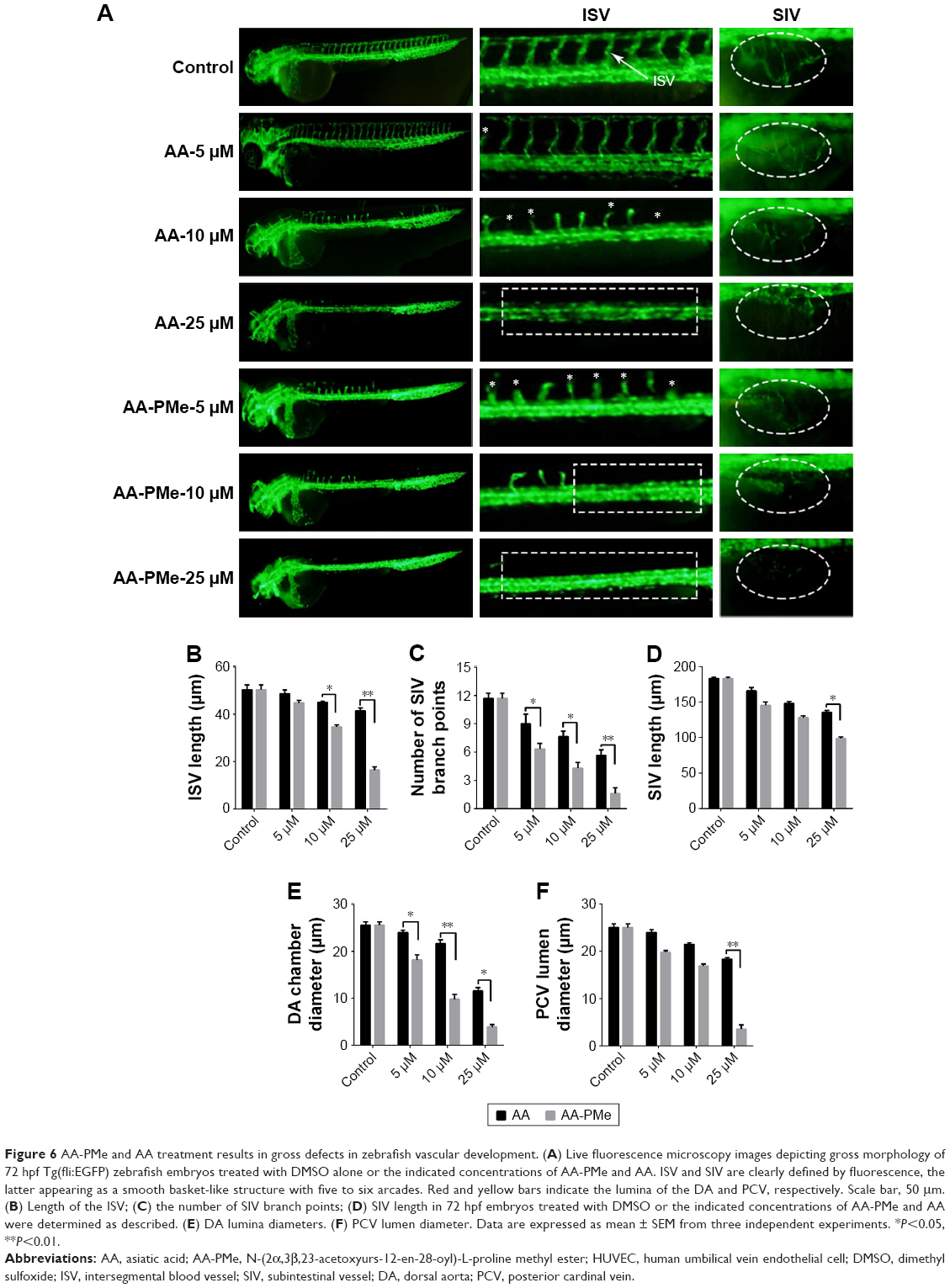 | Figure 6 AA-PMe and AA treatment results in gross defects in zebrafish vascular development. (A) Live fluorescence microscopy images depicting gross morphology of 72 hpf Tg(fli:EGFP) zebrafish embryos treated with DMSO alone or the indicated concentrations of AA-PMe and AA. ISV and SIV are clearly defined by fluorescence, the latter appearing as a smooth basket-like structure with five to six arcades. Red and yellow bars indicate the lumina of the DA and PCV, respectively. Scale bar, 50 μm. (B) Length of the ISV; (C) the number of SIV branch points; (D) SIV length in 72 hpf embryos treated with DMSO or the indicated concentrations of AA-PMe and AA were determined as described. (E) DA lumina diameters. (F) PCV lumen diameter. Data are expressed as mean ± SEM from three independent experiments. *P<0.05, **P<0.01. |
While the SIVs develop first by 72 hpf, the smooth, basket-like structures of the SIVs become apparent and the DA and PCV diameters are measurable. In vehicle-treated embryos, 5–6 SIV arcades were visible, but with 5 μM AA-PMe only partial development occurred and at 10 μM AA-PMe formation failed. AA treatment was slightly less inhibitory, with severe disruption at 10 μM and nearly complete blockage of formation at 25 μM AA (Figure 6A, far right panel). SIV branch points and length were also affected by 25 μM AA-PMe and AA treatment: branch points were reduced by 85.7% and 51.4% (Figure 6C) and SIV length decreased to 98.31±2.67 and 135.69±2.78 μm in response to 25 μM AA-PMe and AA, respectively (Figure 6D).
AA-PMe and AA treatment also decreased DA and PCV diameters (Figure 6A, central panel): DA diameters narrowed to 9.81±1.07 and 21.71±0.72 μM, and PCV diameters shrank to 16.9±0.39 and 21.39±0.39 μm in embryos treated with 10 μM AA-PMe and AA, respectively (Figure 6E and F).
In keeping with these morphometric findings, the quantitative EAP assay revealed a nearly 50% inhibition of blood vessel formation at 10 μM AA-PMe and a 20% inhibition at 10 μM AA (Figure 7). Overall, while both compounds inhibit angiogenesis in zebrafish embryos, we found that AA-PMe has a dramatically more powerful effect than AA.
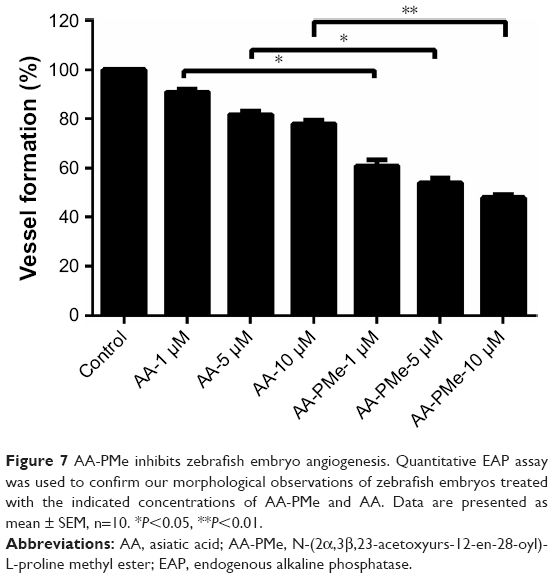 | Figure 7 AA-PMe inhibits zebrafish embryo angiogenesis. Quantitative EAP assay was used to confirm our morphological observations of zebrafish embryos treated with the indicated concentrations of AA-PMe and AA. Data are presented as mean ± SEM, n=10. *P<0.05, **P<0.01. |
AA-PMe changes VEGF-induced VEGFR2 mRNA expression in HUVECs
VEGF is a fundamental mediator of physiological and pathological angiogenesis and acts through two tyrosine kinase receptors, VEGFR1 and VEGFR2.48 However, VEGFR2 has a higher affinity for VEGF and is the major transducer of VEGF signaling in endothelial cells during blood vessel formation, while VEGFR1 modulates VEGFR2 signaling.49 In our study, 10 and 25 μM AA-PMe significantly decreased VEGFR2 mRNA expression, which might reduce the stimulatory effect of VEGF (Figure 8). However, AA treatment had no significant effect at any concentration.
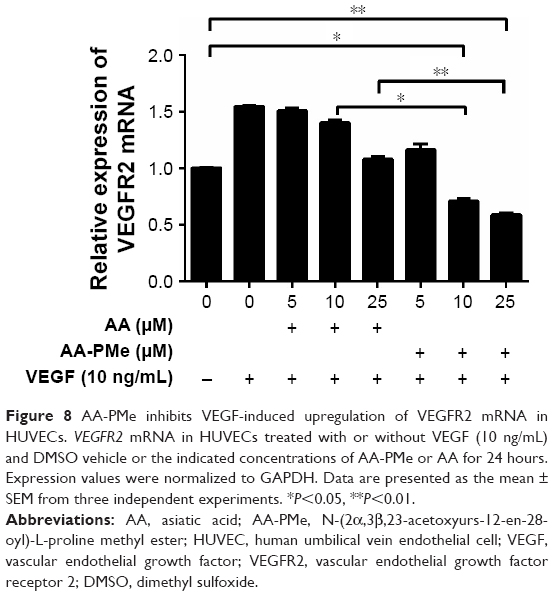 | Figure 8 AA-PMe inhibits VEGF-induced upregulation of VEGFR2 mRNA in HUVECs. VEGFR2 mRNA in HUVECs treated with or without VEGF (10 ng/mL) and DMSO vehicle or the indicated concentrations of AA-PMe or AA for 24 hours. Expression values were normalized to GAPDH. Data are presented as the mean ± SEM from three independent experiments. *P<0.05, **P<0.01. |
AA-PMe inhibits both VEGFR2 phosphorylation and activation of VEGFR2-mediated signaling in HUVECs and zebrafish embryos
Three major tyrosine phosphorylation sites, Tyr951, Tyr1175, and Tyr1214, have been identified on VEGFR2. In developing vessels, pTyr1175 and pTyr1214 have been detected on all VEGFR2 expressing endothelial cells, and autophosphorylation of Tyr1175 on VEGFR2 is crucial for endothelial cell proliferation, making these sites promising targets for antiangiogenic therapeutics.50,51 To dissect the molecular basis underlying the antiangiogenic effects we noted in HUVECs and zebrafish embryos, we followed VEGFR2 protein expression and phosphorylation by Western blot in HUVECs treated with varying concentrations of AA-PMe and AA. While even 10 μM AA-PMe dramatically inhibited phosphorylation of VEGFR2 Tyr1175, AA had little effect on the total VEGFR2 protein expression (Figure 9A) at any dose, but the same inhibitory effect on phosphorylation of VEGFR2 Tyr1175 at 25 μM as AA-PMe at 10 μM. Once again, our results demonstrate the far greater potency of AA-PMe compared to AA.
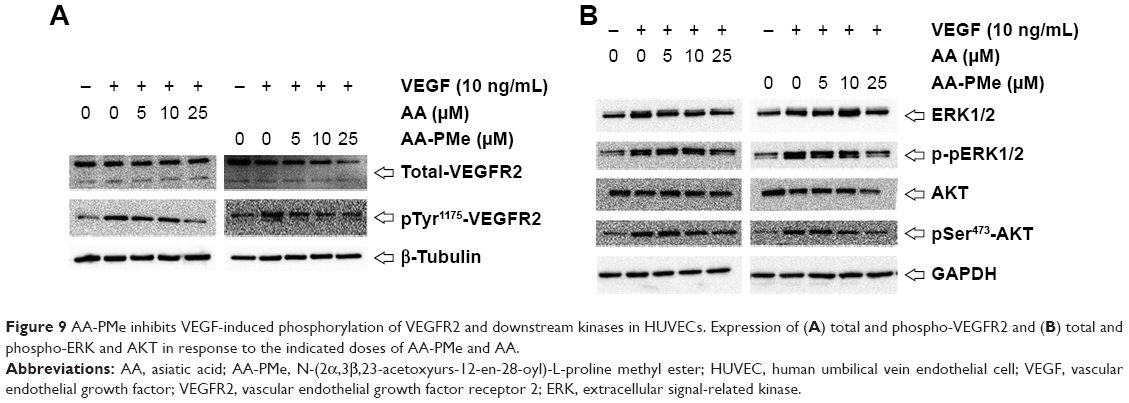 | Figure 9 AA-PMe inhibits VEGF-induced phosphorylation of VEGFR2 and downstream kinases in HUVECs. Expression of (A) total and phospho-VEGFR2 and (B) total and phospho-ERK and AKT in response to the indicated doses of AA-PMe and AA. |
Multiple signaling pathways responsible for endothelial cell migration, proliferation, and survival are activated downstream of VEGF–VEGFR2 interaction;52,53 so, we examined phosphorylation and activation of several of these key kinases. We found that in addition to inhibiting VEGFR2 phosphorylation, AA-PMe significantly suppressed phosphorylation and activation of ERK1/2 and AKT in HUVECs (Figure 9B).
To confirm these VEGFR2 findings in zebrafish embryos, we analyzed total and phospho-VEGFR2 protein and VEGFR2 mRNA expression following AA-PMe and AA treatment. Similar to our HUVEC findings, VEGFR2 phosphorylation was completely inhibited by 25 μM AA-PMe, with lower doses also decreasing p-VEGFR2. There was no effect on total VEGFR2 expression at any dose, resulting in a decreased ratio of phospho to total protein (Figure 10A and B). We noted that 25 μM AA had an effect on VEGFR2 phosphorylation similar to the lower AA-PMe doses. VEGFR2 mRNA expression followed a similar pattern, with all doses of AA-PMe significantly reducing and 25 μM AA partially reducing its expression (Figure 11).
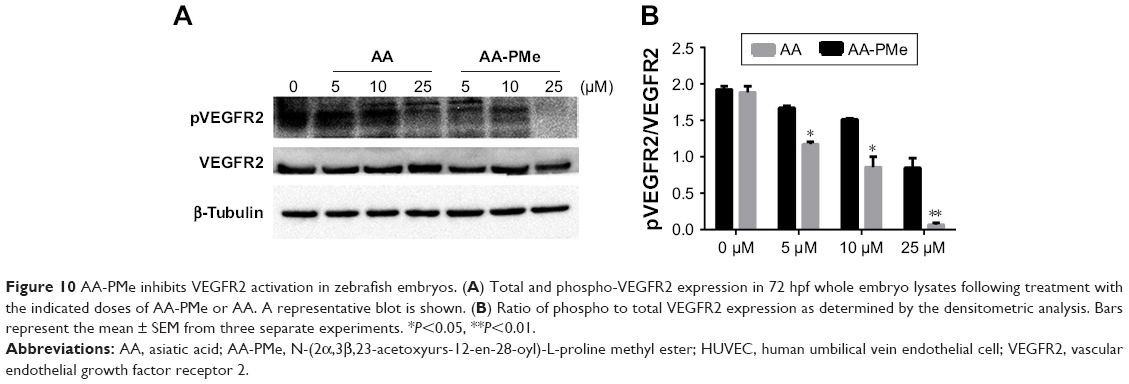 | Figure 10 AA-PMe inhibits VEGFR2 activation in zebrafish embryos. (A) Total and phospho-VEGFR2 expression in 72 hpf whole embryo lysates following treatment with the indicated doses of AA-PMe or AA. A representative blot is shown. (B) Ratio of phospho to total VEGFR2 expression as determined by the densitometric analysis. Bars represent the mean ± SEM from three separate experiments. *P<0.05, **P<0.01. |
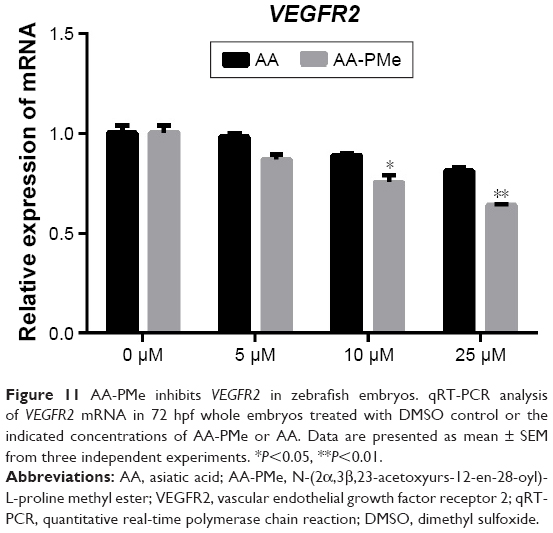 | Figure 11 AA-PMe inhibits VEGFR2 in zebrafish embryos. qRT-PCR analysis of VEGFR2 mRNA in 72 hpf whole embryos treated with DMSO control or the indicated concentrations of AA-PMe or AA. Data are presented as mean ± SEM from three independent experiments. *P<0.05, **P<0.01. |
As a whole, our results suggest that AA-PMe and AA exert their antiangiogenic activity through inhibition of the VEGFR2-mediated signaling cascade in both endothelial cells and zebrafish embryos, with AA-PMe clearly surpassing AA in its activity.
Discussion and conclusion
Angiogenesis is considered to be a key process in many physiological and pathological states, including cancer growth and progression.45,54,55 Complex cellular and extracellular components surround tumor cells at every stage of development to form the tumor microenvironment and this requires a steady blood supply.55 Recent advances suggest that antiangiogenic strategies targeting endothelial cells in the tumor microenvironment could prove particularly valuable because these cells are generally non-transformed and are considered less prone to acquiring drug resistance.45,56 Nontoxic agents that effectively target endothelial cells and reduce pathological angiogenesis in the tumor microenvironment could therefore be important tools in cancer prevention and treatment. Consequently, there is growing interest in the careful analysis of natural compounds derived from traditional medicines like AA, which has been found to influence steps in cancer angiogenesis with very few side effects.34 We have previously reported that AA-PMe, a modified form of AA synthesized in our laboratory, exerts antitumor activity by blocking cell cycling and cell mobility and inducing apoptosis.57 We further demonstrated antiangiogenic effects of AA-PMe in zebrafish.20 In the present study, we extend these findings to demonstrate powerful inhibition of endothelial cell growth, tube formation, and motility, all of which are crucial for blood vessel formation. This work highlighted the importance of AA-PMe as a novel antiangiogenic agent but the mechanism remained unclear.
Although angiogenesis is regulated by a large number of pro- and antiangiogenic cellular factors, the available evidence points to VEGF as a major paracrine mediator that directly contributes to angiogenesis.58 Following VEGF–VEGFR2 binding, signaling pathways controlling endothelial cell survival, proliferation, migration, and tube formation are activated.59,60 The downregulation of VEGFR2 mRNA expression by AA-PMe may contribute to its effect as an angiogenesis inhibitor by decreasing VEGFR2 signaling over time. However, AA-PMe also appears to act immediately by inhibiting phosphorylation of VEGFR2 to block its activation.
In patients, successful antiangiogenic therapy may require the simultaneous blockade multiple pro-angiogenic factor receptors at downstream signaling pathway.61 VEGF–VEGFR2 interaction alone activates numerous signaling pathways, inducing phosphorylation of ERK, c-Jun amino-terminal kinase, phosphatidylinositide 3-kinase, PKB (AKT), and p38 mitogen-activated protein kinase.14,62 Interestingly, AKT signaling has been found to regulate the expression of pro-angiogenic factors at the translational level, which makes this pathway a potential target for antiangiogenic therapy.13 We discovered that AA-PMe blocked two signaling components downstream of VEGFR2, reducing AKT and ERK phosphorylation in endothelial cells. In zebrafish embryos, AA-PMe also decreased VEGFR2. Thus, we suggest that AA-PMe exerts its antiangiogenic activity at least in part by regulating VEGFR2-mediated signaling in HUVECs and zebrafish (Figure 12).
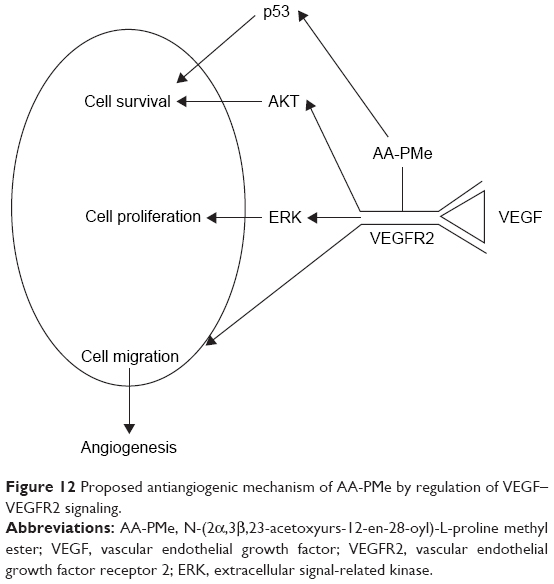 | Figure 12 Proposed antiangiogenic mechanism of AA-PMe by regulation of VEGF–VEGFR2 signaling. |
Our findings begin to elucidate the mechanism underlying the antiangiogenic activity of AA-PMe. We demonstrate here that AA-PMe inhibits phosphorylation of VEGFR2, suppressing the VEGFR2-mediated signaling pathway that plays crucial roles in regulating neovascularization and inducing apoptosis. These data support further development of AA-PMe as a novel VEGFR2 kinase inhibitor for the treatment of angiogenesis-related diseases.
Acknowledgment
This work was supported by grants from the Natural Science Foundation of China (grant no 81573833).
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Quesada AR, Munoz-Chapuli R, Medina MA. Anti-angiogenic drugs: from bench to clinical trials. Med Res Rev. 2006;26:483–530. | ||
Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. | ||
Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. | ||
Zhong H, Bowen JP. Molecular design and clinical development of VEGFR kinase inhibitors. Curr Top Med Chem. 2007;7:1379–1393. | ||
Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. | ||
Dong DF, Li EX, Wang JB, et al. Anti-angiogenesis and anti-tumor effects of AdNT4-anginex. Cancer Lett. 2009;285:218–224. | ||
Simo R, Carrasco E, Garcia-Ramirez M, Hernandez C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr Diabetes Rev. 2006;2:71–98. | ||
Pratheeshkumar P, Budhraja A, Son YO, et al. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR-2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS One. 2012;7:e47516. | ||
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. | ||
Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26:281–290. | ||
Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795–803. | ||
Lin C, Wu MH, Dong JY. Quercetin-4′-O-beta-D-glucopyranoside (QODG) inhibits angiogenesis by suppressing VEGFR2-mediated signaling in zebrafish and endothelial cells. PLoS One. 2012;7:e31708. | ||
Liu B, Faia L, Hu M, Nussenblatt RB. Pro-angiogenic effect of IFNgamma is dependent on the PI3K/mTOR/translational pathway in human retinal pigmented epithelial cells. Mol Vis. 2010;16:184–193. | ||
Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. | ||
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. | ||
Randrup Hansen C, Grimm D, Bauer J, Wehland M, Magnusson NE. Effects and side effects of using sorafenib and sunitinib in the treatment of metastatic renal cell carcinoma. Int J Mol Sci. 2017;18:E461. | ||
Shantakumar S, Nordstrom BL, Djousse L, et al. Occurrence of hepatotoxicity with pazopanib and other anti-VEGF treatments for renal cell carcinoma: an observational study utilizing a distributed database network. Cancer Chemother Pharmacol. 2016;78:559–566. | ||
Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–1805. | ||
Dong D, Ko B, Baumeister P, et al. Vascular targeting and antiangiogenesis agents induce drug resistance effector GRP78 within the tumor microenvironment. Cancer Res. 2005;65:5785–5791. | ||
Jing Y, Wang G, Ge Y, Xu M, Gong Z. Synthesis, anti-tumor and anti-angiogenic activity evaluations of asiatic acid amino acid derivatives. Molecules. 2015;20:7309–7324. | ||
Somboonwong J, Kankaisre M, Tantisira B, Tantisira MH. Wound healing activities of different extracts of Centella asiatica in incision and burn wound models: an experimental animal study. BMC Complement Altern Med. 2012;12:103. | ||
Shukla A, Rasik AM, Jain GK, Shankar R, Kulshrestha DK, Dhawan BN. In vitro and in vivo wound healing activity of asiaticoside isolated from Centella asiatica. J Ethnopharmacol. 1999;65:1–11. | ||
Soo Lee Y, Jin D-Q, Beak S-M, Lee E-S, Kim J-A. Inhibition of ultraviolet-A-modulated signaling pathways by asiatic acid and ursolic acid in HaCaT human keratinocytes. Eur J Pharmacol. 2003;476:173–178. | ||
Mook-Jung I, Shin JE, Yun SH, et al. Protective effects of asiaticoside derivatives against beta-amyloid neurotoxicity. J Neurosci Res. 1999;58:417–425. | ||
Jew SS, Yoo CH, Lim DY, et al. Structure–activity relationship study of asiatic acid derivatives against beta amyloid (Aβ)-induced neurotoxicity. Bioorg Med Chem Lett. 2000;10:119–121. | ||
Lee MK, Kim SR, Sung SH, et al. Asiatic acid derivatives protect cultured cortical neurons from glutamate-induced excitotoxicity. Res Commun Mol Pathol Pharmacol. 2000;108:75–86. | ||
Krishnamurthy RG, Senut MC, Zemke D, et al. Asiatic acid, a pentacyclic triterpene from Centella asiatica, is neuroprotective in a mouse model of focal cerebral ischemia. J Neurosci Res. 2009;87:2541–2550. | ||
Tang LX, He RH, Yang G, et al. Asiatic acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in vivo and in vitro. PLoS One. 2012;7:e31350. | ||
Hsu YL, Kuo PL, Lin LT, Lin CC. Asiatic acid, a triterpene, induces apoptosis and cell cycle arrest through activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways in human breast cancer cells. J Pharmacol Exp Ther. 2005;313:333–344. | ||
Park BC, Bosire KO, Lee ES, Lee YS, Kim JA. Asiatic acid induces apoptosis in SK-MEL-2 human melanoma cells. Cancer Lett. 2005;218:81–90. | ||
Tang X-L, Yang X-Y, Jung H-J, et al. Asiatic acid induces colon cancer cell growth inhibition and apoptosis through mitochondrial death cascade. Biol Pharm Bull. 2009;32:1399–1405. | ||
Cho CW, Choi DS, Cardone MH, Kim CW, Sinskey AJ, Rha C. Glioblastoma cell death induced by asiatic acid. Cell Biol Toxicol. 2006;22:393–408. | ||
Lee YS, Jin D-Q, Kwon EJ, et al. Asiatic acid, a triterpene, induces apoptosis through intracellular Ca2+ release and enhanced expression of p53 in HepG2 human hepatoma cells. Cancer Lett. 2002;186:83–91. | ||
Kavitha CV, Agarwal C, Agarwal R, Deep G. Asiatic acid inhibits pro-angiogenic effects of VEGF and human gliomas in endothelial cell culture models. PLoS One. 2011;6:e22745. | ||
Zhao DX, Qin CJ, Fan XH, Li YC, Gu BH. Inhibitory effects of quercetin on angiogenesis in larval zebrafish and human umbilical vein endothelial cells. Eur J Pharmacol. 2014;723:360–367. | ||
Xu Z, Li Y, Xiang Q, et al. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J Med Chem. 2010;53:4642–4653. | ||
Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. 2002;248:307–318. | ||
Yu XB, Tong Y, Han XQ, et al. Anti-angiogenic activity of Herba Epimedii on zebrafish embryos in vivo and HUVECs in vitro. Phytother Res. 2013;27:1368–1375. | ||
Truong L, Harper SL, Tanguay RL. Evaluation of embryotoxicity using the zebrafish model. Methods Mol Biol. 2011;691:271–279. | ||
Mu XY, Pang S, Sun XZ, et al. Evaluation of acute and developmental effects of difenoconazole via multiple stage zebrafish assays. Environ Pollut. 2013;175:147–157. | ||
He MF, Huang YH, Wu LW, Ge W, Shaw PC, But PP. Triptolide functions as a potent angiogenesis inhibitor. Int J Cancer. 2010;126:266–278. | ||
Lam HW, Lin HC, Lao SC, et al. The angiogenic effects of Angelica sinensis extract on HUVEC in vitro and zebrafish in vivo. J Cell Biochem. 2008;103:195–211. | ||
Lu XL, Xu ZL, Yao XL, et al. Marine cyclotripeptide X-13 promotes angiogenesis in zebrafish and human endothelial cells via PI3K/Akt/eNOS signaling pathways. Mar Drugs. 2012;10:1307–1320. | ||
Lu XL, Luo D, Yao XL, et al. dl-3n-Butylphthalide promotes angiogenesis via the extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase/Akt-endothelial nitric oxide synthase signaling pathways. J Cardiovasc Pharmacol. 2012;59:352–362. | ||
Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene. 2005;24:1188–1202. | ||
Parng C, Seng WL, Semino C, McGrath P. Zebrafish: a preclinical model for drug screening. Assay Drug Dev Technol. 2002;1:41–48. | ||
van Moorselaar RJ, Voest EE. Angiogenesis in prostate cancer: its role in disease progression and possible therapeutic approaches. Mol Cell Endocrinol. 2002;197:239–250. | ||
Zachary I. Signaling mechanisms mediating vascular protective actions of vascular endothelial growth factor. Am J Physiol Cell Physiol. 2001;280:C1375–C1386. | ||
Roberts DM, Kearney JB, Johnson JH, Rosenberg MP, Kumar R, Bautch VL. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004;164:1531–1535. | ||
Matsumoto T, Bohman S, Dixelius J, et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005;24:2342–2353. | ||
Takahashi T, Yamaguchi S, Chida K, Shibuya M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 2001;20:2768–2778. | ||
Pang X, Yi Z, Zhang J, et al. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 2010;70:1951–1959. | ||
Edirisinghe I, Yang SR, Yao H, et al. VEGFR-2 inhibition augments cigarette smoke-induced oxidative stress and inflammatory responses leading to endothelial dysfunction. FASEB J. 2008;22:2297–2310. | ||
Crawford TN, Alfaro DV 3rd, Kerrison JB, Jablon EP. Diabetic retinopathy and angiogenesis. Curr Diabetes Rev. 2009;5:8–13. | ||
Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7:139–147. | ||
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. | ||
Jing Y, Wang G, Ge Y, Xu M, Tang S, Gong Z. AA-PMe, a novel asiatic acid derivative, induces apoptosis and suppresses proliferation, migration, and invasion of gastric cancer cells. Onco Targets Ther. 2016;9:1605–1621. | ||
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. | ||
Gu Q, Wang D, Wang X, et al. Basic fibroblast growth factor inhibits radiation-induced apoptosis of HUVECs. I. The PI3K/AKT pathway and induction of phosphorylation of BAD. Radiat Res. 2004;161:692–702. | ||
Gingis-Velitski S, Zetser A, Flugelman MY, Vlodavsky I, Ilan N. Heparanase induces endothelial cell migration via protein kinase B/Akt activation. J Biol Chem. 2004;279:23536–23541. | ||
Kanda S, Miyata Y, Kanetake H, Smithgall TE. Non-receptor protein-tyrosine kinases as molecular targets for antiangiogenic therapy (review). Int J Mol Med. 2007;20:113–121. | ||
Amoroso F, Capece M, Rotondo A, et al. The P2X7 receptor is a key modulator of the PI3K/GSK3beta/VEGF signaling network: evidence in experimental neuroblastoma. Oncogene. 2015;34:5240–5251. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.