")
Back to Journals » Vascular Health and Risk Management » Volume 13
Anti-PCSK9 antibodies for the treatment of heterozygous familial hypercholesterolemia: patient selection and perspectives
Authors Catapano AL, Pirillo A, Norata GD
Received 8 June 2017
Accepted for publication 6 July 2017
Published 4 September 2017 Volume 2017:13 Pages 343—351
DOI https://doi.org/10.2147/VHRM.S130338
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Naga Venkata Amarnath Kommuri
Alberico Luigi Catapano,1,2 Angela Pirillo,1,2 Giuseppe Danilo Norata1,3,4
1Department of Pharmacological and Biomolecular Sciences, Università degli Studi di Milano, 2IRCCS Multimedica Hospital, Sesto San Giovanni, 3Center for the Study of Atherosclerosis, E. Bassini Hospital, Cinisello Balsamo, Milan, Italy; 4School of Biomedical Sciences, Curtin Health Innovation Research Institute, Curtin University, Perth, WA, Australia
Abstract: Heterozygous familial hypercholesterolemia (FH) is a genetic disorder characterized by high low-density lipoprotein cholesterol levels from birth, which exposes the arteries to high levels of atherogenic lipoproteins lifelong and results in a significantly increased risk of premature cardiovascular events. The diagnosis of FH, followed by an appropriate and early treatment is critical to reduce the cardiovascular burden in this population. Phase I–III clinical trials showed the benefit of proprotein convertase subtilisin kexin 9 inhibitors, both alirocumab and evolocumab, in these patients with an average low-density lipoprotein cholesterol reduction ranging from −40% to −60%. The aim of this review is to address the unmet needs in cholesterol management, elucidate the biology and the clinical benefit of proprotein convertase subtilisin kexin 9 inhibition and finally discuss the open gaps and future directions in the treatment of patients with heterozygous FH.
Keywords: HeFH, dyslipidemia, cholesterol, alirocumab, evolocumab
Unmet needs in cholesterol management: focus on heterozygous familial hypercholesterolemia
Heterozygous familial hypercholesterolemia (HeFH) is a genetic disorder characterized by high low-density lipoprotein cholesterol (LDL-C) levels from birth, which exposes the arteries to high levels of atherogenic lipoproteins lifelong and results in a significantly increased risk of premature cardiovascular events.1 The diagnosis of familial hypercholesterolemia (FH), followed by appropriate and early treatments aimed at reducing LDL-C levels are therefore critical to reduce the cardiovascular burden in this population.
FH is caused primarily by mutations in the gene encoding for the LDL receptor (LDLR) which, by affecting its structure and function, cause decreased removal of LDL from the circulation. Mutations in the LDLR gene account for ~90% of genetically determined HeFH, the remaining is due to mutations which alter the binding site of apoB to the LDLR, or to other mutations increasing the activity of proprotein convertase subtilisin kexin 9 (PCSK9), which cause a decrease in LDLR expression/activity.2 Heterozygous subjects present with elevated plasma LDL-C levels (200–500 mg/dL). The frequency of HeFH in the general population has been estimated in 1:200–250,3 and is higher in selected populations such as patients with premature cardiovascular disease.4 Despite that, HeFH is still underdiagnosed and, as a consequence, undertreated. The homozygous form of FH is much rarer (1:160,000–300,000).5 These patients present with very high LDL-C levels (untreated levels >500 mg/dL) and are at extremely elevated risk of cardiovascular events.5
The diagnosis of FH can be done relatively easily by using clinical tools such as the Dutch Lipid Clinic Network (DLCN) criteria,6 the Make Early Diagnosis to Prevent Early Death (MEDPED) criteria,7 or the Simon Broome (SB) criteria (Table 1).8 Depending on the specific criteria, the score is calculated based on the presence of high LDL-C levels, on patient history of premature coronary heart disease (CHD) or cerebral or peripheral vascular disease, on family history of premature CHD or hypercholesterolemia and on the presence of physical signs such as tendon xanthomas or corneal arcus. The MEDPED criteria rely on age-specific and family relative-specific levels of total cholesterol, but do not integrate this information with the clinical characteristics of the subjects or the identification of a FH mutation. The DLCN score takes into account a family or personal history of premature CHD, physical signs, and high LDL-C levels, and suggests the genetic analysis if the score is >5; a definite FH diagnosis is given when the score is >8. SB criteria are similar in terms of parameters evaluated for the score calculation, giving a definite FH diagnosis in the presence of high LDL-C (or total cholesterol) levels plus tendon xanthomas in the patient or a first or second-degree relative or in the presence of a functional mutation in one of the 3 candidate genes (Table 1).
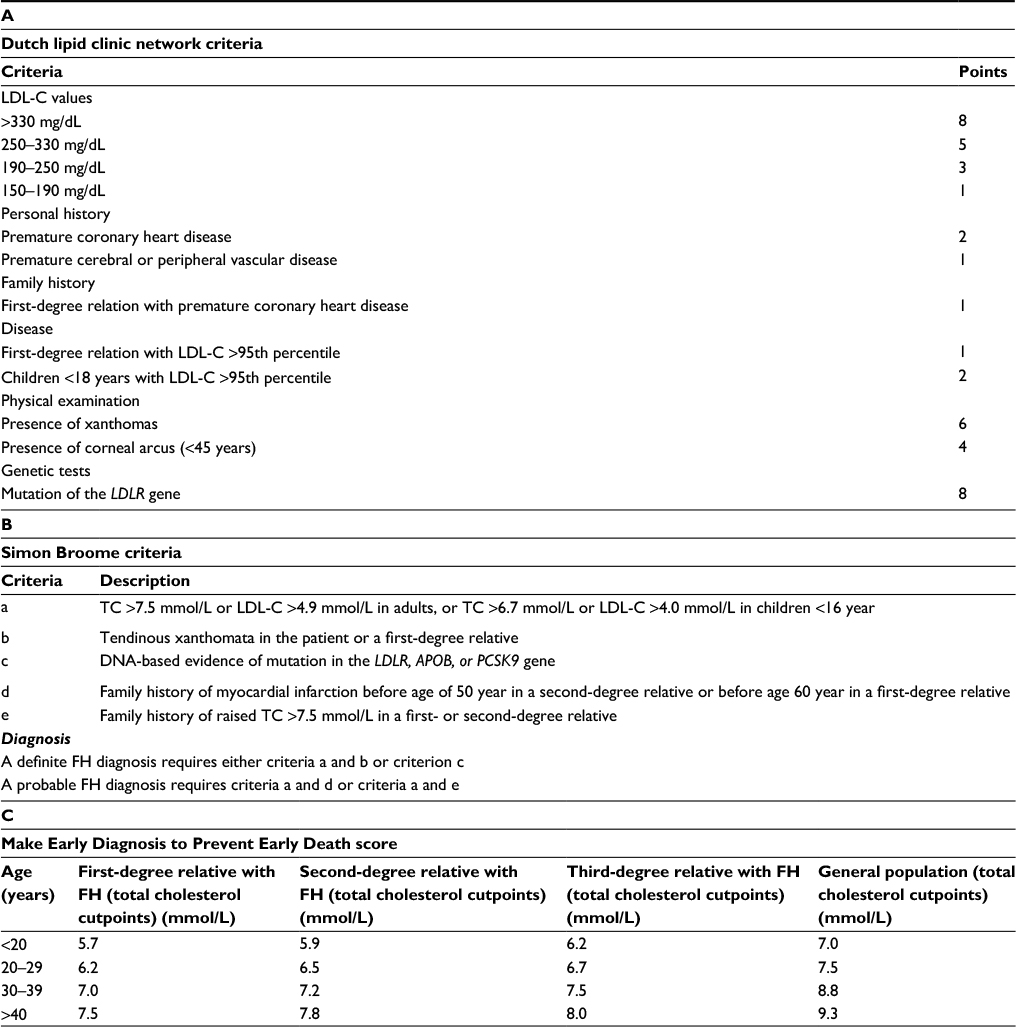 | Table 1 Clinical criteria for the diagnosis of familial hypercholesterolemia Note: Diagnosis: FH is diagnosed if total cholesterol levels exceed the cut point. Abbreviations: FH, familial hypercholesterolemia; LDL-C, low-density lipoprotein cholesterol; TC, total cholesterol. |
Current guidelines suggest to lower LDL-C as early as possible in HeFH patients, to delay the onset of the first cardiovascular event.3,9 Recommended LDL-C goals for these patients are levels <100 mg/dL (<2.5 mmol/L) for adults, or <70 mg/dL (<1.8 mmol/L) for adults with CHD or diabetes, and <135 mg/dL (<3.5 mmol/L) for children.3,9 To this aim, several cholesterol-lowering drugs are available; statins are the first choice in adults but also in children, in whom only those proven to be relatively safe should be used.3,10,11 Statins effectively reduce CHD risk in FH subjects,12 however, increasing the dose of statins did not result in further LDL-C level reduction due to a plateau effect,13,14 which supports the need for drugs with a complementary mechanism of action to be used in combination with statins to achieve recommended LDL-C targets.3,15 These drugs include ezetimibe, mipomersen and lomitapide, or PCSK9 inhibitors.16 In addition, some patients are statin-intolerant, and this has a special relevance in FH patients who need high doses of statins to achieve the recommended LDL-C level target. Mipomersen and lomitapide are registered for patients with homozygous familial hypercholesterolemia (HoFH), while patients with HeFH will benefit from the combination statins/ezetimibe, which yields an additional reduction of 15%–18%17,18 and, more recently, from the monoclonal antibodies directed against PCSK9.
PCSK9 biology and strategies to inhibit PCSK9
PCSK9 is a serine protease that binds the LDLR and promotes its lysosomal degradation19. Through this post-transcriptional mechanism, PCSK9 limits LDL catabolism and increases plasma LDL-C levels.20 PCSK9 also targets lipoprotein synthesis in the intestine20 further contributing to a disturbed plasma lipid profile. Thus, patients homozygous for gain-of-function mutations in PCSK9 gene present the clinical phenotype of FH with tendon xanthomas, history of CHD, early myocardial infarction, and stroke. On the contrary, subjects with loss-of-function mutations in PCSK9 gene present with lower plasma LDL-C levels and are protected from coronary artery diseases.21–23 Of note, PCSK9 plasma levels predict cardiovascular events in statin-treated patients with well-controlled LDL levels and documented stable coronary artery disease,24 further linking PCSK9 to cardiovascular outcomes.
PCSK9 production is mainly regulated by changes in cholesterol levels in the liver via the modulation of the nuclear translocation of the sterol-responsive element-binding protein 2 transcription factor.25,26 Once secreted, mature PCSK9 protein undergoes post-translational modifications that can modulate its function, including the cleavage to a truncated protein of about 60 kDa by furin or PC5/6A, 2 members of the proprotein convertase family.
More importantly, PCSK9 plasma levels increase following cholesterol-lowering treatments, a finding observed not only with statins but also with ezetimibe.27–29 This mechanism contributes to limiting the pharmacological efficacy of statins and other lipid-lowering strategies as well as provides a mechanisms for understanding the poor correlation between PCSK9 and LDL in circulation.28,29 Therefore, given the role of PCSK9 as chaperone in directing the LDLR toward degradation,30 the possibility of inhibiting PCSK9 represents a key approach to enhance the lipid-lowering effect of conventional agents.30 From a pharmacological perspective, PCSK9 could be targeted at different levels from the gene transcription (small interfering RNAs, antisense oligonucleotides) to the circulating protein (anti-PCSK9 monoclonal antibodies or PCSK9 vaccine).30
PCSK9 gene silencing
Gene-silencing approaches are under clinical development, and the results from the first Phase II study, ORION-1, with a siRNA designed to target PCSK9 (inclisiran) were recently published.31 A single injection of the drug results in LDL-C reduction up to −36% while the injection of 2 doses (days 0 and 90) yielded up to −47.2% LDL-C reduction after 240 days.
Anti-PCSK9 antibodies
Monoclonal antibodies targeted against circulating PCSK9 have been approved for the treatment of patients at very high cardiovascular risk, patients with FH and statin intolerance.
Three antibodies targeting PCSK9 have been developed (evolocumab, alirocumab, bococizumab) and one, LY3015014 is under development.32 Evolocumab and alirocumab are commercially available while the development of bococizumab has been halted as a reduction in the efficacy was observed as a consequence of the induction of neutralizing antibodies.33 Results from Phase II studies indicated that PCSK9 therapy reduces LDL-C cholesterol up to 60%–70%,16 setting the stage for further evaluating the benefit of anti-PCSK9 monoclonal antibodies in larger cohorts. Anti-PCSK9 therapies not only reduce LDL-C levels but also favor atherosclerotic plaque regression, as evidenced by the GLAGOV trial, where in patients with angiographic coronary disease treated with statins, addition of evolocumab resulted in a −0.95% decrease in percent atheroma volume after 76 weeks of treatment.34 More recently, the FOURIER trial demonstrated that the inhibition of PCSK9 with evolocumab, on a background of statin therapy, reduced the risk of cardiovascular events (median duration of follow-up 26 months) further supporting the benefit of this pharmacological strategy.35 The clinical trial for cardiovascular outcomes with the other approved antibody, alirocumab, is ongoing and the results are expected to be released in early 2018 (ODYSSEY OUTCOMES). Of note, results on cardiovascular benefits are also available for bococizumab, which, in high-risk patients (SPIRE-2 trial), resulted in a significant reduction of major cardiovascular events, an effect which could not be detected in the trial involving lower risk patients (SPIRE-1 trial).36
These data further reinforce the efficacy of lowering LDL-C by inhibiting PCSK9 and point to the investigation of the efficacy in patients with HeFH.
Clinical trials supporting the use of PCSK9 inhibitors in HeFH
Phase I–III clinical trials clearly showed the benefit of PCSK9 inhibitors, both alirocumab and evolocumab in patients with HeFH (Table 2). Data on HeFH patients are available for both.
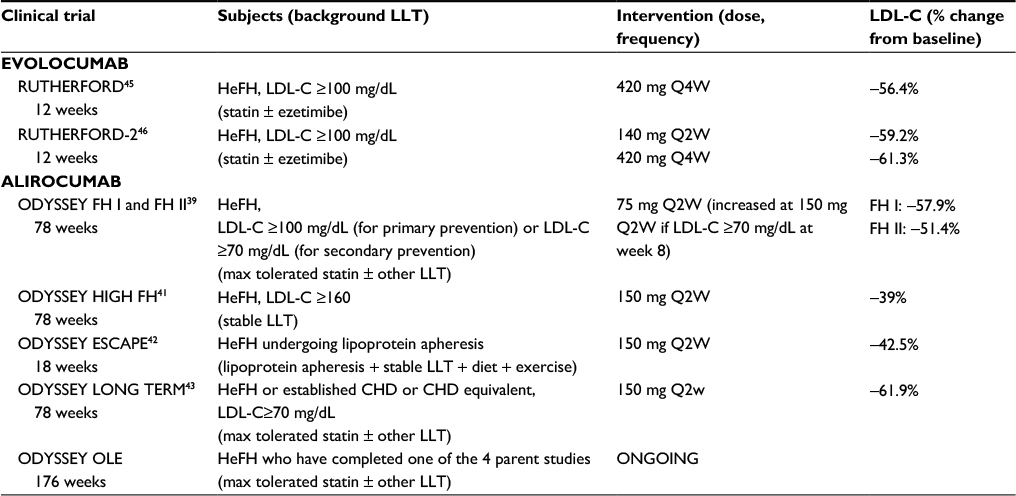 | Table 2 Effects of PCSK9 inhibitors evolocumab and alirocumab in HeFH Abbreviations: CHD, coronary heart disease; FH, familial hypercholesterolemia; HeFH, heterozygous familial hypercholesterolemia; LDL-C, low-density lipoprotein cholesterol; LLT, lipid-lowering therapy; PCSK9, proprotein convertase subtilisin kexin 9; Q4W, every 4 weeks; Q2W, every 2 weeks. |
In the first Phase I trial, alirocumab was tested in 21 HeFH patients on atorvastatin therapy and LDL-C >100 mg/dL.37 The average reduction of LDL-C levels was −41% at the dose of 50 mg and up to −55% at the dose of 150 mg. In the 5 patients receiving 150 mg of alirocumab, the degree of LDL-C reduction observed, ranged from ~−35% to ~−75%. No serious adverse events were reported, with no subjects showing increase in alanine transferase or aspartate transferase levels and 13% of subjects taking also atorvastatin reported an elevation of creatine kinase 3-time higher the upper limit of the normal range.
In a larger multicenter, randomized, placebo-controlled, Phase II trial, alirocumab was assessed in 77 HeFH patients receiving statins with or without ezetimibe and with LDL-C >100 mg/dL.38 Patients were randomized to alirocumab 150, 200, or 300 mg every 4 weeks, 150 mg every 2 weeks, or placebo every 2 weeks, and were stratified by concomitant use of ezetimibe at baseline. At week 12, LDL-C reductions from baseline to week 12 ranged from 29% (150 mg every 4 weeks) to 68% for alirocumab (150 mg every 2 weeks), compared with ~−11% for placebo. Adverse event profiles were similar for alirocumab and placebo; the most common adverse event was injection-site reaction with one patient in the group of alirocumab 300 mg terminating the treatment.
Among Phase III trials with alirocumab, HeFH patients were investigated in the ODYSSEY FH I and FH II,39 ODYSSEY JAPAN,40 ODYSSEY HIGH FH41 and ODYSSEY ESCAPE.42 In ODYSSEY FH I and FH II 735 patients with HeFH receiving maximum tolerated statin doses with or without other lipid-lowering therapy were enrolled.39 Patients were randomized to alirocumab 75 mg every 2 weeks (with possible increase to 150 mg at week 12, if LDL-C goal was not reached at week 8), or placebo. At week 24, mean reductions in LDL-C compared with placebo were 58% in FH I and 51% in FH II. Safety and tolerability were generally comparable in the alirocumab and placebo groups. The ODYSSEY JAPAN study included 41 HeFH patients and alirocumab treatment resulted in LDL-C reduction similar to what was observed with the other studies.40 The ODYSSEY HIGH FH trial included 107 patients with severe HeFH, (LDL-C levels >160 mg/dL on maximally tolerated statins with or without other lipid-lowering therapies).40 Patients were randomized to placebo or alirocumab 150 mg every 2 weeks. At week 24, the relative reduction observed in LDL-C was −45.7% compared with −6.6% in the placebo group. As seen in other trials, alirocumab was generally well tolerated and adverse event rates were generally similar to those of the placebo group. The ODYSSEY ESCAPE trial evaluated the effect of 12-week treatment with alirocumab in HeFH patients undergoing regular lipoprotein apheresis every 1 or 2 weeks.42 More than 90% patients on alirocumab at least halved the standardized rate of apheresis (−53.7% change from baseline at week 6), compared with +1.6% in the placebo group,42 and 63.4% of patients on alirocumab had not apheresis procedures during 12 weeks.42 In contrast to apheresis procedures, characterized by a rapid increase of LDL-C levels toward pre-apheresis levels, alirocumab maintained LDL-C levels persistently low,42 which may translate into a clinical benefit over the long term.
In agreement with this last observation, available data indicate that the effect of alirocumab persists over time and indeed after 78 weeks of treatment, the results of the ODYSSEY LONG TERM study43 showed that LDL-C levels were still significantly lower compared with what was observed in the placebo group; of note, this study included 415 patients with HeFH where the benefit observed was similar to that of the entire cohort and in line with what was reported for the other studies in HeFH patients.44
A pooled analysis of the safety data for alirocumab studies in HeFH patients of treatment was recently published. Data are available for more than 800 HeFH patients treated with alirocumab versus 418 with placebo (both groups were on the maximal tolerated dose of statins + other lipid-lowering therapies) up to 78 weeks. Regardless of the alirocumab dose, rates of treatment-emergent adverse events (TEAEs) were similar in alirocumab (80.5%) and placebo-treated patients (83.0%). TEAEs reported in ≥2% of patients receiving alirocumab were generally comparable with the placebo group, except for a higher proportion of patients experiencing injection-site reactions in the alirocumab group (11.4%) compared with placebo (8.6%).44
The efficacy and safety of evolocumab in HeFH patients were also tested in 2 studies; the RUTHERFORD study was a Phase II trial designed to evaluate the effect of evolocumab 350 or 420 mg versus placebo every 4 weeks in 111 patients with clinical diagnosis of FH (SB criteria) and LDL-C levels above 100 mg/dL despite statin therapy with or without ezetimibe.45 After 12 weeks of treatment, LDL-C was reduced by ~55% with evolocumab 420 mg, compared with a 1% increase with placebo (P<0.001).
Four patients in the evolocumab 350 mg group and one patient in the 420 mg group were considered poor responders based on a <15% reduction in LDL-C at week 12.45 The 3 most common treatment-related adverse events were injection-site pain (reported in 3.6% of evolocumab-treated patients), headache (1.8%), and skin burning sensation (3.6%).45
Evolocumab was further investigated in the Phase III RUTHERFORD-2 trial, in which 331 patients with HeFH were randomly assigned to evolocumab 140 mg twice a month, 420 mg monthly, placebo twice a month, or placebo monthly.46 After 12 weeks, evolocumab was associated with LDL-C reductions of ~60% compared with the placebo group.46 The rate of adverse events was similar between the evolocumab and placebo groups and overall evolocumab was well tolerated.46
As described above, patients who will benefit from anti-PCSK9 therapy are, without any doubt, those with HeFH while for those with HoFH, the benefit will be linked to the mutation underlying the disease. Indeed, based on the mechanism of action (a robust induction of LDLR-mediated clearance of LDL), the benefit of anti-PCSK9 therapy should be more marked in FH patients who retain a partial functionality of the LDLR (LDLR defective), while it would be less effective in those with a severe mutation of the LDLR (LDLR null) or those with other mutations impacting the LDLR axis (such as LDLRAP1). On the contrary, it is expected that the drug would be highly effective in patients with gain of function mutations of PCSK9.
For the treatment of HoFH, the TESLA clinical trials have specifically addressed the benefit of evolocumab in these patients.47,48 A similar observation is available also for alirocumab on HoFH patients, although in a limited cohort: alirocumab normalizes LDLR expression in receptor-defective HoFH fibroblasts, while no effect is observed in receptor-negative HoFH.49 An interim subset analysis of the TAUSSIG trial confirms the robust and durable reductions in LDL-C levels obtained with evolocumab in HoFH patients; in addition, it provides positive results for subjects receiving apheresis in addition to stable lipid-lowering therapy and, even more important, for adolescent patients.50 The data indicate a reduction of LDL-C levels on the top of available therapies of approximately −26%. Although relevant, other drugs with different mechanisms of action, such as lomitapide, have produced a greater magnitude of reduction in HoFH patients51 while for mipomersen, the reduction in LDL-C achieved is closer to that observed for PCSK9 inhibitors.52
Conclusions and future directions
Available data suggest that HeFH patients would benefit most from additional lipid lowering with an anti-PCSK9 antibody. Data on cardiovascular benefit of evolocumab have been recently published (FOURIER trial),35 reporting data on overall and cardiovascular mortality as well as the incidence of myocardial infarction. The study included patients with atherosclerotic cardiovascular disease and LDL-C ≥70 mg/dL who were receiving statin therapy.35 A sub-analysis of cardiovascular outcome in FH patients enrolled in the study is not yet available but it is reasonable to believe that the drugs will be as effective in this cohort. However, given the mechanism of action of anti-PCSK9, it should be taken into account that some HeFH patients with null mutation on the LDL receptor will probably experience a minor benefit following monoclonal antibody therapy, although the RUTHERFORD-2 study did not observe different LDL-C lowering response to evolocumab in patients with defective or negative LDLR.46 All the studies of PCSK9 inhibition in HeFH subjects, in addition, reported a good tolerability and rate of adverse events were similar between the evolocumab/alirocumab and placebo groups (Table 3), as also reported by a recent pooled analysis of studies with alirocumab in HeFH patients.44
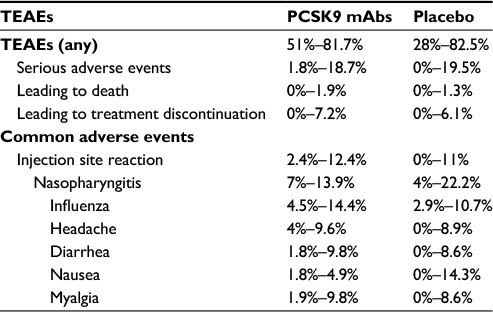 | Table 3 TEAE frequency in PCSK9 mAbs clinical trials in HeFH patients Abbreviations: HeFH, heterozygous familial hypercholesterolemia; mAb, monoclonal antibody; PCSK9, proprotein convertase subtilisin kexin 9; TEAEs, treatment-emergent adverse events. |
PCSK9 inhibition was proven to also reduce the levels of lipoprotein(a) (Lp(a)), an LDL-like particle, which represents a cardiovascular risk factor independently of LDL-C levels.53 Indeed FH patients, especially those with cardiovascular disease, exhibit significantly higher plasma levels of Lp(a) compared with their non-affected relatives.54 Lp(a) was proposed to play causal role in aortic valve calcification development in asymptomatic statin-treated HeFH.55 These observations support the need to asses Lp(a) levels in FH patients to identify those who could benefit from more aggressive lipid-lowering treatments.
In contrast to statins, PCSK9 mAbs reduce Lp(a) levels by up to 30%,56,57 but the exact mechanism by which such reduction takes place is still unclear. LDL-C and Lp(a) levels decrease concomitantly during therapy with PCSK9 mAbs, and patients achieving lower LDL-C levels also show larger Lp(a) reductions.58 Based on these observations, the effect on Lp(a) levels in HeFH patients treated with evolocumab or alirocumab was evaluated. The RUTHERFORD studies reported reductions ranging from −23% up to −31.6% versus placebo;45,46 similar reductions were observed in the studies of the ODYSSEY program performed in FH subjects (−14.8% to −25.6% versus placebo),39,41,43 with the exception of the ODYSSEY ESCAPE trial that reported a −4.1% Lp(a) level reduction versus placebo.42 Also HoFH showed significant Lp(a) level reductions, apparently independent of the genetic defect.47,48 The clinical relevance of Lp(a) reduction remains to be addressed, however.
Do the achieved very low LDL-C levels pose problems of safety? Recently, an analysis of pooled data from 14 trials on alirocumab indicated that LDL-C levels <25 or <15 mg/dL were not associated with a higher incidence of treatment-emergent event rates or neurocognitive events,59 although the incidence of cataract appeared to be increased in subjects achieving LDL-C levels <25 mg/dL.59 This finding, however, might be the consequence of the comparison of non-randomized subgroups. Due to the need of lifelong therapy in patients with FH, the safety of long-term exposure to pharmacologically induced very low levels of LDL-C remains to be determined.
A final aspect to be considered is the cost-effectiveness of anti-PCSK9 therapy in HeFH patients. To date, an analysis is available for the US market.60 Adding PCSK9 inhibitors to statins in HeFH was estimated to prevent 316,300 major adverse cardiovascular events at a cost of $503.000 per quality-adjusted life year (QALY) gained compared with adding ezetimibe to statins.60 Reaching the desired QALY threshold of $100.000 will need an important reduction of annual drug price. However, a more precise determination of the cost-effectiveness of PCSK9 therapy can only be performed once long-term data on clinical outcomes on HeFH patients for PCSK9 inhibitors becomes available.
Acknowledgments
The work of the authors is supported by: Fondazione Cariplo 2015–0524 and 2015–0564 (ALC), and 2016–0852 (GDN); H2020 REPROGRAM PHC-03-2015/667837-2 (ALC); Telethon Foundation (GGP13002) (GDN), Ministero della Salute GR-2011-02346974 (GDN); Aspire Cardiovascular Grant 2016-WI218287 (GDN).
Disclosure
The authors have received research funding, and/or honoraria for advisory boards, consultancy or speaker bureau from Aegerion (ALC, GDN), Amgen (ALC, GDN), AstraZeneca (ALC), Eli Lilly (ALC), Genzyme (ALC), Mediolanum (ALC), Merck or MSD (ALC), Pfizer (ALC, GDN), Recordati (ALC, GDN), Rottapharm (ALC), Sanofi-Regeneron (ALC, GDN), Sigma-Tau (ALC). AP reports no conflicts of interest in this work.
References
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.