")
Back to Journals » International Journal of Nanomedicine » Volume 15
Anti-Oxidant and Anti-Endothelial Dysfunctional Properties of Nano-Selenium in vitro and in vivo of Hyperhomocysteinemic Rats
Authors Zheng Z, Liu L, Zhou K , Ding L, Zeng J, Zhang W
Received 31 March 2020
Accepted for publication 25 May 2020
Published 23 June 2020 Volume 2020:15 Pages 4501—4521
DOI https://doi.org/10.2147/IJN.S255392
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Zeqi Zheng,1,* Lijuan Liu,1,* Kaiwen Zhou,2,* Lu Ding,1,3 Junyi Zeng,1,3 Wan Zhang1,3
1Department of Cardiology, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, People’s Republic of China; 2The First Clinical Medical College, School of Medicine, Nanchang University, Nanchang, Jiangxi 330006, People’s Republic of China; 3Jiangxi Hypertension Research Institute, Nanchang, Jiangxi 330006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wan Zhang
Department of Cardiology, The First Affiliated Hospital of Nanchang University; Jiangxi Hypertension Research Institute, Nanchang, Jiangxi 330006, People’s Republic of China
Tel +86-15907086833
Fax +86-0791-88692748
Email [email protected]
Purpose: Elevation of blood homocysteine (Hcy) level (hyperhomocysteinemia) is a risk factor for cardiovascular disorders and is closely associated with endothelial dysfunction. The present study aims to investigate the protective effect and underlying mechanism of nanoscale selenium (Nano-Se) in Hcy-mediated vascular endothelial cell dysfunction in vitro and in vivo.
Materials and Methods: By incubating vascular endothelial cells with exogenous Hcy and generating hyperhomocysteinemic rat model, the effects of Nano-Se on hyperhomocysteinemia-mediated endothelial dysfunction and its essential mechanisms were investigated.
Results: Nano-Se inhibited Hcy-induced mitochondrial oxidative damage and apoptosis by preventing the downregulation of glutathione peroxidase enzyme 1 and 4 (GPX1, GPX4) in the vascular endothelial cells, thus effectively prevented the vascular damage in vitro and in vivo in the hyperhomocysteinemic rats. Nano-Se possessed similar protective effects but lower toxicity against Hcy in vascular endothelial cells when compared with other forms of Se.
Conclusion: The application of Nano-Se could serve as a novel promising strategy against Hcy-mediated vascular dysfunction with reduced risk of Se toxicity.
Keywords: nano-selenium, Nano-Se, homocysteine, endothelium dysfunction, glutathione peroxidase enzymes, GPXs, reactive oxygen species, ROS
Introduction
Homocysteine (Hcy) is a sulfur-containing amino acid produced by the demethylation of methionine. Nutritional and genetic factors such as deficiencies of vitamins B6, B12, folic acid, and mutations of Hcy metabolic enzymes are associated with the increase of Hcy levels.1 Accumulating epidemiologic studies have shown that moderately elevated plasma Hcy levels are associated with increased risk of cardiovascular disease, and hyperhomocysteinemia has been recognized as an independent risk factor for atherosclerosis.2 Moreover, hyperhomocysteinemia combined with other risk factors like hypertension may cause more severe outcomes in cardiovascular disease.3
Selenium (Se) is an essential trace element for various aspects of human health including immune response, cancer prevention and therapy. Se is incorporated as selenocysteine in various antioxidant enzymes like glutathione peroxidase (GPX) and thioredoxin reductase (TrxR). The antioxidant properties of these selenoproteins may be particularly important in carcinogenesis and cardiovascular disease. Se is a double-edged sword that has both antioxidant and pro-oxidant effects which depend on its dose and chemical form. Bioavailable Se from dietary sources includes both inorganic (eg selenate, selenite) and organic (eg selenomethionine and selenocysteine) forms. These seleno-compounds comprise redox states of +6, +4 and +2. In recent years, nanoscale Se (Nano-Se) using Se0 has attracted more and more attention. It has been shown that Nano-Se has robust cytotoxicity to tumor cells but can be well tolerated by normal cells.4 Many preclinical and clinical studies have reported the preventive and therapeutic effects of Nano-Se in cancer development and progression.5 Nano-Se also has antiviral and antibacterial activities,6 protective effects against metal intoxication,7 immunostimulatory effects,8 etc. However, the potential of Nano-Se in the prevention and treatment of cardiovascular disease has not been fully explored.
Endothelial dysfunction plays an important role in Hcy-mediated vascular injury.9 However, the mechanisms by which Hcy participates in the pathogenesis of endothelial dysfunction are unclear. Studies have supported close relationships among the elevated levels of Hcy, excessive concentration of reactive oxygen species (ROS) in vascular cells, and the occurrence of multiple cardiovascular disease,10,11 suggesting that ROS may serve as a link between Hcy and endothelial dysfunction in cardiovascular disease. Reduced glutathione (GSH) is the most abundant antioxidant in the cells, acting as an important antioxidant defense system in mammals. GPXs are essential factors involved in the GSH antioxidant defense system. Among the four major GPX isozymes, GPX1 is localized in cytosol and GPX4 is identified in the cytosol, nucleus and mitochondria, contributing to antioxidant defense in mammalian cells.12 Notably, GPX4, the phospholipid hydroperoxide glutathione peroxidase (PHGPX), is a unique antioxidant enzyme in the protection of biomembranes exposed to oxidative stress. Especially, mitochondrial GPX4 can suppress the release of cytochrome c from the mitochondria during apoptosis which is induced through mitochondrial death pathway.13 Studies have shown that elevated Hcy decreases the expression of GPX1 in endothelial cells in vitro and in hyperhomocysteinemic mice in vivo.14,15 However, the effect of Hcy on GPX4 and mitochondrial redox status is still not clear, and the importance of manipulating GPX1 and GPX4 by Se compound in preventing Hcy-mediated endothelial dysfunction needs further investigations.
In brief, this study aims to detect the protective role of Nano-Se in Hcy-mediated vascular endothelial cell damage and dysfunction in vitro and in vivo, to investigate the underlying mechanism of Nano-Se’s protective effect against Hcy, and to compare the toxicity among different forms of Se in the presence of Hcy. Our results indicated that the use of Nano-Se could serve as a novel promising strategy in the treatment of Hcy-mediated cardiovascular disease.
Materials and Methods
Reagents
Homocysteine, sodium selenite, selenomethionine, BSA, glutathione, superoxide dismutase, catalase, zVADfmk, rhodamine 123, propidium iodide (PI), Cytotoxicity detection kit, anti-β-actin, thiobarbituric acid (TBA), NADH, ubiquinone, succinate, DCPIP, decylubiquinone oxidized/reduced cytochrome c, antimycin A, DTNB, acetyl coenzyme A and KCN were purchased from Sigma Aldrich (St. Louis, MO, USA). CM-H2DCF-DA, MitoSOX™ Red superoxide indicator, nonyl acridine orange (NAO) and dihydroethidium were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The annexin V fluorescein isothiocyanate (FITC), Z VAD, caspase 3 activity assay kit were purchased from BD Biosciences (San Jose, CA, USA). Cytochrome c release assay kit was from EMD Biosciences-Calbiochem (San Diego, CA, USA). The GSH assay kit was from Cayman Chemical Company (Ann Arbor, MI, USA). The GPX1 overexpressing plasmid (cat. no. SC119804), GPX4 overexpressing plasmid (cat. no. SC324086) and pCMV6-A carrier vector (cat. no. PS10020) were purchased from OriGene (Rockville, MD, USA). Anti-human γ glutamyl cysteine synthetase (GSH1; cat. no. sc-28965) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-caspase-3 (cat. no. 9664) was from Cell Signaling Technology (Danvers, MA, USA). Anti-GPX1 (ab22604) and anti-GPX4 (ab125066) was from Abcam (Cambridge, MA, USA).
Preparation of Nano-Se
The Nano-Se was fabricated based on a modified method.16 Briefly, 40 mL 10 mM sodium selenite was mixed with 160 mL 10 mM glutathione (GSH) solution containing 1.25 mg/mL BSA. The mixture was adjusted to pH 7.1 with 1.0 M sodium hydroxide, instantly forming red Nano-Se and oxidized glutathione (GSSG). The red solution was dialyzed against double distilled water for 96 h, the water was changed every 24 hours to separate GSSG from the Nano-Se. The final solution containing Nano-Se and BSA was lyophilized and stored at room temperature. Transmission Electron Microscopy (TEM) showed the size of red elemental Se was between 20–40 nm (Figure 1A).
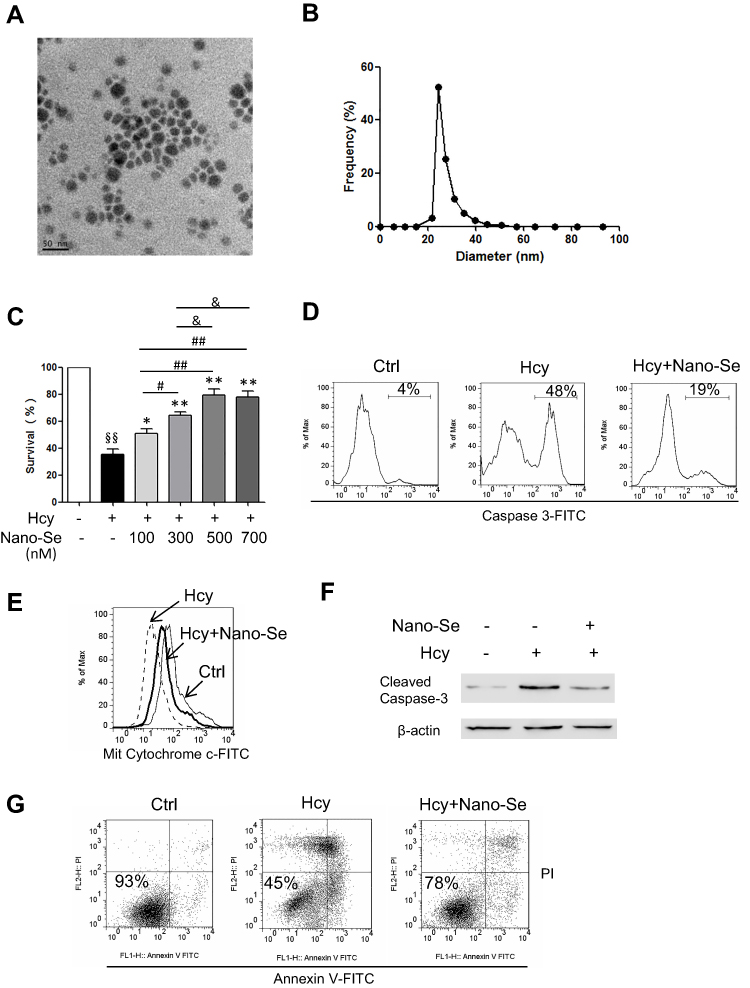 |
Figure 1 Nano-Se protected HUVECs against Homocysteine-mediated apoptosis. (A) Transmission electron microscopy image of Nano-Se. (B) Dynamic light scattering measurement. (C) Dose-dependent effect of Nano-Se on HUVEC viability with Hcy. The cells were pre-treated with 100–700 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 24 hours. Cell viability was analyzed by LDH release assay. The histograms show the mean ± SEM of three separate experiments, each measured in triplicate (§§p<0.01 vs Ctrl; *p<0.05, **p<0.01 vs Hcy treatment; #p<0.05 and ##p<0.01 vs Hcy+100 nM Nano-Se treatment; &p<0.05 vs Hcy+300 nM Nano-Se treatment). (D) Caspase 3 activation in HUVECs treated with Hcy in the presence or absence of Nano-Se. The cells were pre-treated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours. Caspase 3 activation was detected by flow cytometry analysis. The numbers indicate the gating of a subpopulation of cells with positive caspase 3 activation. Representative histograms of three separate experiments are shown. The numbers indicate the gating of the subpopulation of cells with positive caspase 3 activation. (E) Determination of mitochondrial cytochrome c in HUVECs with 1.5 mM Hcy in the presence or absence of 500 nM Nano-Se for 20h, detected by flow cytometry analysis. The cells were pre-treated with 500 nM Nano-Se 8 hours before 1.5 mM Hcy was added. The overlays show the distribution of mitochondrial cytochrome c fluorescence intensity of each cell population. Representative histograms of three separate experiments are shown. (F) Expression of cleaved caspase-3 in HUVECs with Hcy in the presence or absence of 500 nM Nano-Se. The cells were pre-treated with 500 nM Nano-Se for 8 hour before incubation with 1.5 mM Hcy for 20 hours. Cell lysates were assayed for cleaved caspase-3 using Western blot analysis. The representative Western blot results were shown, with β-actin expression as an internal control. The experiments were performed three times. (G) Effect of Nano-Se on HUVEC viability with Hcy for 24 hours. The cells were preincubated with 500 nM Nano-Se for 8 hours before 1.5 mM Hcy was added. Cell viability was measured using annexin V/PI double staining. Representative dot plots of a CLL sample are shown, with numbers indicating the percentage of viable cells (annexin V/PI double negative). The experiments were performed three times. Abbreviations: Ctrl, control; Hcy, homocysteine. |
Cell Culture
Human umbilical vein cells (HUVECs) were isolated by collagenase treatment of vessels as previously described,17 and cultured in endothelial cell medium (ScienCell, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum, 1% endothelial cell growth supplement (ScienCell, Carlsbad, CA, USA) and penicillin (100 U/mL) + streptomycin (100 μg/mL). HUVECs were used in 2th- 6th passage. Each subject provided signed written informed consent and the study was approved by the ethics committee of the First Affiliated Hospital of Nanchang University.
Animals and Treatment
Male Sprague-Dawley (SD) rats weighing between 200–240g and male Kunming mice weighing between 18–24g from the Animal Center, Jiangxi University of Traditional Chinese Medicine, PR China, and male 10-week-old spontaneously hypertensive rats (SHR) and age- and sex-matched normal Wistar rats from Beijing Vital River Laboratory Animal Technology Co., Ltd. were used for the experiments. The animals were maintained under normal laboratory condition of humidity (50±10%), temperature (25±2 °C) and a 12-h light/dark cycle for 7 days, and allowed free access to food and water ad libitum. The care and use of the laboratory animals followed the guidelines and all animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanchang University, PR China. In the experiments of proving the effect of Nano-Se on hyperhomocysteinemic rats, SD rats or SHRs or Wistar rats were randomly divided into a Hcy group and a Hcy+Nano-Se group with 6 rats per group, respectively. The protocol of the study, including doses and duration of treatment for Nano-Se, were all designed according to our pilot studies and previous reports,18–20 which have demonstrated its protective effects. Chronic hyperhomocysteinemia was induced as described previously,21,22 with a minor modification. Hcy dissolved in 0.9% NaCl was buffer to pH7.4, and administering subcutaneously twice a day at 8h intervals from day 1 to day 21 of male SD rats or from 13th to 16th week-of-age of male hypertensive rats and Wistar rats. During the first week, rats receive 0.3 μmol Hcy/g body weight, and 0.4 and 0.6 μmol Hcy/g body weight during the second and third weeks, respectively. Rats subjected to this treatment achieved plasma Hcy levels similar to those found in homocystinuric patients (Figure S3). Meanwhile, rats were given oral Nano-Se at the dose of 1 mg Se/kg. Control rats received a saline solution in the same volumes. The rats were sacrificed after 21 days’ treatment. In the toxicity experiments of Se-containing compounds in the presence of Hcy, L-Methionine was orally administered to male Kunming mice at a dose of 150 mg/kg to induce hyperhomocysteinemia.23 Mice were randomly divided into 6 groups with 8 mice per group. Sodium selenite, SeMet or Nano-Se was administered orally at the doses indicated in Table 1. Cumulative mortality within 20 days after the treatment was used for the calculation of LD50. In the short-term toxicity experiments, mice were randomly divided into 4 groups with 8 mice per group, and were orally administered 5 mg Se/kg sodium selenite, SeMet or Nano-Se for 10 consecutive days; or Hcy mice were randomly divided into 6 groups with 8 mice per group, and were orally administered 0.5, 1, 2, 4, 6 mg Se/kg of Nano-Se for 10 consecutive days. The basal diet was shown by analysis to contain 0.02 mg Se/kg. In the bioavailability experiments, Se-deficient (0.01 ppm Se) diet was obtained from Guangdong Medical Laboratory Animal Center, PR China. Mice were fed with a Se-deficient diet for 6 weeks, then were randomly divided into 7 groups with 8 mice per group. They were orally administered saline (as control) or sodium selenite, SeMet and Nano-Se at doses of 0.05 mg Se/kg and 1 mg Se/kg for 10 days. The Se-deficient diet was still used during this period. At the end of each experiment, mice were sacrificed by decapitation and trunk blood was collected; rats were euthanized with pentobarbital sodium and blood samples were drawn by cardiac puncture. Liver tissue from mice was excised for the determination of GPX1 activity and a part was fixed in cold 4% formaldehyde for histopathological assessment of liver damage. The thoracic aortas from rats were excised and carefully cleaned of periadventitial tissue for detection of vascular ROS production and aortic relaxation; or fixed in 4% formaldehyde for histopathological assessment of endothelium damage or in 2.5% buffered glutaraldehyde for electron microscopy; or kept in −80 °C until use.
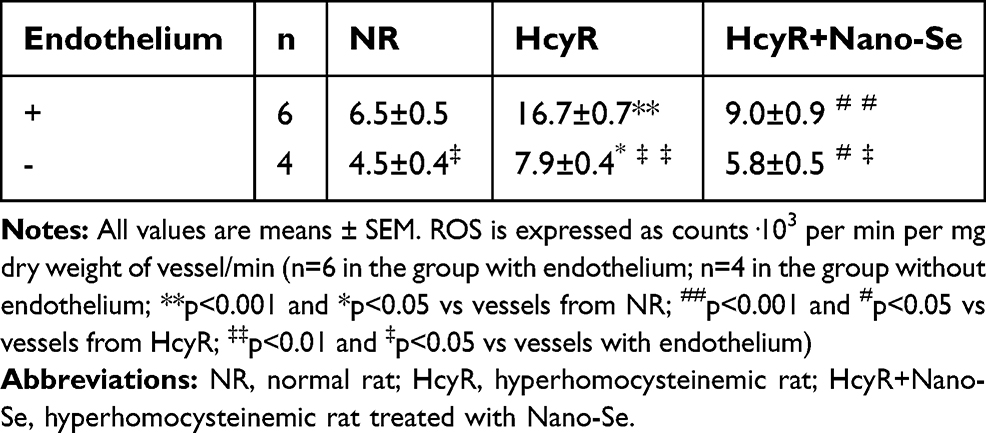 |
Table 1 ROS Production in Intact Aortic Ring Segments from NR, HcyR and HcyR+Nano-Se with or Without Endothelium |
Cell Viability Assays
Cell death was assessed by the measurement of lactate dehydrogenase (LDH), released from dead cells or determined by flow cytometry after double staining cells with annexin V-FITC and PI as described previously.24 HUVECs were treated with various compounds under the conditions indicated in the figure legends. LDH release into culture supernatants was detected by colorimetric enzyme-linked immunosorbent assay, using the cytotoxicity detection kit (LDH) from Sigma, according to manufacturers’ instructions. All assays were carried out at least three times.
Detection of Cellular and Mitochondrial ROS Levels
The cellular and mitochondrial ROS levels were detected with a fluorescent probe of CM-H2DCF-DA and MitoSOX Red, respectively. In brief, the cells were cultured under various experimental conditions and then incubated with 1 μM CM-H2DCF-DA for 60 minutes or with 2 μg/mL MitoSOX Red for 30 minutes at 37°C in the dark. After washing twice, the resulting samples were measured using flow cytometry, and the results were analyzed based on forward scatter/side scatter gating to differentiate between dead and viable cells using the built-in software.
Determination of Thiobarbituric Acid Reactive Substances (TBARS)
The TBARS fluorescence method was used to measure membrane lipid peroxidation in both cells and aortic tissue.25 HUVECs were washed, scraped on ice; the aortic rings were cleaned of adherent tissue in an ice-cold PBS, flash-freezed in liquid nitrogen, and homogenized in 10 vol of ice-cold 0.05 M phosphate buffer, pH 7.0, previously bubbled with gas nitrogen; aliquots were removed for protein determination. Cell suspensions and tissue homogenates were added to a solution of 0.3 mL of 10% TCA, and 0.15 mL of TBARS reagent (0.34% 2-thiobarbituric acid in 50% glacial acetic acid) was added. The solution was incubated at 90°C for 45 min, cooled and centrifuged at 1300 g for 15 min, and fluorescence was measured at 532 nm. The concentrations of TBARS were calculated from a standard calibration curve of malondialdehyde (MDA) equivalents generated by acid-catalyzed hydrolysis of known amounts 1,1,3,3-tetramethoxypropane.
Assessment of Mitochondrial Respiratory Chain Enzymatic Activities
Mitochondria of endothelial cells were isolated and the electron transport chain complex activities were assessed as described previously.26 Briefly, HUVECs were detached by trypsin-EDTA, flash-freezed in liquid nitrogen and homogenized in ice-cold hypotonic Tris buffer (pH 7.6). After adding 1.5 M sucrose, the mixture was centrifuged at 600 g for 10 min at 2 °C, and the supernatant containing mitochondria was collected and centrifuged at 14,000 g for 10 min at 2 °C. The mitochondrial fraction was stored at −80 °C. The total protein concentration was quantified using a BCA Protein Assay Kit (Pierce Biotechnology, #23225). Activities of complex I–IV and mitochondrial matrix marker enzyme citrate synthase were determined by an enzyme kinetic analysis.26 For measuring complex activities, 20–50 μg isolated mitochondria were resuspended in respective respiration buffers of complex I–IV in the presence of specific substrates/electron acceptors and inhibitors. These substrates were NADH and ubiquinone for complex I; succinate, DCPIP and decylubiquinone for complex II; decylubiquinol and oxidized cytochrome c for complex III; and reduced cytochrome c for complex IV, and was calculated as the faction sensitive to the specific inhibitors rotenone (complex I) and antimycin A (complex III). Citrate synthase activity was measured in buffer containing DTNB and acetyl coenzyme A. The values expressed as the ratio of activities of complex I–IV and citrate synthase.
Mitochondrial Respiration Activity
Mitochondrial respiration in whole cells was measured by an oxygen consumption assay, as described.27 Following incubating with 1.5 mM Hcy for 20 hours, HUVECs were resuspended in 1 mL of fresh culture medium pre-equilibrated with 21% oxygen at 37°C, followed by applying the cells to the sealed respiration chamber of a Clark-type oxygen measuring system (Oxytherm; Hansatech Instruments, Cambridge, UK) with constant stirring.
Analysis of Mitochondrial Oxidative Damage, Cytochrome C Release, Caspase 3 Activation and Mitochondrial Transmembrane Potential
Cardiolipin is the predominant lipid component of the internal mitochondrial membrane, and it contributes to the maintenance of its structure.28 The fluorescent dye, NAO, which specifically binds to cardiolipin, is used to measure the oxidation of cardiolipin.29 Following incubating with 1.5 mM Hcy for 20 hours, HUVECs were labeled with 50 nM NAO for 15 min and analyzed using flow cytometry, as previously described.27 A cytochrome c release kit (EMD Millipore, San Diego, CA, USA) and caspase 3 activation assay kit (BD Biosciences, San Jose, CA, USA) were used to measure the loss of mitochondrial cytochrome c and the levels of activated caspase 3, according to the manufacturer’s instructions. Rhodamine 123 (Invitrogen Life Technologies) was used to evaluate mitochondrial transmembrane potential. Following incubating with 1.5 mM Hcy for 20 hours, HUVECs were labeled with 1 μM Rhodamine 123 for 60 min and analyzed using flow cytometry, as previously described.27
Western Blot Analysis
After being cultured under various experimental conditions, the cells were harvested and washed in cold PBS, and directly solubilized in buffered solution containing 10 mM pH 7.6 Tris–HCl, 1% SDS and the complete protease inhibitor cocktail (Roche Diagnostics Ltd, Mannheim, Germany). Frozen thoracic aortas were homogenized in ice-cold lysis buffer containing 50 mM pH 7.4 Tris–HCl, 150 mM NaCl, 1 mM EDTA, 1% TritonX-100, 0.5% Na-deoxycholate, 0.1% SDS, 1 mM PMSF, 1mg/mL aprotinin, leupeptin and pepstatin, 1 mM Na3VO4, 1 mM NaF. The total protein concentrations were quantified using a BCA Protein Assay Kit (Pierce Biotechnology, #23,225), and then adjusted to 2 μg/mL with sample buffer containing 250 mM pH 6.8 Tris–HCl, 4% SDS, 10% glycerol, 0.006% bromophenol blue and 2% mercaptoethanol. The lysates were heated at 95°C for10 minutes, and equal amounts of proteins were separated on SDS–PAGE in a Mini-Protean II Dual Slab Cell (Bio-Rad Laboratories, Hercules, CA, USA). The proteins were then transferred on to nitrocellulose membranes using a Mini Trans-Blot Transfer Cell (Bio-Rad Laboratories). The transfer was performed at 4°C for 2 hours at a constant voltage setting of 110 V. The blots were blocked in 5% skimmed milk for 1 hour at room temperature. The membranes were then probed with the following primary antibodies: GCLC, caspase-3, GPX1 and GPX4, all at 1:1000 dilution, and β-actin at 1:10,000 dilution. After incubation for 2 hours at room temperature, the blots were washed three times for 10 minutes in PBS containing 0.1% Tween-20, and then incubated for 1 hour at room temperature in the following secondary antibodies: goat anti-rabbit polyclonal antibody for GCLC, caspase-3, GPX1 and GPX4 detection, all at 1:3,000 dilution, and goat anti-mouse polyclonal antibody for β-actin detection at 1:20,000 dilution. The blots were then washed three times for 10 minutes with the same buffer as above and incubated in enhanced chemiluminescence detection reagents (GE Healthcare Life Sciences, Chalfont, UK) for 1 minute. The blots were then exposed to an X-OMAT AR X-ray film (Kodak, Rochester, NY, USA) for between 10 seconds and 5 minutes.
GPX1 and GPX4 Enzyme Activity
After being cultured under various experimental conditions, the cells were washed, scraped on ice; around 100 mg tissues were flash-freezed in liquid nitrogen. The cell pellet or tissue were homogenized in cold buffer containing 50 mM Tris-HCl, pH 7.5, 5 mM EDTA and 1 mM DTT, and centrifuge at 10,000 x g for 15 minutes at 4°C. The blood was centrifuged at 1000 x g for 10 minutes at 4°C, and the plasma was collected. Lysates or plasma were stored at −20°C prior to protein estimation and activity assay. The total protein concentrations were quantified using a BCA Protein Assay Kit (Pierce Biotechnology, #23,225). GPX1 or GPX4 enzyme activities were determined using hydrogen peroxide or phospholipid hydroperoxide as the substrate and a coupled enzymatic assay with glutathione reductase and oxidized glutathione (GSSG) as previously described.30 Briefly, 10 μL of cell lysate were added to 1 mL buffer containing 0.1 M pH 7.6 Tris-HCl, 5 mM EDTA, 1 mM azide, 3 mM GSH, 0.1% peroxide-free Triton X-100, 0.1 mM NADPH and 1.2 U glutathione reductase. The reaction was started at 37 °C by the addition of either` hydrogen peroxide or phosphatidylcholine hydroperoxide (for GPX1 and GPX4 activities, respectively), and the change in absorption at 340 nm was recorded.
GPX1 and GPX4 Transfection
HUVECs were transfected using the Amaxa nucleofection system (Amaxa, Gaithersburg, MD, USA) in procedures described by the manufacturer. Briefly, 1×106 cells were transfected with 2 μg of GPX1 or GPX4 expression vector DNA (Origene, MD, USA) or empty vector (as control, Origene, Rockville, MD, USA) per cuvette in 100 μL of HUVEC Nucleofector solution using the A034 setting on the nucleofector. Cells were supplemented with 100 nM sodium selenite and used between 1 and 3 days after Nucleofection.
Biochemical Parameters
An aspartate aminotransferase (AST) assay kit, an alanine aminotransferase (ALT) assay kit and a homocysteine assay kit from Nanjing Jiancheng Bioengineering Institute, Nanjing, P.R. China, were used to detect ALT activity, AST activity and Hcy concentration, according to the manufacturer’s instructions.
In situ Detection of Vascular ROS Production
ROS generation was measured by detection of fluorescent dihydroethidium (DHE) oxidation products as described.31 Briefly, unfixed aortic rings were cryopreserved by incubation with PBS containing 30% sucrose for 1–2 h, included in OCT, frozen, and 10 μm cross sections were obtained in a Leica CM1850 cryostat (Leica, Germany). Sections were incubated in a humidified chamber at 37 °C for 30 min in HEPES-buffered solution containing 130 mM NaCl, 5 mM KCl, 1.2 mM MgCl2, 10 mM glucose, 10 mM HEPES, buffered to pH 7.3. Then the sections were further incubated for 30 min in HEPES solutions containing DHE in the dark. Images were obtained with an Olympus BX51 microscope (Tokyo, Japan). This fluorescence was evaluated in at least three sections of each preparation.
Estimation of Vascular ROS Production
The vascular ROS production was measured using lucigenin chemiluminescence as described with minor modification.32 Briefly, the thoracic aortas were carefully cleaned of periadventitial tissue, and dissected into 2–3 mm each and longitudinal cut to expose endothelium. For the endothelium-independent examination, the endothelium was removed by inserting the closed tips of a metal hemostat into the ring segment and rolling it gently on moistened filter paper. Aortic rings were then placed in organ chambers filled with Krebs-Hepes buffer maintained at 37 °C and gassed with 95% O2 and 5% CO2. After equilibration for 10 min, the aortic ring segment was transferred to a scintillation vial containing 250 μM lucigenin in a total volume of 2 mL of Krebs-Hepes buffer for determining the basal ROS level. The chemiluminescence was recorded every minute for 15 min by a liquid scintillation counter (Wallac 1409, Turku, Finland) switched to the out-of-coincidence mode. The respective background was subtracted from the total count. The thoracic aortas rings were then dried by placing them in a 90 °C for 24 h, for determination of dry weight. Lucigen counts were expressed as counts · 103 per min per mg dry weight.
Transmission Electron Microscopic Investigation
A small piece of the thoracic thoracic aortas was removed from the rat immediately after sacrifice, placed in fixative, and minced into 3–4 mm rings. The rings were fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) at 4°C for 3 hours, then rinsed in buffer, post fixed in 1% OsO4, dehydrated in ethanol and embedded in Epon 812. Thin sections after staining with 2% uranyl acetate and lead citrate were examined with TECNAI G2 20 TWIN (FEI, Eindhoven, The Netherlands).
Histomorphological Examination
The thoracic aortas were cut into 1 cm rings and a part of the liver was excised. The tissues were fixed in cold 4% formaldehyde for more than 24 hours, processing, and embedding in paraffin, 5 μm thick slices were obtained that were sectioned at equally spaced intervals, and stained with hematoxylin and eosin (H&E) staining for morphometry. The samples were examined with an Olympus BX51 microscope (Tokyo, Japan).
Endothelium-Dependent Relaxations
Aortic rings (2–3 mm) were mounted in a wire myograph (Multi Myograph System DMT-620, Danish Myotechnologies, Denmark) in Krebs solution at 37 °C and gassed with 95% O2 and 5% CO2. Rings were stretched to 2g of tension and equilibrated for 90 min. The aortic rings were then activated with 120 mM KCl and secondly with 1 μM phenylephrine (PE) for 30 min. After a stable contraction plateau was reached, the rings were exposed to endothelium-dependent vasodilator acetylcholine (Ach, 1 nM–3 μM), and a concentration-response curve was constructed.
Blood Hcy Detection, Blood Pressure Measurement and Analysis of Cellular GSH
Please refer to the Supplemental Methods for details.
Statistical Analysis
All experiments were performed at least three times. Statistical significance was estimated by using Student’s t-test. P < 0.05 was considered to possess a statistically significant difference. Bar graphs and plots were produced on GraphPad Prism 5 software (GraphPadSoftware, Inc., La Jolla, CA, USA).
Results
Nano-Se Inhibited Homocysteine-Induced Apoptosis of Vascular Endothelial Cells
We produced Nano-Se by reducing selenite with GSH in the presence of BSA that aggregate into particles 20–40 nm in size (Figure 1A), which was further confirmed by dynamic light scattering (DLS) measurement (Figure 1B). We first analyzed the survival of HUVECs in the presence of Hcy. Analysis of LDH release indicated that increased concentration of Hcy lead to a decrease in cell viability, and 1.5 mM Hcy remarkably decreased cell survival to 40% (Figure S1). However, the decrease in cell viability due to 1.5 mM Hcy treatment was reduced by Nano-Se preincubation in a dose-dependent manner (Figure 1C). Nano-Se significantly improved cell survival against Hcy when used at a concentration of ≥ 100 nM, and reached the maximal protection at 500 nM. To explore the mechanism of Nano-Se-mediated protection against Hcy in vascular endothelial cells, we first analyzed the cell death manner induced by Hcy. The prevention of Hcy-induced cell death by Z-VAD-fmk, a pan-caspase inhibitor, indicated that cell death induced by Hcy was caspase-mediated and Hcy was likely to induce apoptosis in HUVECs (Figure S2). Consistently, we found that exposure to Hcy induced the activation of caspase-3 in HUVECs (Figure 1D). Importantly, 500 nM Nano-Se decreased Hcy-mediated caspase-3 activation from 48% to 19% (Figure 1D). Meanwhile, detected by both flow cytometry (Figure 1E) and Western blot analysis (Figure 1F), 1.5 mM Hcy led to mitochondrial cytochrome c release, and can be greatly prevented by preincubation with 500 nM Nano-Se. The protective effect of the Nano-Se against Hcy-induce apoptosis was further examined by annexin V/PI assay. As shown in Figure 1G, the survival rate of HUVECs treated with 1.5 mM Hcy was decreased from 93% to 45%, and Nano-Se increased the viability from 45% to 78% after Hcy treatment. Overall, the results above indicated that Nano-Se inhibited Hcy-induced apoptosis in vascular endothelial cells.
Nano-Se Prevented Vascular Dysfunction in Hyperhomocysteinemic and Hypertensive Hyperhomocysteinemic Rat
We further investigated the effect of Nano-Se on Hcy-mediated vascular endothelial cell damage and vascular dysfunction in vivo. Since hyperhomocysteinemic rat model was previously described and regularly used, we generated a chronic hyperhomocysteinemic rat model (HcyR) based on previous reports (Figure S3).21,22 Figure S4 shows that HcyR with or without Nano-Se treatment did not differ significantly from saline-treated rats (NR) in body weight, indicating that Hcy and Nano-Se administration did not cause malnutrition in the animals. However, the thoracic aortas of HcyR exhibited significant histological and functional changes, which were inhibited by Nano-Se treatment. As shown in Figure 2A, the thoracic aorta sections from NR showed unbroken endothelial monolayer with regularly shaped and arranged endothelial cells. However, various alterations of the thoracic aortas appeared in HcyR, including focal loss of endothelial cells, exfoliation from the vessel wall and expanded junctions between endothelial cells. In contrast, the integrity of aortic endothelial cells from HcyR was largely retained after Nano-Se treatment (Figure 2A). Meanwhile, the endothelium-dependent aortic relaxations to acetylcholine were markedly blunted in HcyR when compared with NR, whereas Nano-Se administration greatly improved endothelium-dependent aortic relaxation responses of HcyR (Figure 2B, Table S1A). Since elevated Hcy levels are associated with other risk factors like hypertension, we generated hyperhomocysteinemia in spontaneously hypertensive rat (SHR), and further examined the effect of Nano-Se in hypertensive hyperhomocysteinemic rat (SHHcyR). Hcy and Nano-Se administration did not cause malnutrition (Figure S4B and C) or change of blood pressure (Figure S5) in both SHR and Wistar rat control. As shown in Figure 2C, the aortic endothelial cells in SHR were retained; after Hcy treatment, there was a heavy loss of endothelial cells, and the arrangement of smooth muscle fiber and the elastic membrane in the media were deranged. However, the Nano-Se administration greatly prevented the loss of aortic endothelial cells in SHHcyR (Figure 2C). Similar to the SD rat, in the Wistar rat control, Hcy administration induced focal loss of endothelial cells, which were largely prevented by Nano-Se treatment (Figure 2C). Furthermore, acetylcholine-induced endothelium-dependent relaxation of the SHR thoracic aortas was much smaller than that from Wistar rat control, and the relaxation effects were further suppressed in the thoracic aortas from SHHcyR (Figure 2D, E, Table S1B, C, D). Importantly, in thoracic aortas of SHHcyR, Nano-Se treatment resulted in a 2-fold increase in relaxation at the dose of 10−6 M (Figure 2D and E, Table S1C). Overall, these results demonstrated that Nano-Se can effectively prevent Hcy-mediated vascular damage in vivo.
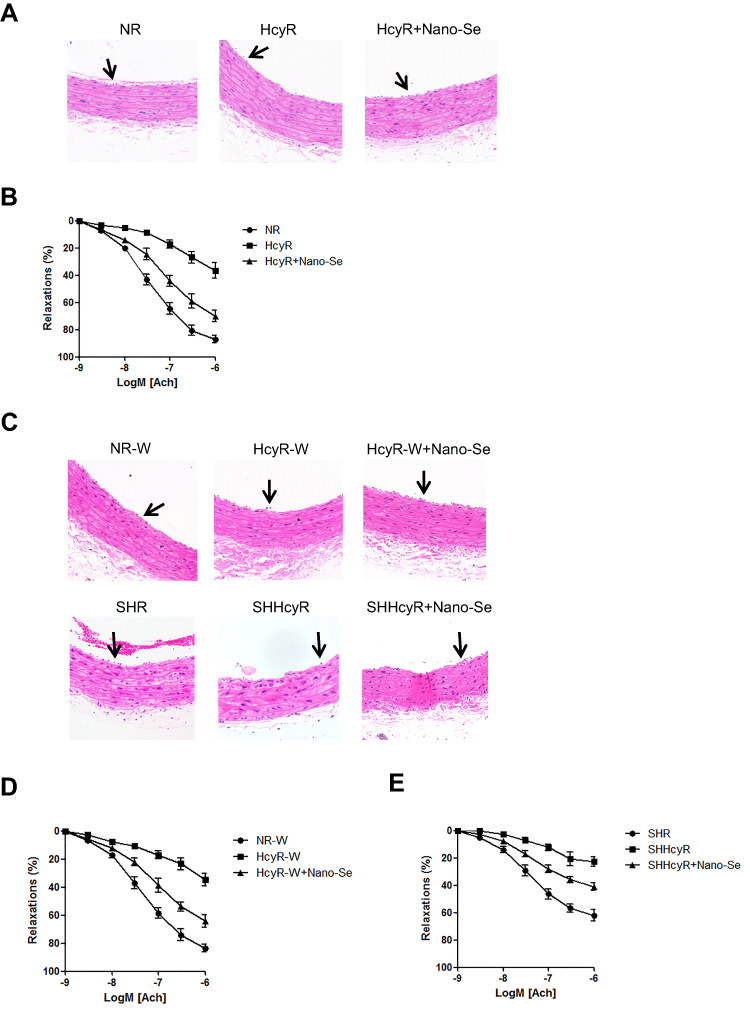 |
Figure 2 Nano-Se reduced the severity of vascular dysfunction in hyperhomocysteinemic and hypertensive hyperhomocysteinemic rats. (A) The histological structure of aorta of NR, HcyR and HcyR+Nano-Se. The representative photographs of hematoxylin and eosin (H&E) staining of aorta sections (X200) were shown. The arrows indicate endothelial cells. (B) Relaxant response of aorta to acetylcholine in NR, HcyR and HcyR+Nano-Se. (C) The histological structure of aorta and (D, E) Relaxant response of aorta to acetylcholine in NR-W, HcyR-W, HcyR-W+Nano-Se, SHR, SHHcyR and SHHcyR+Nano-Se. The representative photographs of hematoxylin and eosin (H&E) staining of aorta sections (X200) were shown. The arrows indicate endothelial cells. Abbreviations: NR, normal rat; HcyR, hyperhomocysteinemic rat; HcyR+Nano-Se, hyperhomocysteinemic rat treated with Nano-Se; SHR, spontaneously hypertensive rat; SHHcyR, spontaneously hypertensive hyperhomocysteinemic rat; SHHcyR+Nano-Se, spontaneously hypertensive hyperhomocysteinemic rat treated with Nano-Se. |
Nano-Se Counteracted Hcy-Mediated Oxidative Damage to Mitochondria in Vascular Endothelial Cells
We next investigated the mechanism of the protective effect of Nano-Se against Hcy. Se is an important component of Se-containing antioxidant enzymes including GPX, and Nano-Se protected HUVECs against Hcy-induced cell death in a similar manner to the antioxidant catalase or SOD (Figure 3A). We measured the ROS levels in HUVECs, and found that cellular and mitochondrial ROS had increased after incubation with 1.5 mM Hcy for 8 hours and 16 hours, respectively (Figure S6). Notably, mitochondrial ROS increased in the later stage, indicating that mitochondria are not the initial source of ROS induced by Hcy. Importantly, 500 nM Nano-Se decreased both cellular and mitochondrial ROS dramatically (Figure 3B and C). Since oxidation of mitochondrial cardiolipin can lead to cytochrome c release, caspase activation and apoptosis,33 we analyzed the levels of membrane lipid peroxidation. As shown in Figure 3D, incubation of HUVECs with 1.5 mM Hcy for 20 hours led to oxidation of cardiolipin detected by loss of interaction with fluorescent dye NAO, which has affinity to mitochondrial lipid cardiolipin. However, 500 nM Nano-Se pretreatment prevented the loss of NAO from 64% to 32%. Consistently, Hcy increased TBARS fluorescence in HUVECs and was decreased by Nano-Se preincubation (Figure 3E). Meanwhile, we examined whether mitochondrial electron transport chain (ETC) complex function was influenced by mitochondrial ROS with Hcy incubation. As shown in Figure 3F, the activities of ETC complex I, III, IV in total mitochondria were decreased in Hcy-treated HUVECs, and a significant decrease of O2 consumption was also observed (Figure 3G). However, 500nM Nano-Se pretreatment greatly preserved mitochondrial ETC complex function (Figure 3F, G). Furthermore, Nano-Se prevented the loss of mitochondrial transmembrane potential, an indicator of early induction of apoptosis, from 56% to 27% in HUVECs by Hcy treatment (Figure 3H). The results above indicated that the protective effect of Nano-Se was associated with the prevention against Hcy-induced mitochondrial oxidative damage and the preservation of mitochondrial ETC function in endothelial cells.
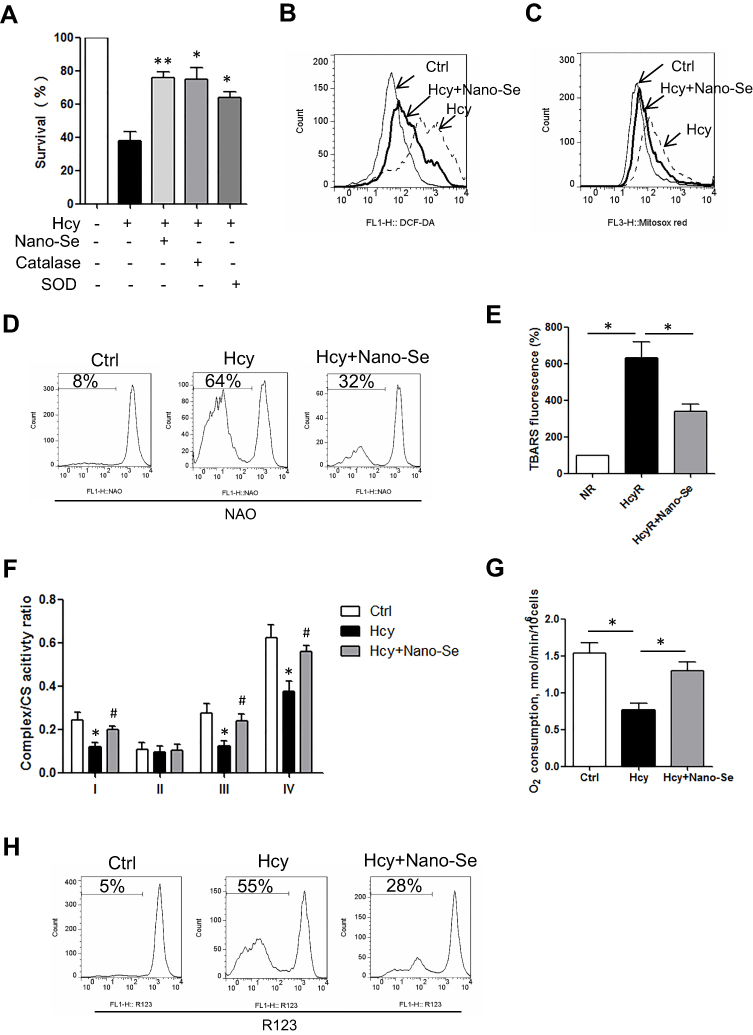 |
Figure 3 Nano-Se protected HUVECs against Hcy-mediated oxidative damage to mitochondria. (A) Effect of Nano-Se or catalase or SOD on HUVEC viability with Hcy. The cells were preincubated with 500 nM Nano-Se for 8 hours, or with 500 U/mL catalase or 150 U/mL SOD for 1 hour before incubation with 1.5 mM Hcy for 24 hours. Cell viability was analyzed by LDH release assay. The histograms show the mean ± SEM of three separate experiments, each measured in triplicate (*p<0.05; **p<0.01). (B) Determination of cellular and (C) mitochondrial ROS in HUVECs preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 16 hours. ROS was detected by flow cytometry analysis. Representative histograms of three separate experiments are shown. (D) Change of cardiolipin oxidation in HUVECs preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours. Cardiolipin oxidation was detected by flow cytometry analysis. Representative histograms of three separate experiments are shown. The numbers indicate the gating of the subpopulation of cells exhibiting loss of cardiolipin signal due to oxidation. (E) TBARS content in HUVECs preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours, measured by TBARS fluorescence method. The histograms show the mean ± SEM of three separate experiments (*p<0.05). (F) Effect of Nano-Se on mitochondria electron transport chain (ETC) function in HUVEC with Hcy. The cells were preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours. ETC complex activities were determined by an enzyme kinetic analysis. The histograms show the mean ± SEM of three separate experiments (*p<0.05 vs Ctrl; #p<0.05 vs Hcy treatment). (G) The oxygen consumption rate of HUVECs preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours, measured by an oxygen consumption assay. The histograms show the mean ± SEM of three separate experiments (*p<0.05). (H) Determination of mitochondrial transmembrane potential in HUVECs preincubated with 500 nM Nano-Se for 8 hours before incubation with 1.5 mM Hcy for 20 hours. Mitochondrial transmembrane potential was detected by flow cytometry analysis. Representative histograms are shown. The numbers indicate the gating of the subpopulation of cells exhibiting loss of mitochondrial transmembrane potential. The experiments were performed three times. Abbreviations: Ctrl, control; Hcy, homocysteine; CS: citrate synthase; TBARS: thiobarbituric acid reactive substances. |
Hcy Disabled GPX Systems and Was Prevented by Nano-Se
Because Hcy induced oxidative stress and glutathione (GSH) is the most abundant antioxidant in cells, we examined GSH system in endothelial cells after Hcy treatment. To our surprise, the GSH level in HUVECs did not change much in the Hcy-cultured group when compared with the control group (Figure S7A), and Western blot analysis even showed an increase in GSH synthesis key enzyme (GSH1) after Hcy treatment (Figure S7B). As glutathione peroxidases (GPXs) are the major enzymes that use GSH as the substrate to scavenge peroxides, we then tested if Hcy affected GPX activity and compromised the cell’s ability to utilize GSH. As shown in Figure 4A, the expression of classic GPX (GPX1) in HUVECs was downregulated after 1.5 mM Hcy treatment in a time-dependent manner, which was consistent with other reports.34,35 Since we found that Hcy-treated HUVECs exhibited an increase of mitochondrial ROS and damage, we also examined GPX4, which is mainly located in mitochondria and is important for the defense of mitochondrial ROS. We found that the expression of GPX4 was decreased in endothelial cells in the presence of 1.5 mM Hcy (Figure 4A). Further analysis of the GPX1 and GPX4 enzyme activities illustrated in Figure 4B showed that both enzyme activities were decreased in HUVEC in the presence of Hcy, confirming the inhibition of GPX1 and GPX4 in endothelial cells by Hcy. To examine the effect of Hcy-mediated GPX inhibition on apoptosis, we overexpressed GPX1 or GPX4 in HUVECs, respectively (Figure S8), and determined whether this level of overexpression could prevent mitochondrial oxidative stress and cell death. Compared with HUVECs that are transfected with empty vector, the HUVECs that overexpressed GPX1 or GPX4 had less loss of NAO with 1.5 mM Hcy treatment (Figure S9), and the extent of Hcy-mediated mitochondrial ROS upregulation was greatly reduced (Figure S10). As shown in Figure 4C, the survival rate of HUVECs transfected with empty vector was decreased from 75% to 32% after Hcy treatment; however, GPX1 or GPX4 overexpression increased the survival rate of HUVECs from 32% to 58% or 52% after Hcy incubation. These results indicated that inhibition of GPX systems may play an important part in Hcy-mediated oxidative stress and apoptosis, and increasing the expression of GPXs may serve as a promising strategy against Hcy-induced apoptosis of endothelial cells. Since GPXs are Se-containing enzymes, supplementation of Se can increase the expression of GPXs. As shown in Figure 4D, incubation of HUVECs with 1.5 mM Hcy for 16 hours greatly reduced the expression of GPX1 and GPX4, whereas 100 nM and 500 nM Nano-Se pretreatment upregulated the expression of both GPX1 and GPX4 in a dose-dependent manner. Consistently, Nano-Se increased the activities of GPX1 and GPX4 in HUVECs with Hcy incubation at the same conditions (Figure 4E). Given all this, we demonstrated that Hcy might increase cellular and mitochondrial ROS through downregulating antioxidant GPX1 and GPX4, whereas Nano-Se upregulated the expression of both GPX1 and GPX4, and thus counteracted Hcy-mediated oxidative damage of mitochondria and the following apoptosis of vascular endothelial cells.
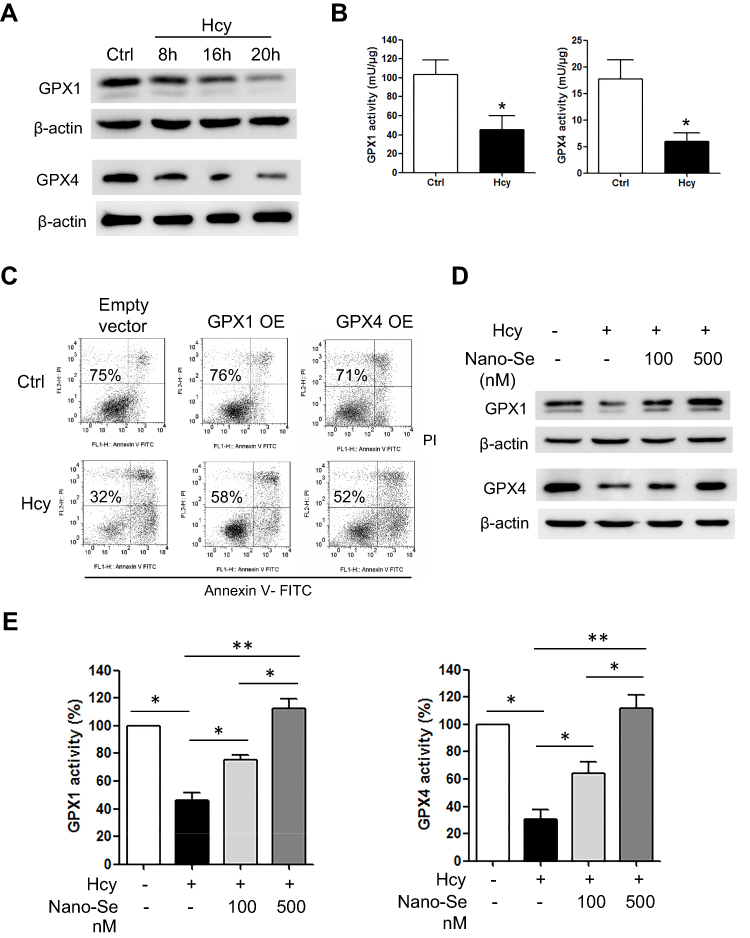 |
Figure 4 Hcy-mediated GPX1 and GPX4 inhibition and apoptosis were prevented by Nano-Se treatment. (A) Expressions of GPX 1 and GPX4 in HUVECs with Hcy incubation (1.5 mM) at given time points, as assessed by Western blot analysis. The representative Western blot results were shown, with β-actin expression as an internal control. The experiments were performed three times. (B) Determination of activities of GPX1 and GPX4 in HUVECs with Hcy incubation (1.5 mM) for 20 hours, detected by a coupled enzymatic assay. The histograms show the mean ± SEM of three separate experiments (*p<0.05). (C) Effect of GPX1 or GPX4 overexpression on HUVEC viability with Hcy for 24 hours. The cells were transfected with empty vector pCMV6 or pCMV-GPX1 or pCMV-GPX4 for 24 hours, followed by 1.5 mM Hcy treatment for another 24 hours. Cell viability was measured using annexin V/PI double staining. Representative dot plots of a CLL sample are shown, with numbers indicating the percentage of viable cells (annexin V/PI double negative). (D) Effect of Nano-Se on expressions of GPX1 and GPX4 in HUVECs with Hcy for 20 hours. The cells were preincubated with 100 nM or 500 nM Nano-Se for 8 hours before 1.5 mM Hcy was added. The expressions of GPX1 and GPX4 were assessed by Western blot analysis. The representative Western blot results were shown, with β-actin expression as an internal control. The experiments were performed three times. (E) Effect of Nano-Se on activities of GPX1 (left panel) and GPX4 (right panel) in HUVECs with Hcy for 20 hours. The cells were preincubated with 100 nM or 500 nM Nano-Se for 8 hours before 1.5 mM Hcy was added. The enzyme activities were examined by a coupled enzymatic assay. The histograms show the mean ± SEM of three separate experiments, each measured in triplicate (*p<0.05, **p<0.01). Abbreviations: Ctrl, control; Hcy, homocysteine; GPX1, glutathione peroxidase 1; GPX4, glutathione peroxidase 4; GPX1 OE, GPX1 overexpression; GPX4 OE, GPX4 overexpression. |
Effect of Nano-Se on Vascular Redox Status in Hyperhomocysteinemic Rats
We further investigated the mechanism of Nano-Se-mediated protection against Hcy in vivo. As shown in Figure 5A, we consistently found the expressions of GPX1 and GPX4 were significantly downregulated in thoracic aortas of hyperhomocysteinemic rat model (HcyR) when compared with those in normal SD rat control (NR). However, treatment with 1 mg Se/kg Nano-Se can efficiently increase the expressions of GPX1 and GPX4. Meanwhile, thoracic aortas from HcyR showed a marked increase in total ROS production, especially the endothelium-originated ROS, which was detected by dihydroethidium (DHE) fluorescence, whereas Nano-Se administration greatly inhibited ROS accumulation in HcyR thoracic aortas (Figure 5B). Aortic production of basal ROS was also detected by chemiluminescence assay. As shown in Table 1, ROS production by aortic rings of NR averaged 6.5±0.5 c.p.m.·103/mg and increased more than 2-fold in the rings of HcyR (16.7±0.7 c.p.m.·103/mg). The same results were observed in aortic rings without endothelium but to a much smaller extent, indicating that Hcy induced large amounts of endothelium-dependent ROS in the aorta. Meanwhile, removal of the endothelium decreased the aortic ROS in HcyR by 56% from 16.7±0.7 c.p.m.·103/mg to 7.9±0.4 c.p.m.·103/mg, and decreased the aortic ROS in NR by 30% from 6.5±0.5 c.p.m.·103/mg to 4.5±04 c.p.m.·103/mg, which further proved that Hcy induced a considerable amount of ROS in vascular endothelial cells. Importantly, Nano-Se treatment significantly reduced aortic ROS of HcyR but to a greater extent with endothelium integrity (Table 1). We next examined aortic lipid oxidation in HcyR. As shown in Figure 5C, the concentration of TBARS in aortic homogenates of NR was 1.1±0.3 nmol/mg protein, whereas Hcy administration induced an increase by 191% from 1.1±0.3 nmol/mg protein to 3.2±0.4 nmol/mg protein. However, after Nano-Se administration, the concentration of TBARS decreased by 41% from 3.2±0.4 nmol/mg protein to 1.9±0.3 nmol/mg protein. In addition, the ultrastructure of endothelium showed that the mitochondria in aortic endothelium cells from HcyR exhibited morphological changes (Figure 5D). The major alterations involved increased pallor of the inner compartment and swollen, signaled by an increase in size and a disrupted outer membrane, indicating that the mitochondria in aortic endothelium cells from HcyR were damaged. Consistently, Nano-Se treatment greatly preserved mitochondria morphology (Figure 5D). All these data suggested that HcyR exhibited a significant decrease in aortic GPX1 and GPX4 expressions, as well as a remarkable increase in aortic oxidative stress and the mitochondrial damage of vascular endothelial cells. Importantly, Nano-Se administration upregulated GPX expressions and relieved aortic oxidative stress in HcyR.
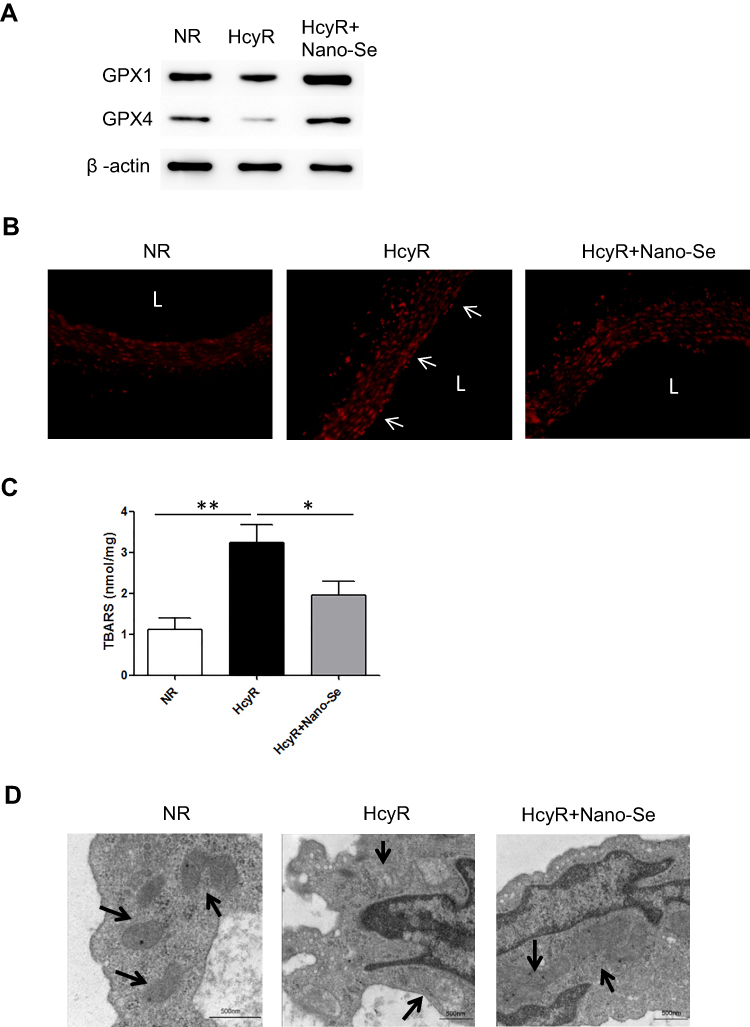 |
Figure 5 Effect of Nano-Se on vascular GPX expression, ROS and endothelial mitochondrial damage in hyperhomocysteinemic rats. (A) Expression of GPX 1 and GPX4 in the aorta of NR, HcyR and HcyR+Nano-Se as assessed by Western blot analysis. The representative Western blot results were shown. (B) Dihydroethidium (DHE) staining of the aorta of NR, HcyR and HcyR+Nano-Se. The arrows indicate endothelial cells. L, Lumen (C) TBARS content in the aorta of NR, HcyR and HcyR+Nano-Se. Representative histograms of three separate experiments are shown (*p<0.05; **p<0.01). (D) Ultrastructure of aortic endothelial cells of NR, HcyR and HcyR+Nano-Se. The representative transmission electron microscopic (TEM) images were shown. The arrows indicate mitochondria structures. Abbreviations: NR, normal rat; HcyR, hyperhomocysteinemic rat; HcyR+Nano-Se, hyperhomocysteinemic rat treated with Nano-Se; SHR, spontaneously hypertensive rat; SHHcyR, spontaneously hypertensive hyperhomocysteinemic rat; SHHcyR+Nano-Se, spontaneously hypertensive hyperhomocysteinemic rat treated with Nano-Se. |
Comparison of Nano-Se and Se-Containing Chemicals (Selenite, SeMet)
We compared the effects of inorganic (selenite), organic (SeMet) and nano form (Nano-Se) of Se on the activities of GPX1 and GPX4 in endothelial cells after Hcy treatment. As shown in Figure S11A, B, Hcy decreased GPX1 and GPX4 activities in HUVECs, whereas selenite, SeMet and Nano-Se increased GPX activities in a dose-dependent manner that reached a maximal response plateau with 500 nM to all compounds. Notably, Nano-Se has a comparable effect in increasing GPX1 and GPX4 activities in endothelial cells at 100 nM and 500 nM when compared with other forms of Se, whereas SeMet has a compromised effect at 100 nM (Figure 6A), which might due to the nonspecifically incorporating of SeMet into other proteins at lower concentrations. Meanwhile, selenite, SeMet and Nano-Se consistently decreased Hcy-mediated ROS accumulation in HUVECs (Figure 6B). We next tested the mitochondrial transmembrane potential, and found that treatment with 1.5 mM Hcy for 22 hours resulted in a 50% loss of the transmembrane potential, whereas 500 nM selenite, SeMet and Nano-Se prevented the loss to 28%, 25%, 27%, respectively (Figure 6C). The protective effects of the three forms of Se-containing reagents were also examined by annexin V/PI assay. As shown in Figure 6D, incubation with 1.5 mM Hcy for 24 hours was toxic to HUVECs, and consistently, induction of apoptosis by Hcy was significantly inhibited by all three Se-containing reagents in a similar manner, indicating the comparable protective effect of Nano-Se to other chemical forms of Se. Since Se compounds are toxic which depends on its chemical forms, we then compared the toxicity of selenite, SeMet and Nano-Se in Hcy mice simultaneously. As shown in Table 2, selenite and SeMet caused 90% mortality at the dose of 13.3 mg Se/kg and 26.2 mg Se/kg, respectively. However, Nano-Se caused only 10% mortality at the dose of 31.6 mg Se/kg and 90% mortality at a dose as high as 197 mg Se/kg. Based on these data, the LD50 of selenite, SeMet and Nano-Se in Hcy mice were 10.7 mg Se/kg (with 95% confidence limits of 8.9–13), 21.9 mg Se/kg (with 95% confidence limits of 20–24.2) and 89.2 mg Se/kg (with 95% confidence limits of 61–125.2), respectively. The acute toxicity of selenite and SeMet was 8- and 4-fold greater than that of Nano-Se in Hcy mice. These results were consistent with that in the normal mice.36–38 Since liver is the major target organ of Se toxicity, we next compared the short-term toxicity of the Se-containing reagents at a nonlethal dose (5 mg Se/kg) in Hcy mice. As shown in Figure 7A, all three forms of Se-containing reagents at 5 mg Se/kg caused growth retardation; however, selenite and SeMet retarded growth more pronouncedly than Nano-Se after 10 days treatment. In addition, serum ALT in SeMet- and selenite-treated Hcy mice were significantly decreased, as well as the serum AST in all three forms of Se-treated Hcy mice. The increase of ALT and AST was most obvious in the selenite group, whereas Nano-Se treatment either did not increase ALT or induced much less increase of AST when compared with selenite or SeMet treatment (Figure 7B). Consistently, by comparing the liver architecture, we found that selenite and SeMet caused serious pathological changes in the forms of liver cell chaos, neutrophil and lymphocyte infiltration, and appearance of necrosis regions and apoptotic bodies, whereas Nano-Se caused moderate pathological changes (Figure 7C). Therefore, Nano-Se has a lower toxicity than the inorganic and organic Se in Hcy mice, and the inorganic form of Se is even more toxic than the organic one. To exclude the possibility that the lower toxicity of Nano-Se is due to the biological inert property of elemental Se, we compared its bioavailability by administering different forms of Se at nutritional (0.05 mg Se/kg) and supranutritional (1 mg Se/kg) levels in Hcy mice for 10 days. All three forms of Se increased plasma and hepatic GPX1 activity at the dose of 0.05 mg Se/kg, and further increased at 1 mg Se/kg (Figure 7D). Nutritional SeMet was less effective than the other two forms of Se in enhancing the hepatic GPX1 activity in Hcy mice, which is consistent with the results in HUVECs (Figure 6A). To our surprise, 0.05 mg Se/kg Nano-Se treatment induced even higher hepatic GPX1 activity when compared with selenite and SeMet treatment in Hcy mice, which was consistent with the observation in vitro.39 We further examined the toxicity of Nano-Se in Hcy mice at supranutritional and nonlethal doses. As shown in Figure 7E, the absolute changes in body weight were significantly decreased in mice only when received doses above 4 mg Se/kg Nano-Se after 10 days. Consistently, serum ALT and AST activities were not changed at doses less than 6 or 4 mg Se/kg Nano-Se (Figure 7F). Overall, these results indicated that Nano-Se has a comparable protective effect and slightly higher bioavailability against Hcy while possessing a lower toxicity, and the supranutritional levels of Nano-Se had no obvious toxic effects in Hcy mice.
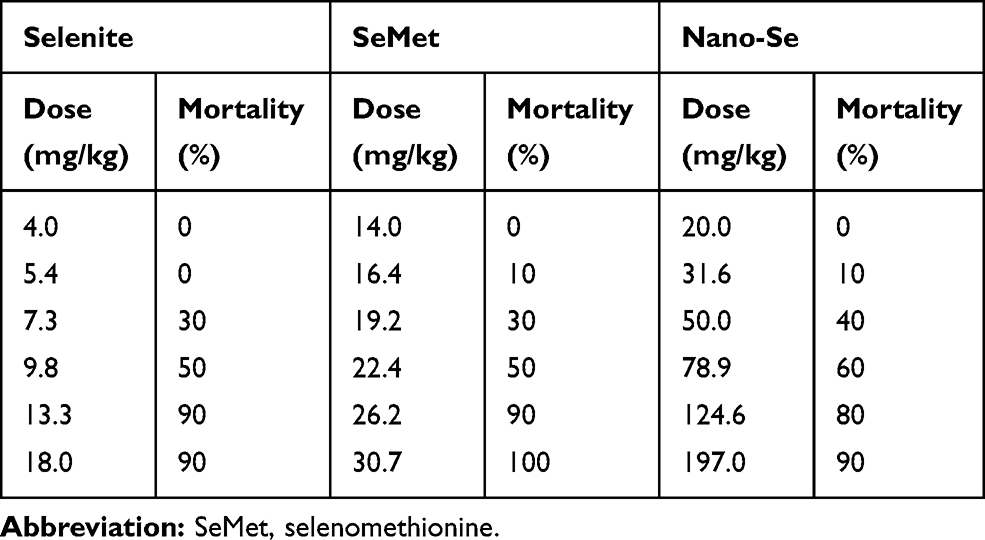 |
Table 2 Acute Lethal Effect of Selenite, SeMet and Nano-Se in Hcy Mice |
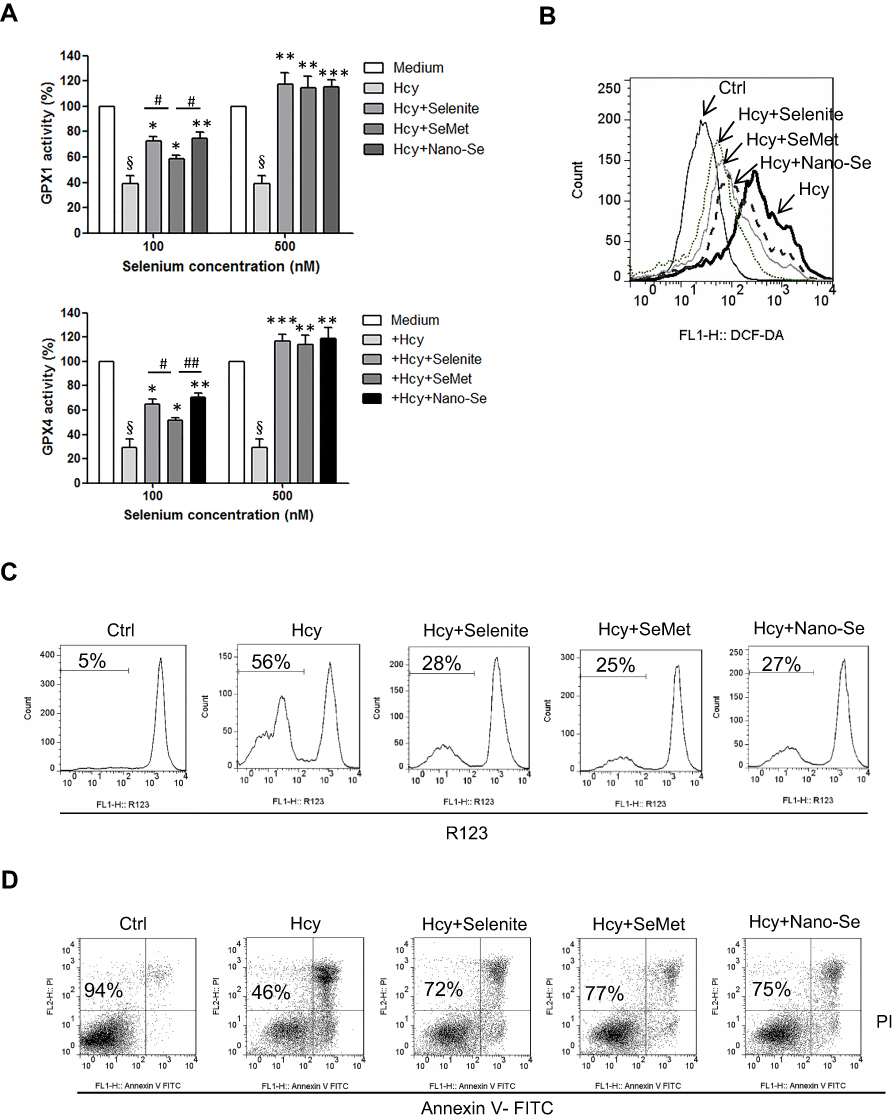 |
Figure 6 Nano-Se, selenite and SeMet increased GPX1 and GPX4 activities and protected HUVECs against Hcy-induced oxidative stress and apoptosis. (A) Effects of selenite, SeMet and Nano-Se on the activities of GPX1 and GPX4 in HUVECs with Hcy. The cells were pre-treated with 100 nM or 500 nM of selenite, SeMet or Nano-Se for 8h before 1.5 mM Hcy was added, and the enzyme activities were examined by a coupled enzymatic assay. The histograms show the mean ± SEM of three separate experiments (§p<0.05 vs Medium; *p<0.05, **p<0.01, ***p<0.001 vs Hcy treatment; #p<0.05 and ##p<0.01 vs Hcy+SeMet treatment). (B) Determination of cellular ROS in HUVECs with Hcy in the presence or absence of selenite, SeMet or Nano-Se for 16 hours, detected by flow cytometry analysis. The cells were pre-treated with 500 nM selenite, SeMet or Nano-Se 8 hours before 1.5 mM Hcy was added. Representative histograms of three separate experiments are shown. (C) Determination of mitochondrial transmembrane potential in HUVECs with Hcy in the presence or absence of selenite, SeMet or Nano-Se for 20 hours, detected by flow cytometry analysis. The cells were pre-treated with 500 nM selenite, SeMet or Nano-Se 8 hours before 1.5 mM Hcy was added. Representative histograms are shown. The numbers indicate the gating of the subpopulation of cells exhibiting loss of mitochondrial transmembrane potential. The experiments were performed three times. (D) Effect of selenite, SeMet or Nano-Se on HUVEC viability with Hcy for 24 hours. The cells were pre-treated with 500 nM selenite, SeMet or Nano-Se 8 hours before 1.5 mM Hcy was added. Cell viability was measured using annexin V/PI double staining. Representative dot plots of a CLL sample are shown, with numbers indicating the percentage of viable cells (annexin V/PI double negative). The experiments were performed three times. Abbreviations: Ctrl, control; Hcy, homocysteine; SeMet, selenomethionine. |
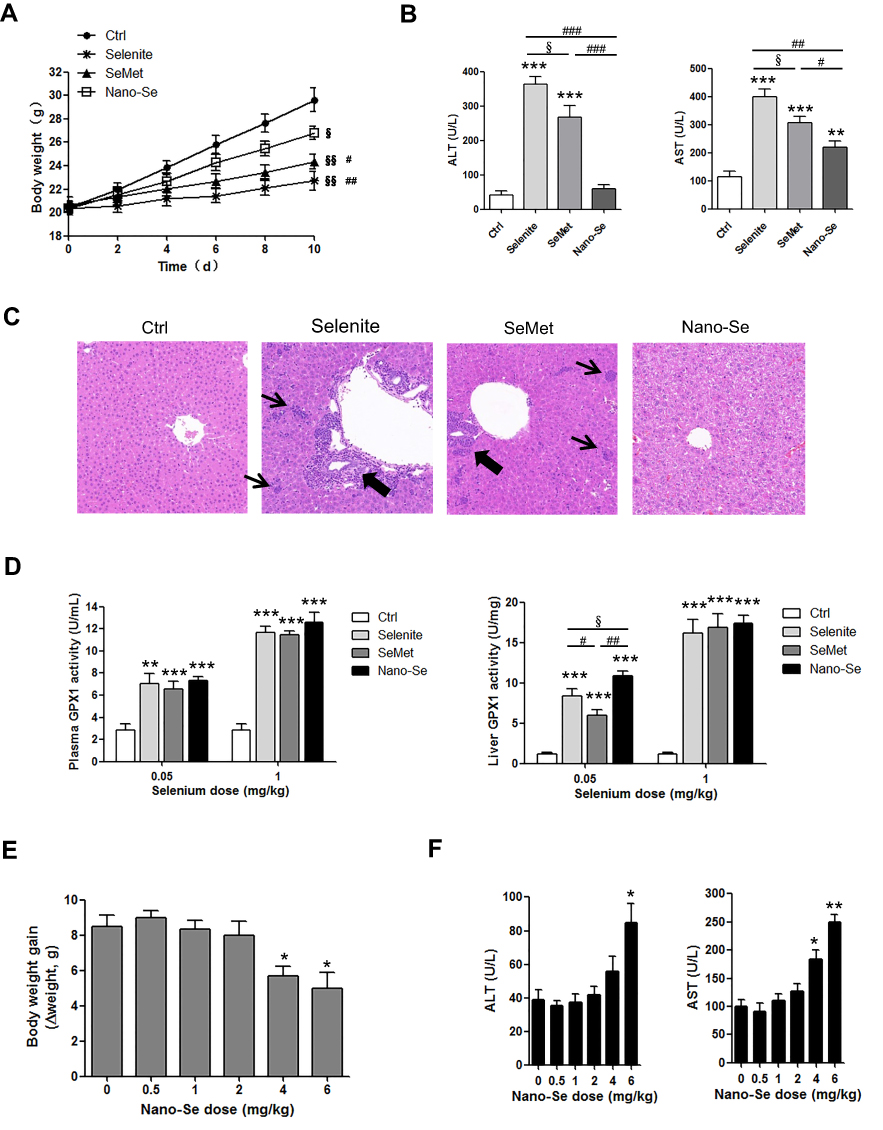 |
Figure 7 Toxicity and bioavailability of Senite, SeMet and Nano-Se in Hcy mice. (A) Growth curve, (B) Activities of hepatic metabolic enzymes ALT and AST in serum and (C) Representative sections of H&E stained of thoracic aorta of mice orally administered 150 mg/kg L-methionine and 5 mg Se/kg sodium selenite, SeMet or Nano-Se for 10 days (The arrows and thick arrows indicate the focal necrosis of liver tissues and neutrophil and lymphocyte infiltration, respectively; n=8; (A) §p<0.05 and §§p<0.01 vs Ctrl; #p<0.05 and ##p<0.01 vs sodium selenite Treatment; (B) **p<0.01 and ***p<0.001 vs Ctrl; #p<0.05, ##p<0.01 and ###p<0.001 vs Nano-Se treatment; §p<0.05 vs SeMet treatment). (D) Activities of plasma (left panel) and liver (right panel) GPX1 of mice orally administered 150 mg/kg L-methionine and sodium selenite, SeMet and Nano-Se at doses of 0.05 mg Se/kg and 1 mg Se/kg for 10 days (n=8; **p<0.01 and ***p<0.001 vs Ctrl; #p<0.05 and ##p<0.01 vs SeMet treatment; §p<0.05 vs Nano-Se treatment). (E) Body weight gain (Δweight) and (F) activities of ALT and AST in serum of mice orally administered 150 mg/kg L-Methionine and Nano-Se at doses of 0.5, 1, 2, 4, 6 mg Se/kg for 10 days (n=8; *p<0.05, **p<0.01 vs Ctrl). Abbreviations: Ctrl, control; SeMet, selenomethionine; ALT, alanine aminotransferase; AST, aspartate aminotransferase; GPX1, glutathione peroxidase 1. |
Discussion
Increased plasma level of Hcy is an independent risk factor for atherosclerosis.1 Lowering blood Hcy levels by supplementation with folic acid, vitamin B6 and B12 has proven to be an effective therapy in delaying the occurrence of vascular events.40 However, other clinical trials showed that vitamin intervention failed to lower the risk of cardiovascular disease,41 indicating that understanding the action of Hcy and searching for new approaches are urgently needed to prevent Hcy-induced events. In the present study, we show promising results that Nano-Se effectively combated Hcy-induced endothelial oxidative stress and vascular dysfunction in vitro and in vivo in hyperhomocysteinemic rats. We also exhibit the comparable protective effect of Nano-Se over other chemical forms of Se but with much lower toxicity, promising the possible therapeutic applications of Nano-Se against Hcy-induced cardiovascular disease.
The trace mineral, Se, is an essential nutrient of fundamental importance to human biology.42 The best-known role of Se is attributed to its presence in a serial of selenoprotein-containing antioxidant enzymes including GPX and TrxR. Since GPX and TrxR are the main selenoproteins expressed by endothelial cells, the role of Se in cardiovascular disease is gradually recognized, and an inverse relationship between Se concentration and incident cardiovascular disease and mortality was observed in most studies.43 Se acts as an important element in the prevention and treatment of cardiovascular disease but its health-related effects have not been fully explored. Increased plasma level of Hcy is an independent risk factor for atherosclerosis.1 The association between Se and Hcy is complicated. An animal study shows that Se deficiency decreases total Hcy levels.44 On the contrary, the studies in human suggest that there are either no correlation or inverse association between Hcy and serum Se concentrations.45–48 The discrepancy might be due to the different status of Se being considered. Besides the importance of Se’s influence on Hcy levels, the role of Se in the treatment of Hcy-mediated vascular damage is critical and remains largely unknown. In the present study, we synthesized nanoform of Se (Nano-Se) by reducing selenite with GSH in the presence of BSA that aggregate into particles 20–40 nm in size. We utilized it as a protective agent against Hcy-induced endothelial injury in both cultured endothelial cells and hyperhomocysteinemic rats. Moreover, since studies have shown a synergistic effect of Hcy and hypertension, and patients with both represent a high-risk group of cardiovascular disease,3,49 we also proved that Nano-Se administration can greatly improve the vascular function in spontaneously hypertensive hyperhomocysteinemic rats, suggesting the protective effect of Nano-Se in hyperhomocysteinemia with hypertension.
Elevated levels of Hcy contribute to endothelial dysfunction, an early marker of vascular injury in atherogenesis.1 The underlying mechanism responsible for the cytotoxic effects of Hcy is not clear and may involve the imbalance between ROS producing and scavenging.50,51 GPX is the primary antioxidant system against oxidative damage. In general, intracellular GPX comprises classical GPX (GXP1) and phospholipid hydroperoxide GPX (GPX4, PHGPX), the latter exists in the nucleus, mitochondria and cytosol.12 Since the release of cytochrome c from mitochondria, which is the initiator of mitochondrial apoptosis, requires the generation of cardiolipin hydroperoxide,28 mitochondrial GPX4 might play a critical role as a new anti-apoptotic factor by preventing cardiolipin oxidation and inhibiting cytochrome c release in mitochondrial oxidative death pathway. Earlier works have demonstrated that Hcy decreases the activity of GPX1,34,35 in our study, we consistently found the decreased expression and activities of GPX1 in HUVECs and aorta after Hcy treatment. Importantly, to our knowledge, we are the first to show the concomitant downregulation of GPX4 by Hcy in both cultured endothelial cells and aorta of animal models. In agreement with it, we found the increase of mitochondrial ROS level and a series of mitochondrial dysfunctions including oxidation of mitochondrial cardiolipin oxidation, inhibition of mitochondrial ETC function and loss of mitochondrial transmembrane potential, all of which might be correlated with oxidative damage to mitochondria induced by Hcy-mediated GPX4 downregulation in vascular endothelial cells.
ROS including superoxide, hydrogen peroxide and lipid hydroperoxides can be produced at different sites of the cells. Many kinds of ROS cannot penetrate the organelle membranes, indicating that each antioxidant enzyme at different organelle has a functionally distinct role or cooperates with others. It has been shown that mitochondrial GPX4-overexpressing rat basophile leukemia 2H3 (RBL-2H3) cells are more resistant than non-mitochondrial GPX-overexpressing cells to cell death, induced either by direct mitochondria damage or by extracellular oxidative stress like H2O2,52 indicating that mitochondria is a target for intracellular and extracellular oxidative stress, and that mitochondrial GPX4 might play a primary role in protecting cells against oxidative stress. In our study, we found that Hcy firstly increased cellular and then mitochondrial ROS, and overexpression of GPX1 and GPX4 has comparable effect to protect vascular endothelial cells against Hcy. Our results suggest that Hcy-induced cellular ROS, likely through activating NADPH oxidase,53 might be the initial source that can further induce mitochondrial ROS in endothelial cells; Overexpression of GPX4 could directly prevent lipid oxidation of mitochondria and suppress mitochondrial apoptosis pathway, whereas overexpression of GPX1 inhibits cellular ROS and thus prevents subsequent mitochondrial oxidation and apoptosis. In this regard, upregulating GPX1 and GPX4 is crucial to inhibit Hcy-mediated vascular endothelial cell damage, which explains the important protective effect of Nano-Se against Hcy. By incorporating into selenoproteins as selenocysteine, Nano-Se increases the expressions and activities of both GPX1 and GPX4, which make up the loss induced by Hcy and prevent mitochondrial oxidative stress and apoptosis of vascular endothelial cells.
Despite the positive role of Se in the prevention and treatment of cardiovascular disease, prospective epidemiological studies have had mixed findings to date.54 The disparity between studies may be explained to some extent by unique antioxidant and pro-oxidant effects of Se depending on the dose, duration and forms. The biological function of Se is dependent on its chemical form, and different forms of Se have distinctive antioxidant properties. Meanwhile, Se is antioxidant at sub-nutritional doses but becomes pro-oxidant at supra-nutritional doses. Therefore, the toxicity of Se is of great interest. Nanomedicine has become a new trend in the area of nutrition and clinical medicine, as this novel platform offers unique advantages and addresses the complex issues of conventional forms of drugs. A widely accepted advantage of nanomedicine is the enhanced safety, studies have focused on the use of Se in nanoparticles with substantially reduced toxicity.5 Experiments in mice show that compared with selenite and SeMet, Nano-Se has a comparable efficiency in upregulating plasma GPX activity while exhibiting a much lower toxicity.36–38 Consistently, we found that Nano-Se has a comparable protective effect but lower toxicity and slightly higher bioavailability against Hcy, and the supranutritional levels of Nano-Se had no obvious toxic effects in Hcy mice, indicating that Nano-Se can be administered as an antioxidant against Hcy toxicity with a reduced risk of Se toxicity. Besides as an antioxidant, Nano-Se is demonstrated to target multiple signaling pathways. A study utilizes OX26 antibody to target transport of Nano-Se to brain for stroke therapy, and shows that Nano-Se contributes to neuronal survival by targeting different cellular signaling pathways that regulate cellular metabolic state, oxidative defense system, inflammatory reactions, autophagy and apoptotic cell death, as well as functional properties of the hippocampal neurons.55 Therefore, there might be multiple mechanisms that contribute to the protective effects of Nano-Se that need further research.
To date, Nano-Se has been used in many disease conditions including cancer, inflammatory disorders, metal poisoning, infection, etc.6–8,55,56 However, the potential of Nano-Se in the prevention and treatment of cardiovascular disease have not been fully explored. In the present study, we proved that the application of Nano-Se could serve as a novel promising strategy against Hcy-mediated vascular dysfunction with reduced risk of Se toxicity. Furthermore, the surface of Nano-Se is negatively charged.57,58 Studies have found that nanoparticles with positive charge exhibited strong affinity for the negatively charged cell membrane, thus achieving selective uptake and anticancer action in cancer cells.58 However, the enhanced cellular uptake of positively charged nanoparticles induces toxicity to normal tissues. A recent study indicates that positively charged nanoparticles are not suitable for the treatment of ischemic stroke under oxidative stress in vivo.59 Altogether, the characters of comparable protective effect against Hcy to other forms of Se, remarkably reduced toxicity, and slightly higher bioavailability as well as the negatively charged surface, guarantee the therapeutic applications of Nano-Se against Hcy-induced vascular damage. Moreover, Nano-Se can be given by various routes and is convenient for administration.16 This advantage enables designing strategies that combined Nano-Se with other therapeutic compounds to treat cardiovascular disease complicated with hyperhomocysteinemia, and further investigation is needed.
Conclusion
In summary, as a new form of Se, Nano-Se can effectively protect Hcy-mediated vascular endothelial cell damage and dysfunction both in vitro and in vivo in hyperhomocysteinemic rats with lower toxicity. The beneficial effects of Nano-Se are ascribed to the prevention of GPX1 and GPX4 downregulation, thus inhibiting Hcy-induced mitochondrial oxidative damage and apoptosis. With comparable protective effect against Hcy but much lower toxicity than other chemical forms of Se, we believe the application of Nano-Se may serve as an important therapeutic strategy against Hcy-induced cardiovascular disease with decreased side effects.
Acknowledgments
The present study was supported by National Natural Science Foundation of China (grant no. 81760052) and the Jiangxi Science and Technology Department (grant no. 20171BAB215080, 20192BCBL23019)
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ganguly P, Alam SF. Role of homocysteine in the development of cardiovascular disease. Nutr J. 2015;14(1):6. doi:10.1186/1475-2891-14-6
2. Jensen MK, Bertoia ML, Cahill LE, Agarwal I, Rimm EB, Mukamal KJ. Novel metabolic biomarkers of cardiovascular disease. Nat Rev Endocrinol. 2014;10(11):659–672. doi:10.1038/nrendo.2014.155
3. Yang B, Fan S, Zhi X, et al. Interactions of homocysteine and conventional predisposing factors on hypertension in Chinese adults. J Clin Hypertens (Greenwich). 2017;19(11):1162–1170. doi:10.1111/jch.13075
4. Sieber F, Daziano JP, Gunther WH, et al. Elemental selenium generated by the photobleaching of selenomerocyanine photosensitizers forms conjugates with serum macromolecules that are toxic to tumor cells. Phosphorus Sulfur Silicon Relat Elem. 2005;180(3–4):647–657. doi:10.1080/10426500590907200
5. Hosnedlova B, Kepinska M, Skalickova S, et al. Nano-selenium and its nanomedicine applications: a critical review. Int J Nanomedicine. 2018;13:2107–2128. doi:10.2147/IJN.S157541
6. Piacenza E, Presentato A, Zonaro E, et al. Antimicrobial activity of biogenically produced spherical Se-nanomaterials embedded in organic material against Pseudomonas aeruginosa and Staphylococcus aureus strains on hydroxyapatite-coated surfaces. Microb Biotechnol. 2017;10(4):804–818. doi:10.1111/1751-7915.12700
7. Trabelsi H, Azzouz I, Ferchichi S, Tebourbi O, Sakly M, Abdelmelek H. Nanotoxicological evaluation of oxidative responses in rat nephrocytes induced by cadmium. Int J Nanomedicine. 2013;8:3447–3453. doi:10.2147/IJN.S49323
8. Faghfuri E, Yazdi MH, Mahdavi M, et al. Dose-response relationship study of selenium nanoparticles as an immunostimulatory agent in cancer-bearing mice. Arch Med Res. 2015;46(1):31–37. doi:10.1016/j.arcmed.2015.01.002
9. Lai WK, Kan MY. Homocysteine-induced endothelial dysfunction. Ann Nutr Metab. 2015;67(1):1–12. doi:10.1159/000437098
10. Au-Yeung KK, Woo CW, Sung FL, Yip JC, Siow YL. O K. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ Res. 2004;94(1):28–36. doi:10.1161/01.RES.0000108264.67601.2C
11. Martinez Y, Li X, Liu G, et al. The role of methionine on metabolism, oxidative stress, and diseases. Amino Acids. 2017;49(12):2091–2098. doi:10.1007/s00726-017-2494-2
12. Margis R, Dunand C, Teixeira FK, Margis-Pinheiro M. Glutathione peroxidase family - an evolutionary overview. FEBS J. 2008;275(15):3959–3970. doi:10.1111/j.1742-4658.2008.06542.x
13. Nomura K, Imai H, Koumura T, Kobayashi T, Nakagawa Y. Mitochondrial phospholipid hydroperoxide glutathione peroxidase inhibits the release of cytochrome c from mitochondria by suppressing the peroxidation of cardiolipin in hypoglycaemia-induced apoptosis. Biochem J. 2000;351(Pt 1):183–193. doi:10.1042/bj3510183
14. Outinen PA, Sood SK, Pfeifer SI, et al. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood. 1999;94(3):959–967. doi:10.1182/blood.V94.3.959.415k20_959_967
15. Weiss N, Heydrick S, Zhang YY, Bierl C, Cap A, Loscalzo J. Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine beta-synthase-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22(1):34–41. doi:10.1161/hq1201.100456
16. Gao F, Yuan Q, Gao L, et al. Cytotoxicity and therapeutic effect of irinotecan combined with selenium nanoparticles. Biomaterials. 2014;35(31):8854–8866. doi:10.1016/j.biomaterials.2014.07.004
17. Baudin B, Bruneel A, Bosselut N, Vaubourdolle M. A protocol for isolation and culture of human umbilical vein endothelial cells. Nat Protoc. 2007;2(3):481–485. doi:10.1038/nprot.2007.54
18. Rezvanfar MA, Rezvanfar MA, Shahverdi AR, et al. Protection of cisplatin-induced spermatotoxicity, DNA damage and chromatin abnormality by selenium nano-particles. Toxicol Appl Pharmacol. 2013;266(3):356–365. doi:10.1016/j.taap.2012.11.025
19. He Y, Chen S, Liu Z, Cheng C, Li H, Wang M. Toxicity of selenium nanoparticles in male Sprague-Dawley rats at supranutritional and nonlethal levels. Life Sci. 2014;115(1–2):44–51. doi:10.1016/j.lfs.2014.08.023
20. Atteia HH, Arafa MH, Prabahar K. Selenium nanoparticles prevents lead acetate-induced hypothyroidism and oxidative damage of thyroid tissues in male rats through modulation of selenoenzymes and suppression of miR-224. Biomed Pharmacother. 2018;99:486–491. doi:10.1016/j.biopha.2018.01.083
21. Streck EL, Matte C, Vieira PS, et al. Reduction of Na(+),K(+)-ATPase activity in hippocampus of rats subjected to chemically induced hyperhomocysteinemia. Neurochem Res. 2002;27(12):1593–1598. doi:10.1023/A:1021670607647
22. Streck EL, Bavaresco CS, Netto CA, Wyse AT. Chronic hyperhomocysteinemia provokes a memory deficit in rats in the Morris water maze task. Behav Brain Res. 2004;153(2):377–381. doi:10.1016/j.bbr.2003.12.013
23. Da CK, Gaffney CE, Fischer LM, Zeisel SH. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am J Clin Nutr. 2005;81(2):440–444. doi:10.1093/ajcn.81.2.440
24. Zhang W, Trachootham D, Liu J, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276–286. doi:10.1038/ncb2432
25. Yavuzer H, Yavuzer S, Cengiz M, et al. Biomarkers of lipid peroxidation related to hypertension in aging. Hypertens Res. 2016;39(5):342–348. doi:10.1038/hr.2015.156
26. Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc. 2012;7(6):1235–1246. doi:10.1038/nprot.2012.058
27. Ding L, Zhang W, Yang Let al,. Targeting the autophagy in bone marrow stromal cells overcomes resistance to vorinostat in chronic lymphocytic leukemia. Onco Targets Ther. 2018;11:5151–5170. doi:10.2147/OTT.S170392
28. Iverson SL, Orrenius S. The cardiolipin-cytochrome c interaction and the mitochondrial regulation of apoptosis. Arch Biochem Biophys. 2004;423(1):37–46. doi:10.1016/j.abb.2003.12.002
29. Zhang W, Pelicano H, Yin R, et al. Effective elimination of chronic lymphocytic leukemia cells in the stromal microenvironment by a novel drug combination strategy using redox-mediated mechanisms. Mol Med Rep. 2015;12(5):7374–7388. doi:10.3892/mmr.2015.4364
30. Weitzel F, Ursini F, Wendel A. Phospholipid hydroperoxide glutathione peroxidase in various mouse organs during selenium deficiency and repletion. Biochim Biophys Acta. 1990;1036(2):88–94. doi:10.1016/0304-4165(90)90018-R
31. Androwiki AC, Camargo LL, Sartoretto S, et al. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front Chem. 2015;3:24. doi:10.3389/fchem.2015.00024
32. Wu R, Millette E, Wu L, de Champlain J. Enhanced superoxide anion formation in vascular tissues from spontaneously hypertensive and desoxycorticosterone acetate-salt hypertensive rats. J Hypertens. 2001;19(4):741–748. doi:10.1097/00004872-200104000-00011
33. Gonzalvez F, Schug ZT, Houtkooper RH, et al. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183(4):681–696. doi:10.1083/jcb.200803129
34. Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J Biol Chem. 2005;280(16):15518–15525. doi:10.1074/jbc.M501452200
35. Lubos E, Loscalzo J, Handy DE. Homocysteine and glutathione peroxidase-1. Antioxid Redox Signal. 2007;9(11):1923–1940. doi:10.1089/ars.2007.1771
36. Zhang J, Wang X, Xu T. Elemental selenium at nano size (Nano-Se) as a potential chemopreventive agent with reduced risk of selenium toxicity: comparison with se-methylselenocysteine in mice. Toxicol Sci. 2008;101(1):22–31. doi:10.1093/toxsci/kfm221
37. Shakibaie M, Shahverdi AR, Faramarzi MA, Hassanzadeh GR, Rahimi HR, Sabzevari O. Acute and subacute toxicity of novel biogenic selenium nanoparticles in mice. Pharm Biol. 2013;51(1):58–63. doi:10.3109/13880209.2012.710241
38. Zhang J, Wang H, Yan X, Zhang L. Comparison of short-term toxicity between Nano-Se and selenite in mice. Life Sci. 2005;76(10):1099–1109. doi:10.1016/j.lfs.2004.08.015
39. Zhang JS, Gao XY, Zhang LD, Bao YP. Biological effects of a nano red elemental selenium. Biofactors. 2001;15(1):27–38. doi:10.1002/biof.5520150103
40. Clarke R, Lewington S. Homocysteine and coronary heart disease. Semin Vasc Med. 2002;2(4):391–399. doi:10.1055/s-2002-36768
41. Cesari M, Rossi GP, Sticchi D, Pessina AC. Is homocysteine important as risk factor for coronary heart disease? Nutr Metab Cardiovasc Dis. 2005;15(2):140–147. doi:10.1016/j.numecd.2004.04.002
42. Wrobel JK, Power R, Toborek M. Biological activity of selenium: revisited. IUBMB Life. 2016;68(2):97–105. doi:10.1002/iub.1466
43. Lopes JE, Leite HP, Konstantyner T. Selenium and selenoproteins: from endothelial cytoprotection to clinical outcomes. Transl Res. 2019;208:85–104. doi:10.1016/j.trsl.2019.01.004
44. Uthus EO, Yokoi K, Davis CD. Selenium deficiency in Fisher-344 rats decreases plasma and tissue homocysteine concentrations and alters plasma homocysteine and cysteine redox status. J Nutr. 2002;132(6):1122–1128. doi:10.1093/jn/132.6.1122
45. Gonzalez S, Huerta JM, Alvarez-Uria J, Fernandez S, Patterson AM, Lasheras C. Serum selenium is associated with plasma homocysteine concentrations in elderly humans. J Nutr. 2004;134(7):1736–1740. doi:10.1093/jn/134.7.1736
46. Angelova EA, Atanassova PA, Chalakova NT, Dimitrov BD. Associations between serum selenium and total plasma homocysteine during the acute phase of ischaemic stroke. Eur Neurol. 2008;60(6):298–303. doi:10.1159/000157884
47. Bekaert B, Cooper ML, Green FR, et al. Effect of selenium status and supplementation with high-selenium yeast on plasma homocysteine and B vitamin concentrations in the UK elderly. Mol Nutr Food Res. 2008;52(11):1324–1333. doi:10.1002/mnfr.200700353
48. Venn BJ, Grant AM, Thomson CD, Green TJ. Selenium supplements do not increase plasma total homocysteine concentrations in men and women. J Nutr. 2003;133(2):418–420. doi:10.1093/jn/133.2.418
49. Cacciapuoti F. Hyper-homocysteinemia: a novel risk factor or a powerful marker for cardiovascular diseases? Pathogenetic and therapeutical uncertainties. J Thromb Thrombolysis. 2011;32(1):82–88. doi:10.1007/s11239-011-0550-4
50. Zou CG, Banerjee R. Homocysteine and redox signaling. Antioxid Redox Signal. 2005;7(5–6):547–559. doi:10.1089/ars.2005.7.547
51. Tian X, Zhao L, Song X, et al. HSP27 inhibits homocysteine-induced endothelial apoptosis by modulation of ROS production and mitochondrial caspase-dependent apoptotic pathway. Biomed Res Int. 2016;2016:4847874. doi:10.1155/2016/4847874
52. Arai M, Imai H, Koumura T, et al. Mitochondrial phospholipid hydroperoxide glutathione peroxidase plays a major role in preventing oxidative injury to cells. J Biol Chem. 1999;274(8):4924–4933. doi:10.1074/jbc.274.8.4924
53. Edirimanne VE, Woo CW, Siow YL, Pierce GN, Xie JY. O K. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can J Physiol Pharmacol. 2007;85(12):1236–1247. doi:10.1139/Y07-112
54. Zhang X, Li X, Zhang W, Song Y. Selenium and cardiovascular disease: epidemiological evidence of a possible U-shaped relationship. In: Michalke B, editor. Selenium. Cham: Springer International Publishing; 2018:303–316.
55. Amani H, Habibey R, Shokri F, et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci Rep. 2019;9(1):6044. doi:10.1038/s41598-019-42633-9
56. Ramamurthy C, Sampath KS, Arunkumar P, et al. Green synthesis and characterization of selenium nanoparticles and its augmented cytotoxicity with doxorubicin on cancer cells. Bioprocess Biosyst Eng. 2013;36(8):1131–1139. doi:10.1007/s00449-012-0867-1
57. Liu W, Golshan NH, Deng X, et al. Selenium nanoparticles incorporated into titania nanotubes inhibit bacterial growth and macrophage proliferation. Nanoscale. 2016;8(34):15783–15794. doi:10.1039/C6NR04461A
58. Yu B, Zhang Y, Zheng W, Fan C, Chen T. Positive surface charge enhances selective cellular uptake and anticancer efficacy of selenium nanoparticles. Inorg Chem. 2012;51(16):8956–8963. doi:10.1021/ic301050v
59. Ramamurthy ME, Alebouyeh MR, et al. Would colloidal gold nanocarriers present an effective diagnosis or treatment for ischemic stroke? Int J Nanomedicine. 2019;14:8013–8031. doi:10.2147/IJN.S210035
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.