")
Back to Archived Journals » Antibody Technology Journal » Volume 5
Anti-MTG16 antibodies reveal MTG16 subcellular distribution and nucleocytoplasmic transport in erythroleukemia cells
Authors Nguyen H, Mariotti J, Bareyan D, Carnahan R, Cooper T, Williams C, Engel M
Received 16 September 2014
Accepted for publication 9 October 2014
Published 19 February 2015 Volume 2015:5 Pages 27—41
DOI https://doi.org/10.2147/ANTI.S74419
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shixia Wang
Hong Nguyen,1 Jolene Mariotti,1 Diana Bareyan,2 Robert H Carnahan,3 Tracy Cooper,3 Christopher S Williams,4 Michael E Engel2,5
1Department of Pediatrics, Vanderbilt University School of Medicine, Nashville, TN, 2Department of Oncological Sciences, Huntsman Cancer Institute, University of Utah School of Medicine, Salt Lake City, UT, 3Department of Cancer Biology, Vanderbilt University School of Medicine, Nashville, TN, 4Department of Medicine, Vanderbilt University School of Medicine, Nashville, TN, 5Department of Pediatrics, University of Utah School of Medicine and Primary Children's Hospital, Salt Lake City, UT, USA
Abstract: The myeloid translocation gene (MTG) family of transcriptional corepressors consists of three highly conserved members: MTG8, MTG16, and MTGR1, each evolutionarily related to the Drosophila protein NERVY and with orthologs across the mammalian hierarchy. By coordinating coincident interactions between DNA binding proteins, other corepressors, and epigenetic effectors, MTG proteins occupy a critical nexus in transcriptional control complexes to profoundly impact the specification of cell fate. MTG family members are most conserved within nervy homology regions (NHRs) 1–4, with each region fulfilling functions common to the family. Studies of functional differences between MTG proteins require carefully qualified immunologic reagents specific to each family member. We have developed a group of a-MTG16 antibodies and carefully characterized their specificity for MTG16. These tools reveal that MTG16 is concentrated in the cytoplasm of erythroleukemia cell lines from human and mouse. Using the chromosome region maintenance 1 (CRM1) antagonist leptomycin B, we show that MTG16 levels rise in the nucleus of murine erythroleukemia cells and decline in the cytoplasm. Together, these data indicate bidirectional movement of MTG16 between cytoplasmic and nuclear compartments. Our work reveals an unrecognized feature of MTG16 regulation that may impact cell fate specification and provides reagents to address important questions regarding MTG16 functions in vivo.
Keywords: antibody, nucleus, cytoplasm, transport
Introduction
Myeloid translocation gene (MTG) 16, a member of a family of transcriptional corepressors that also includes MTG8 and MTGR1, is a master regulator of cell fate specification in hematopoiesis.1–5 MTG16 was originally identified from its homology to MTG8, the most widely studied member of the MTG family and partner of runt-related transcription factor 1 (RUNX1) in the t(8;21)(q22;q22) translocation of acute myeloid leukemia (AML).6–9 Since its discovery, MTG16 has been found to participate in a translocation with RUNX1, t(16;21)(q24;q22), in de novo and secondary AML [analogous to the t(8;21)(q22;q22) translocation] and to partner with GLIS2 in the inv(16)(p13.3q24.3) inversion acute megakaryoblastic leukemia.10–14 These findings intimate profound implications for MTG16 dysfunction in malignant hematopoiesis.
While viable at normal Mendelian ratios, mice with a constitutional deletion of MTG16 display striking phenotypes within the hematopoietic compartment, including preferential granulocyte/macrophage fate acquisition, reduced megakaryocyte-erythroid progenitor cell mass, and profoundly diminished thymocyte development.1,3,15 Moreover, these phenotypes are significantly exacerbated by stress stimuli, intimating roles for MTG16 in maintaining the stem and multipotent progenitor cell compartments. Notably, neither MTG8 -/- nor MTGR1 -/- mice show hematopoietic phenotypes, suggesting their contributions to hematopoiesis are either minimal or easily compensated.16,17 By extension, the pro-leukemic phenotype promoted by the t(8;21)(q22;q22) translocation may reflect perturbations in functions normally carried out by MTG16.4 Carefully qualified immunologic reagents for each family member are necessary to clarify their relative contributions to lineage allocation and to provide mechanistic insights.
MTG proteins are evolutionarily related to the Drosophila protein, NERVY.7 With NERVY, they share four highly conserved domains known as “nervy homology regions” (NHRs) 1–4. Each NHR fulfills a primary role common to MTG family proteins. The NHR1 domain binds several structurally distinct DNA binding proteins with important roles in hematopoietic cell fate specification.5,18–24 The NHR2 domain contains a hydrophobic heptad repeat that confers MTG protein oligomerization in an antiparallel fashion, and, in combination with nearby sequence elements, contributes to the binding of histone deacetylases (HDACs) and other transcriptional corepressors such as nuclear receptor corepressor (NCoR) and silencing mediator for retinoid or thyroid hormone receptors (SMRT).25–28 The NHR3 domain shares homology to A-kinase anchoring proteins (AKAPs) and interacts with the regulatory subunit of cAMP-dependent protein kinase (PKA).29,30 In MTG16, the NHR3 domain also provides a binding surface for CSL, the DNA binding component of the canonical Notch transcription complex.2 The NHR4 domain provides additional surfaces for corepressor and HDAC recruitment,25 and is essential for MTG-mediated transcriptional repression.31
Many MTG binding partners are critical regulators of gene expression. As such, MTG family members were presumed to reside and act within the nucleus. Indeed, enforced MTG expression typically results in nuclear accumulation.2,32,33 Yet, several reports document cytoplasmic localization of endogenously expressed MTG proteins using immune fluorescence immunohistochemistry approaches.29,30,34 Without confirming nucleocytoplasmic distribution by a second technique, conclusions drawn from these data are inherently reliant upon the specificity of the antibodies employed. To clarify this issue for MTG16, and to facilitate insights into the role of MTG16 subcellular distribution to its functions in cell fate determination, we have prepared polyclonal and monoclonal antibodies to MTG16, carefully qualified them, and leveraged them to address MTG16 subcellular distribution in erythroleukemia cell lines from human and mouse. We show that the α-MTG16 antibody 2D1:1H10 displays tremendous selectivity for MTG16 with no discernible cross-reactivity with MTG8 or MTGR1. The α-MTG16 polyclonal antibodies r332 and r333 are exquisitely sensitive, and in conjunction with 2D1:1H10 permit studies of MTG16 at endogenous levels of expression. Using these antibodies, we show that MTG16 localizes preferentially to the cytoplasm in K562 cells and murine erythroleukemia (MEL) cells. In the presence of the chromosome region maintenance 1 (CRM1) inhibitor leptomycin B, MTG16 accumulates in the nucleus. These data suggest that MTG16 moves dynamically and bidirectionally between the cytoplasm and nucleus and that its distribution may influence its contributions to cell fate.
Materials and methods
Commercial antibodies and reagents
Mouse monoclonal α-HA (12CA5) and α-Myc (9E10) were acquired from the antibody core facility of Vanderbilt University Medical Center. α-tubulin (B-5-1-2), normal mouse serum (NMS), and normal goat serum (NGS) were obtained from Sigma-Aldrich. Rabbit polyclonal α-Myc (A-14) was obtained from Santa Cruz Biotechnology, Inc. α-Histone H3 (ab1791) antibody was bought from Abcam. Horseradish peroxidase (HRP)-conjugated α-mouse and α-rabbit immunoglobulin (Ig) G were obtained from Jackson Immunoresearch. Alexa-488- and Alexa-568-conjugated α-mouse IgG were purchased from Cell Signaling Technologies. Ni-NTA-agarose was obtained from Qiagen. Lipofectamine® and To-Pro®-3 iodide were obtained from Invitrogen. Puromycin, hygromycin, Polybrene®, leptomycin B, dimethyl sulfoxide (DMSO), reduced glutathione, glutathione agarose, Protein G Sepharose®, bovine serum albumin (BSA), Hoechst 33342, and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich. Restriction endonucleases, polymerases, and ligases were purchased from New England Biosciences. Methylcellulose growth medium for hybridoma selection and maintenance was purchased from Stemcell Technologies. All other materials were purchased from commercial suppliers and of reagent grade.
Cell culture, transient transfection, and mouse strains
COS7L and MEL cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, L-glutamine, and penicillin/streptomycin. K562 cells were similarly maintained, but using Roswell Park Memorial Institute (RPMI)-1640 as base medium. COS7L cells were transfected using Lipofectamine for enforced expression of MTG16 or its deletion derivatives as previously described.2 C57BL/6 wild-type mice and the MTG16 null mice derived from them have been previously described.1
Plasmids, subcloning, and protein purification
NHRs 1–4 and their flanking regions, were subcloned into pGEX4T3 in frame with GST (glutathione-S-transferase) by standard methods.2 Plasmids were transformed into DH5α™ and expression of GST-fusion proteins induced with 0.4 mM isopropyl-β-D-thiogalactopyranoside (IPTG). Fusion proteins were bound to glutathione agarose from clarified bacterial extracts then eluted with reduced glutathione as previously described.35 A complementary DNA (cDNA) fragment expressing amino acids 1–242 of murine MTG16 was subcloned in frame with a deca-histidine (His10) tag in pBG (Vanderbilt University Center for Structural Biology). DH5α bacteria were transformed with this plasmid and induced to express His10-MTG16 (1–242) with IPTG as described in materials and methods. His10-MTG16 (1–242) was purified from clarified bacterial extracts by Ni-NTA-agarose chromatography as previously described.36 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining were used to analyze purified proteins.37 pCMV5-based expression plasmids for full-length MTG16 and deletion derivatives Δ1N through Δ10N (Figure 1D) and Δ1C through Δ7C (Figure 2B), each with an N-terminal Myc-epitope tag, have been previously described.2 Expression plasmids for pCMV5-HA:MTG16, pCMV5-HA:MTG8, and pCMV5-HA:MTGR1 have been previously described.25 Expression plasmids for pcDNA3.1+-Myc:MTG16-ΔNHR1, pcDNA3.1+-Myc:MTG16-ΔNHR2, pcDNA3.1+-Myc:MTG16-ΔNHR3, pcDNA3.1+-Myc:MTG16-ΔNHR4, pcDNA3.1+-Myc:MTG16-ΔNHR3/4, and pcDNA3.1+-Myc:MTG16-ΔPST2+ were made using a three-fragment ligation strategy employing EcoR1/Xho1-restricted, linearized vector and with the ΔC and ΔN fragments required to provide the desired N- and C-terminal segments, respectively. A detailed strategy can be provided upon request. Constructs were verified by automated di-deoxy sequencing by the Vanderbilt University Medical Center Sequencing and Oligonucleotide Synthesis Core.
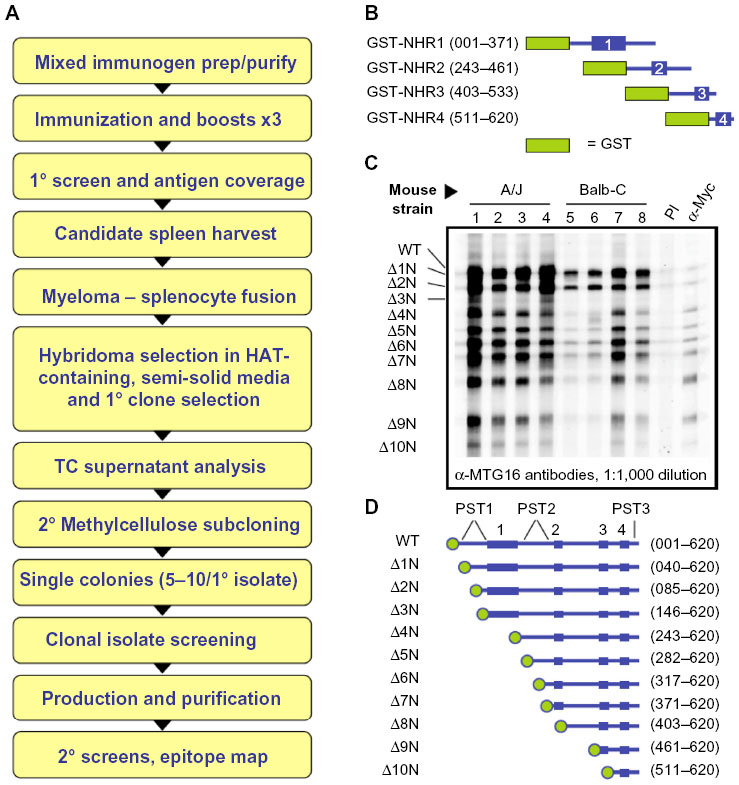 | Figure 1 Monoclonal antibody production strategy and screening of polyclonal sera. |
 | Figure 2 A/J #1 polyclonal serum recognition pattern spans the myeloid translocation gene (MTG) 16 primary structure. |
Monoclonal antibody production
Four A/J and four BALB/c mice were immunized with a mixed antigen composed of purified GST-NHR1, -NHR2, -NHR3, and -NHR4 in equal quantities estimated from Coomassie-stained gels of each purified protein. Before immunization, a pre-immune serum was obtained from each recipient mouse to serve as a negative control for screening of sera. For primary immunization, 50 μg of purified antigen was emulsified in 50% phosphate-buffered saline (PBS)/50% Freund’s complete adjuvant and injected subcutaneously into the nape of the neck (50%) and intramuscularly to the gluteal muscles (50%). For subsequent boost injections, Freund’s incomplete adjuvant was substituted for Freund’s complete adjuvant. Fourteen days after each booster injection, serum was collected from each mouse and assayed for reactivity with the antigen by enzyme-linked immunosorbent assay (ELISA) as previously described.38 Following the third boost, serum samples from each mouse were assayed by Western blot against a ladder of full-length MTG16 and fragments Δ1N through Δ10N (Figure 1C and D). To create this ladder, full-length MTG16 and the ΔN deletion derivatives were expressed individually in COS7L cells, then extracts mixed in equal amounts to create a single volume containing all fragments. The mixture was separated by Laemmli SDS-PAGE using a preparative comb and proteins transferred to a nitrocellulose filter. Recognition of MTG16 and its derivative fragments by each serum was accomplished by Western blot using a slot blot apparatus.
The mouse with the best reactivity to all MTG16 fragments was screened against MTG16, the MTG16-ΔC deletion panel, and the components of the original immunogen (Figure 2). Recipient A/J #1 was chosen for a final boost by intraperitoneal injection of mixed antigen diluted in PBS. After 4 days, spleen cells were harvested and fused to SP2/0 myeloma cells using a standard polyethylene glycol (PEG)-based method.39 The products of the fusion were plated under selection for 14 days in semisolid media (Stemcell Technologies) supplemented with hypoxanthine, aminopterin, and thymidine. Surviving colonies were isolated and distributed individually into 96-well plates. Tissue-culture supernatants from approximately 1,000 hybridomas were screened by ELISA for antigen-specific antibodies. Promising supernatants from 160 primary isolates were then screened for antigen-recognition and gross epitope localization by Western blot. Clones producing antibodies with desired properties were subcloned through methylcellulose to ensure monoclonality, and then their recognized epitope precisely mapped using the ΔN, ΔC, and interstitial deletion derivatives.
Clone 2D1:1H10 was selected for scale up, inoculated into 1 L bioreactors and grown for 3–4 weeks. Bulk monoclonal antibody (mAb) 2D1:1H10 was purified over Protein G Sepharose from the bioreactor supernatant, isotyped, then quantified by both bicinchoninic acid (BCA) assay and Coomassie-stained SDS-PAGE.
α-MTG16 polyclonal antibody generation
Anti-mouse MTG16 polyclonal antibodies were generated by immunization of two New Zealand white rabbits with amino acids 1–242 of MTG16 conjugated to deca-histidine to facilitate Ni-NTA agarose purification. Pre-immune sera were collected prior to primary immunization. Bleeds from sequential boosts were collected from both rabbits and tested by Western blot. The portion of MTG16 recognized by sera from terminal bleeds, termed r332 and r333, was determined using a select group of deletion mutants as shown in Figure 3C. Antisera were subsequently characterized for performance in immune precipitation assays in conjunction with 2D1:1H10 mAb described in the Monoclonal antibody production section.
 | Figure 3 α-myeloid translocation gene (MTG) 16 polyclonal antibodies r332 and r333 recognize the MTG16 N-terminus. |
Enzyme-linked immunosorbent assay
Flat-bottomed 96-well microtiter plates were coated with mixed antigen described in the Monoclonal antibody production section at a total concentration of 10 μg/mL in carbonate-bicarbonate buffer, pH 9.6 (15 mM Na2CO3, 30 mM NaHCO3, 0.001% thimerosal) and incubated for 4 hours at 37°C or overnight at 4°C. Coated plates were washed and blocked with PBS + 0.1% Tween® 20 (PBST). Supernatant – 100 μL per well – was added and plates incubated at 37°C for 1 hour before washing three times with PBST. Peroxidase-conjugated goat-α-mouse IgG Fc fragment-specific secondary antibody (Jackson Immunoresearch) diluted 1:5,000 in PBST/1% BSA was added to the wells and the plates were again incubated for 1 hour at 37°C. Plates were washed three times in PBST and bound antibodies were detected utilizing 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) (Sigma-Aldrich) and H2O2. The reaction was allowed to develop for 30 minutes at room temperature and optical densities determined at 405 nm using a PowerWave™ HT-340 (Bio-Tek).
Immune precipitation
Immune precipitations of transiently or endogenously expressed MTG16 from whole-cell extracts, cytoplasmic or nuclear fractions were performed essentially as described.2 Briefly, clarified extracts, cytoplasmic or nuclear fractions were pre-cleared with Protein G Sepharose followed by the addition of the desired antibody or pre-immune serum as shown in the figures and described in figure captions. Immune complexes were collected on Protein G Sepharose beads, washed extensively then boiled in Laemmli sample buffer (LSB). Immune purified proteins were separated by SDS-PAGE then analyzed by Western blot using the antibodies shown in the figures and described in figure captions.
Western blotting and dot blotting
Aliquots of purified proteins, whole-cell lysates, or immune complexes were adhered directly to nitrocellulose filters using a Bio-Rad dot-blot apparatus or separated by SDS-PAGE and electro-transferred in transfer buffer (25 mM Tris, 192 mM glycine, pH 8.35) to nitrocellulose filters that were then blocked in gelatin blocking buffer (GBB PBST supplemented with 0.25% gelatin and 0.02% NaN3). Filters were incubated in primary antibodies as indicated in the figures, washed in PBST, and probed with HRP-conjugated secondary antibodies. Proteins were visualized by chemiluminescence detection.
Immune fluorescence immunohistochemistry
MEL or K562 cells transferred to glass slides by cytospin were fixed in 3% paraformaldehyde/PBS, washed with PBS, chased with 10 mM glycine, permeabilized with 0.5% Triton™ X-100 then washed extensively. After blocking in 1% BSA/PBS, specimens were incubated in 2D1:1H10 mAb, washed thoroughly, exposed to α-mouse secondary antibody conjugated to either Alexa-488 or -568, washed and mounted in Aqua/Poly-mount (Polybiosciences). Cells were visualized by epifluorescence microscopy.
Nucleocytoplasmic fractionation
Cultured cells were fractionated into nuclear and cytoplasmic components by hypotonic lysis essentially as described.40 Briefly, cells were washed with ice-cold PBS, resuspended in IsoHi+ buffer (10 mM Tris, 140 mM NaCl, 1.5 mM MgCl2, pH 8.4, supplemented with 0.5% Triton X-100, 1 mM ethylenediaminetetraacetic acid [EDTA], 0.2 mM phenylmethanesulfonyl fluoride [PMSF], and 8.5 μg/mL aprotinin) then incubated on ice. The cytoplasmic fraction was collected by centrifugation at 1,000 g, made 1× in LSB, and boiled. The remaining pellet was washed with IsoHi+ buffer, resuspended in LSB, sonicated, and clarified by centrifugation. Samples were either analyzed directly or diluted for immune precipitation.
Results
MTG16-specific monoclonal antibody, 2D1:1H10
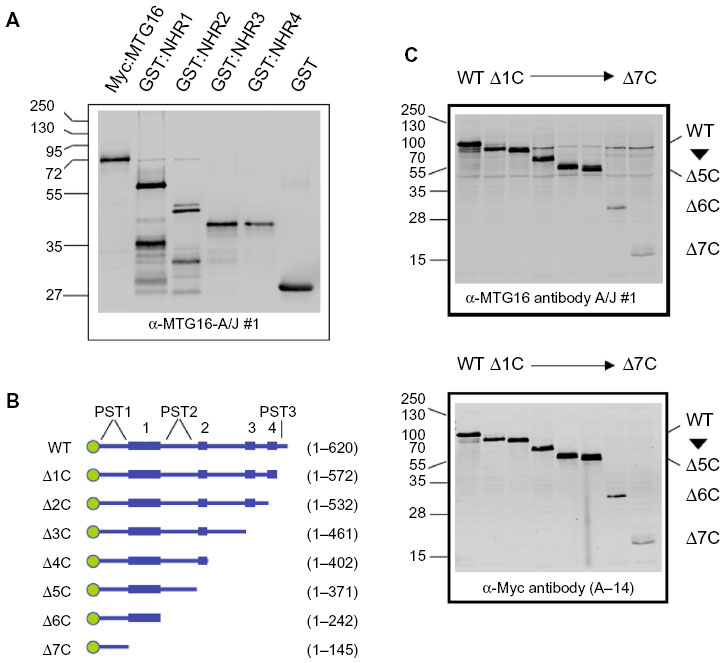
MTG family proteins are master regulators of cell fate specification in diverse tissues, yet their high degree of conservation, particularly within their NHR regions, creates a challenge when specific detection of a given family member is necessary. We employed a strategy to maximize the opportunity for developing, in an unbiased way, mAbs with specificity for MTG16 from a single, mixed immunogen (Figure 1A). GST-fusion proteins (Figure 1B) representing the complete murine MTG16 primary structure were purified from bacterial extracts. Each fusion protein contained an NHR domain and its flanking primary structure, such that the C-terminus of one fragment overlapped with the N-terminal flanking region in the next. We hypothesized that because the junction between GST and structures immediately downstream is often immunogenic, we may develop antibodies preferentially directed at less conserved epitopes between NHR domains. We immunized four A/J and four BALB/c mice with the mixed immunogen, as shown in Figure 1B, and screened serum samples from each recipient at a 1:1,000 dilution after the second boost (Figure 1C) for recognition of MTG16 and a ladder of MTG16 fragments serially deleted from the N-terminus, each containing an N-terminal Myc-epitope tag (Figure 1D). Sera from each mouse were analyzed by Western blot prior to the primary immunization to confirm no antecedent immune reactivity to MTG16 (data not shown). A/J #1 displayed robust immune reactivity toward each fragment in the ladder, intimating but not proving the recognition of multiple epitopes in MTG16. To gain additional insights, we assessed A/J #1 detection of each component of the mixed immunogen, Myc:MTG16, and GST alone by Western blot. Each component of the immunogen was individually recognized by A/J #1, as was GST and Myc:MTG16, indicating antibodies directed toward both MTG16 and GST epitopes (Figure 2A). To confirm detection of more than a single epitope in MTG16 by A/J #1, a secondary Western blot screen was performed against a panel of Myc-tagged MTG16 derivatives serially deleted from the C-terminus and expressed individually in COS7L cells (Figure 2B and C). Immune reactivity was readily demonstrated against each fragment of MTG16 in a pattern matching that observed with the α-Myc polyclonal antibody A-14. These findings show that antibodies capable of recognizing both C-terminal and N-terminal epitopes in MTG16 are represented in the A/J #1 polyclonal serum. Given this, and the robust response relative to other recipients, A/J #1 was selected for further processing of splenocytes to monoclonality.
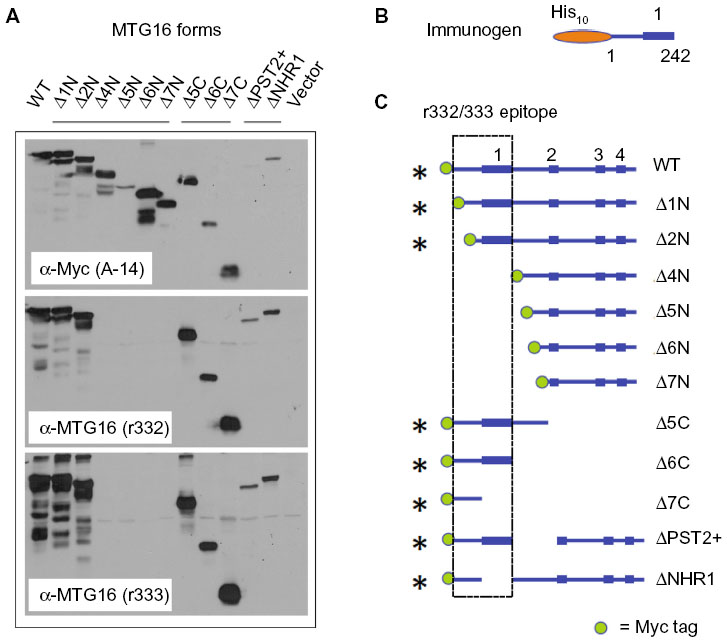
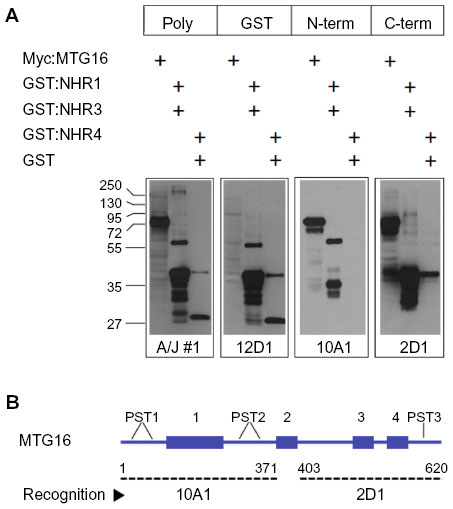
Splenocytes were isolated from A/J #1, fused to myeloma cells using PEG, and grown in semisolid media. Colonies were transferred to 96-well plates and tissue-culture supernatants from approximately 1,000 hybridomas were screened by ELISA. Supernatants from 160 primary isolates were then screened for antigen-recognition and gross epitope localization by Western blot (Figure 4). Using this screening strategy, we identified three recognition patterns, here represented by clones 12D1, 10A1, and 2D1. A/J #1 polyclonal serum was employed as a control that recognizes all component proteins in the screen. Clone 12D1 displays a GST recognition pattern, as it identifies all GST-fusion proteins employed and GST, but does not detect Myc:MTG16. Clone 10A1, which detects Myc:MTG16 and GST-NHR1 but not GST-NHR3, GST-NHR4, or GST, recognizes an epitope located between the N-terminus and amino acid 371 of MTG16. In contrast, 2D1 recognizes an epitope found between amino acids 403 and 620, as it identifies Myc:MTG16, GST-NHR3, and GST-NHR4, but neither GST-NHR1 nor GST. Recognition regions for 2D1 and 10A1 on MTG16 are represented graphically in Figure 4B.
 | Figure 4 Discrete recognition patterns displayed by primary hybridoma isolates derived from A/J #1 splenocytes. |
With rabbit polyclonal antibodies toward the MTG16 N-terminus coincidentally under development, we focused on subcloning the 2D1 primary isolate and characterization of secreted antibodies. The 2D1 primary isolate was subcloned through methylcellulose and six individual colonies were chosen at random (2D1:1C6, 2D1:1F2, 2D1:1F5, 2D1:1F9, 2D1:1F11, and 2D1:1H10) for screening by Western blot as in Figure 4A. Each revealed a recognition pattern toward purified immunogen components comparable to the primary isolate (Figure 5A and data not shown). We chose 2D1:1H10 for further evaluation. To assess clonality, we selected 24 discrete subclones of 2D1:1H10 from secondary passage through methylcellulose for analysis by dot blotting against Myc:MTG16 expressed in COS7L cells or purified GST-NHR1 (Figure 5B). All colonies selected readily detected Myc:MTG16 but showed little signal when presented to GST-NHR1. These findings suggest that the 2D1:1H10 isolate is likely comprised of a single clone producing antibody directed at the C-terminal region of MTG16.
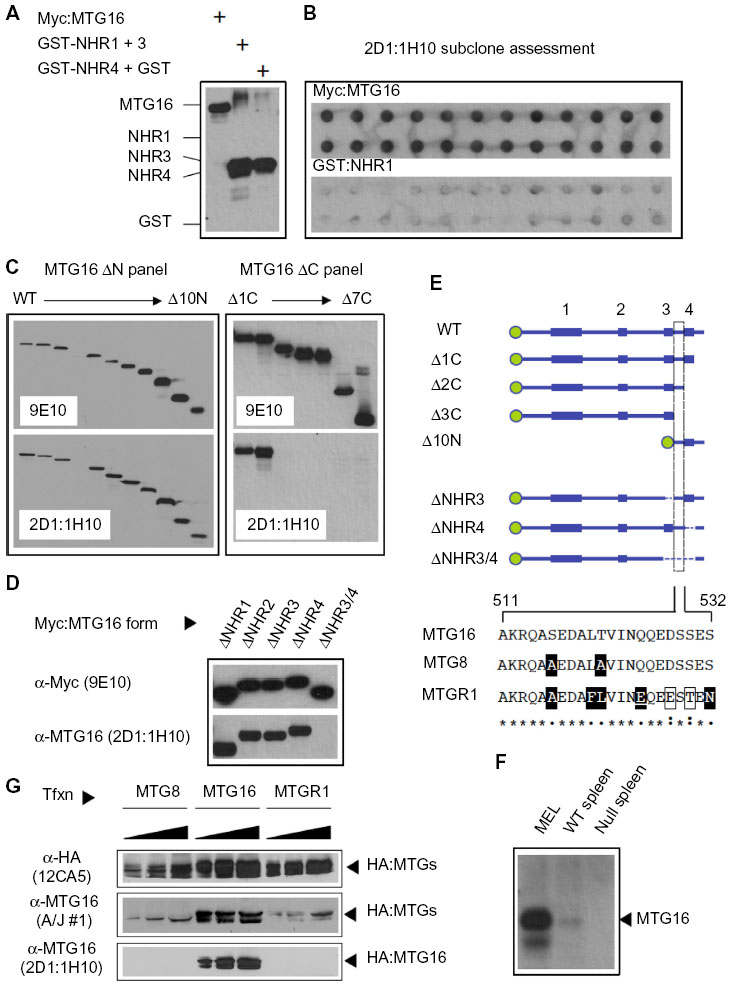 | Figure 5 Clonally purified 2D1:1H10 specifically recognizes myeloid translocation gene (MTG) 16 via an epitope located between the NHR3 and NHR4 domains. |
To precisely map the epitope being recognized, we leveraged our extensive collection of MTG16 deletion derivatives (Figures 1D and 2B). Using the N-terminal deletion panel (Figure 5C), we found that 2D1:1H10 recognizes each MTG16 derivative, defining amino acid 511 as the N-terminal boundary of the 2D1:1H10 epitope. Similarly, using the C-terminal deletion panel, we found no immune reactivity toward MTG16 deletion derivatives past MTG16-Δ2C. This suggests amino acid 532 as the C-terminal boundary of the 2D1:1H10 epitope. To confirm these findings, we created interstitial deletion derivatives of MTG16 specifically lacking each NHR domain, and one lacking NHR3, NHR4, and the region between them (amino acids 511–532) (Figure 5D and E). The 2D1:1H10 antibody clearly recognizes each NHR deletion mutant, yet fails to detect MTG16-ΔNHR3/4. These data indicate that the NHR regions are dispensable for MTG16 recognition by 2D1:1H10, but residues 511 through 532 are required. This motif is absolutely conserved between human and murine MTG16.
Conservation among MTG family members is greatest in the NHR domains, with considerable divergence emerging within intervening primary structure. To address specificity of 2D1:1H10 toward MTG16, we first analyzed extracts prepared from splenocytes of C57BL/6 wild-type and MTG16 –/– mice. Whole-cell extract prepared from MEL cells served as a positive control. A band co-migratory with MTG16 in MEL-cell extract was clearly identified in wild-type but not MTG16 –/– splenocytes, suggesting specific recognition (Figure 5F). To confirm this finding, we transiently expressed HA-tagged versions of MTG8, MTG16, and MTGR1 in COS7L cells then examined their expression by Western blot using α-HA, A/J #1, and 2D1:1H10 antibodies (Figure 5G). Each HA-tagged MTG family protein was readily detected with α-HA antibody 12CA5, and also by the polyclonal α-MTG16 serum from the A/J #1 mouse. However, 2D1:1H10 specifically recognized MTG16, without any obvious immune reactivity toward other family members. In concert, these data indicate that MTG16 is identified by 2D1:1H10 with high fidelity through an epitope found between its NHR3 and NHR4 domains.
Rabbit α-MTG16 polyclonal antibodies r332 and r333
In conjunction with 2D1:1H10, we also raised polyclonal antibodies against amino acids 1–242 of MTG16. The immunogen was His10-conjugated MTG16-(1–242) purified by Ni-NTA agarose chromatography. Sera from the terminal bleeds were designated r332 and r333. To confirm the expected pattern of MTG16 recognition by each antibody, we performed Western blots against Myc-tagged MTG16 and deletion derivatives either with or without sequences from the immunogen (Figure 3). Expression of each derivative was confirmed by Western blot against the Myc-epitope tag. Each MTG16 derivative with primary structural elements between 1 and 242 was recognized by both r332 and r333, yet neither antibody detected derivatives lacking these structures. Additionally, r333 serum and affinity purified r333 yielded the same band pattern in the Western blot analysis of MEL-cell extracts (data not shown). These findings confirm r332 and r333 recognition of MTG16 to the amino terminal 242 residues.
Characterizing α-MTG16 antibodies in immune precipitation
To characterize these antibodies further, we first compared their sensitivities against serial dilutions of an extract from COS7L cells transiently expressing Myc-tagged MTG16 (Figure 6A). Identical dilutions of r332 and r333 showed comparable sensitivities toward MTG16 in Western blot. Likewise, 2D1:1H10 at 2 μg/mL detected MTG16 equal to 9E10 at 0.5 μg/mL, and in both cases were approximately tenfold less sensitive for MTG16 detection relative to either r332 or r333. We then confirmed the ability of each antibody to immune purify MTG16 in the context of enforced expression. Myc-tagged MTG16 was transiently expressed in COS7L cells then immune purified from whole-cell extracts using α-Myc (9E10), 2D1:1H10, or NMS (Figure 6B). Myc:MTG16 was readily detected by Western blot in both α-Myc and 2D1:1H10 immune complexes. The bands recognized in α-Myc (9E10) and 2D1:1H10 immune complexes were the same in α-Myc and 2D1:1H10 Western blots, yet absent from NMS immune complexes and vector control lysates. These data indicate specific immune precipitation of Myc:MTG16 by 2D1:1H10.
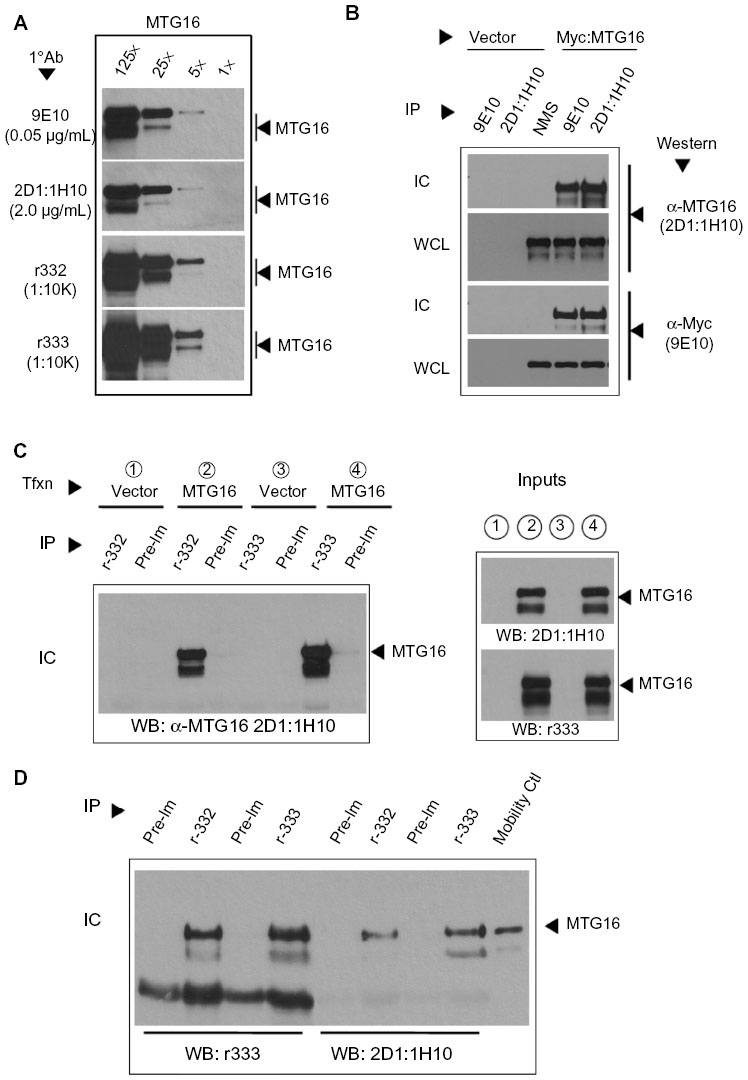 | Figure 6 Specificity and sensitivity assessment of α-myeloid translocation gene (MTG) 16 antibodies. |
The specificity of 2D1:1H10 for MTG16 enabled us to then characterize r332 and r333 in immune purification (Figure 6C). Using r332, r333 or their corresponding pre-immune sera, Myc:MTG16 was immune purified from aliquots of whole-cell extracts of COS7L cells expressing either Myc:MTG16 (samples 2 and 4) or vector control (samples 1 and 3). Expression of Myc:MTG16 was confirmed by Western blot using both 2D1:1H10 and r333 (inputs). 2D1:1H10 readily detected Myc:MTG16 in r332 and r333 immune complexes, but not pre-immune complexes, only in the context of Myc:MTG16 expression. An identical pattern of detection was seen using α-Myc (9E10) for both immune complexes and inputs (data not shown). These data confirm the specific immune purification of Myc:MTG16 by both polyclonal antibodies.
We then extended our analysis to immune purification of MTG16 at endogenous levels of expression from MEL-cell extracts (Figure 6D). MTG16 was collected using r332, r333, or their corresponding pre-immune sera and the immune complexes subjected to Western blot with both r333 and 2D1:1H10. An aliquot of Myc:MTG16 was employed as a mobility control. In Western blots, MTG16 was specifically detected by r333 and 2D1:1H10 in r332 and r333 immune complexes but not their paired pre-immune sera. We then subjected these bands to LC-MS-MS tandem mass spectrometry to confirm the presence of MTG16 (data not shown). Collectively, our analysis confirms that 2D1:1H10, r332, and r333 recognize MTG16 through distinct epitopes, and suggests that by leveraging their specificity and sensitivity, we could begin to explore MTG16 biology at endogenous levels of expression.
MTG16 distributes preferentially to the cytoplasm in erythroleukemia cell lines
Previous reports have intimated endogenously expressed MTG family members are preferentially found in the cytoplasm, yet concentrate in the nucleus with enforced expression.29,30,32–34 While nuclear localization is well aligned with their roles as transcriptional corepressors, any potential role for MTG proteins in the cytoplasm requires confirmation of the finding. With respect to MTG16, our panel of antibodies allowed us to address the possibility of cytoplasmic distribution. Erythroleukemia cells lines from mouse and human (MEL and K562, respectively) display abundant expression of MTG16. To address nucleocytoplasmic distribution of MTG16, we performed immune fluorescence immunohistochemistry (IF-IHC) and subcellular fractionation of MEL and K562 cells in parallel (Figure 7). Using 2D1:1H10, we observed abundant fluorescence signal from the cytoplasm but only modest signal from the nucleus for both MEL and K562 cells. Likewise, when separated into cytoplasmic and nuclear fractions, MTG16 was seen preferentially, but not exclusively, in the cytoplasm.
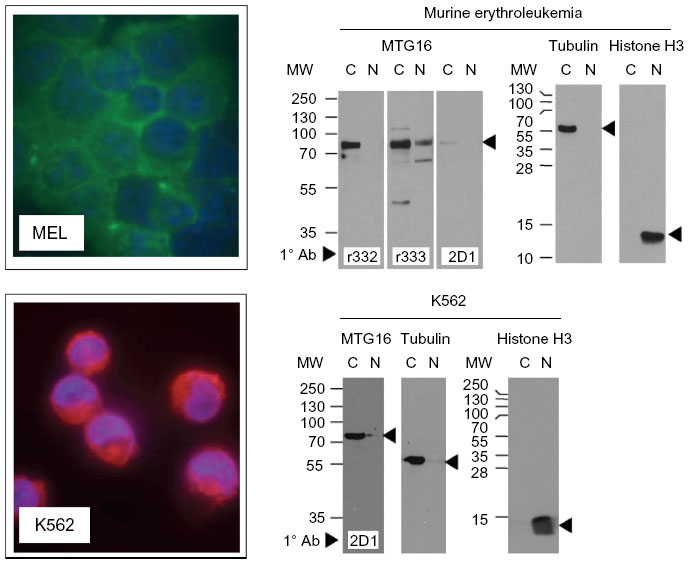 | Figure 7 α-myeloid translocation gene (MTG) 16 antibodies show predominately cytoplasmic localization of MTG16 in murine and human erythroleukemia cells. |
To confirm these findings, we separated MEL cells into cytoplasmic and nuclear fractions, divided these fractions into equal aliquots, and subjected each to reciprocal immune precipitations and Western blots with 2D1:1H10 and r333 (Figure 8A and B). As mobility controls, we included aliquots of the neat cytoplasmic and nuclear fractions (φ) and a sample of transiently expressed Myc:MTG16. Notably, MTG16 was readily detected by 2D1:1H10 and r333 in 2D1:1H10 immune complexes collected from cytoplasmic fractions. Likewise, MTG16 was found preferentially in r333 immune complexes from the cytoplasmic fraction by both 2D1:1H10 and r333. These data provide strong evidence for predominantly cytoplasmic distribution of MTG16 in these erythroleukemia cell lines.
 | Figure 8 Confirming myeloid translocation gene (MTG) 16 subcellular distribution between cytoplasm and nucleus. |
MTG16 undergoes CRM1-dependent nuclear export in erythroleukemia cell lines
MTG16’s function as a transcriptional corepressor, consistent with its presence in the nucleus, has been described.25 Yet, confirming its presence in the cytoplasm in erythroleukemia cells suggests either the failure of MTG16 to enter the nucleus or active export from it. To address the possibility of active nuclear export, we treated MEL cells with the CRM1 antagonist, leptomycin B, and then determined the distribution of MTG16 between cytoplasm and nucleus (Figure 8C). In untreated cells, MTG16 was again found predominantly in the cytoplasmic fraction. However, MTG16 levels in the nucleus increase significantly following exposure to leptomycin B and decline in the cytoplasm. These data suggest that MTG16, produced in the cytoplasm and then entering the nucleus, undergoes active nuclear export in MEL cells.
Discussion
Movement between cytoplasm and nucleus is frequently employed to modulate functions of transcriptional regulators. Both retention in the cytoplasm and export from the nucleus are viable strategies to limit transcriptional potential, and examples of each have been described.41–45 Among closely related proteins, confidently assessing subcellular localization at endogenous levels of expression relies upon antibodies that reliably distinguish between family members. Specificity can be accomplished prospectively using a peptide immunogen with primary structure unique to the protein of interest. While effective, this strategy limits the repertoire of antibodies generated to the peptide employed.
An alternative approach, with the theoretical advantage of yielding multiple monoclonal, target-specific antibodies, is to employ an immunogen that comprises the target’s entire primary structure, and then rely upon screening of hybridoma supernatants using fragments from the target protein. Definition of the epitope for each clone can be determined and refined by the fragments chosen for screening. Moreover, predictions about specificity can be made from conservation of the epitope among family members, and then confirmed by testing the recognition of each. In so doing, antibodies may develope that specifically detect one family member, while others may be discovered that recognize all family members by targeting highly conserved regions of primary structure. Being an unbiased approach, the number of potential antibodies produced should be limited only by the number of clones collected following splenocyte–myeloma cell fusion and the inherent immunogenicity of discrete regions of the target protein.
We employed this strategy to develop monoclonal antibodies specific for MTG16 yet preserve opportunities to identify “pan-specific” antibodies if that became desirable. We cryopreserved thousands of primary isolates cloned out of semisolid media, performed tier-one screening of 1,000 hybridoma supernatants by ELISA and tier-two screening of 160 supernatants by Western blot against components of the mixed immunogen. This approach confirmed MTG16 recognition, generally localized the epitope to N- or C-terminal motifs, and intimated recognition within the linkers separating NHRs. Among these, we chose clone 2D1 for secondary subcloning to 2D1:1H10, repeated tier-two screening, and then confirmed clonality from a collection of tertiary isolates. An extensive panel of MTG16 deletion derivatives permitted fine mapping of the epitope recognized by 2D1:1H10 to a 22-amino acid motif between the NHR3 and NHR4 domains. The specificity of 2D1:1H10 toward MTG16 and its performance in standard assays could then easily be determined, and, in combination with polyclonal antibodies r332 and r333, leveraged to answer questions related specifically to MTG16 biology.
Conclusion
By using this panel of α-MTG16 antibodies, we have confirmed that MTG16 can be found abundantly in the cytoplasm of erythroleukemia cell lines, and, with the CRM1 inhibitor leptomycin B, shown that it can be actively exported from the nucleus to the cytoplasm. Erythroleukemia cells may leverage MTG16’s export from the nucleus to modulate its contributions to transcriptional control, provide opportunity for additional MTG16 functions in the cytoplasm and perhaps coordinate nuclear and cytoplasmic functions that together govern cell fate specification. Moreover, it may be that MTG16 distribution between cytoplasm and nucleus differs for distinct cell types or at distinct stages of differentiation, and that changes in distribution govern lineage allocation programs. We are now actively exploring each of these hypotheses.
Acknowledgments
The authors are indebted to colleagues Jason Singer, Don Ayer, and Mahesh Chandrasekharan for critical review and helpful suggestions regarding the manuscript. We gratefully acknowledge support from the oligonucleotide synthesis and DNA sequencing core facilities of the University of Utah and the Huntsman Cancer Institute. This work was supported by grants from the National Institutes of Health (K08 DK080190) and generous support from the St Baldrick’s Foundation and Hyundai Hope on Wheels Foundation.
Disclosure
Other than the funding support received (outlined in the Acknowledgments), the authors declare no conflicts of interest in this work.
References
Chyla BJ, Moreno-Miralles I, Steapleton MA, et al. Deletion of Mtg16, a target of t(16;21), alters hematopoietic progenitor cell proliferation and lineage allocation. Mol Cell Biol. 2008;28(20):6234–6247. | |
Engel ME, Nguyen HN, Mariotti J, Hunt A, Hiebert SW. Myeloid translocation gene 16 (MTG16) interacts with Notch transcription complex components to integrate Notch signaling in hematopoietic cell fate specification. Mol Cell Biol. 2010;30(7):1852–1863. | |
Hunt A, Fischer M, Engel ME, Hiebert SW. Mtg16/Eto2 contributes to murine T-cell development. Mol Cell Biol. 2011;31(13):2544–2551. | |
Ibanez V, Sharma A, Buonamici S, et al. AML1-ETO decreases ETO-2 (MTG16) interactions with nuclear receptor corepressor, an effect that impairs granulocyte differentiation. Cancer Res. 2004;64(13):4547–4554. | |
Goardon N, Lambert JA, Rodriguez P, et al. ETO2 coordinates cellular proliferation and differentiation during erythropoiesis. EMBO J. 2006;25(2):357–366. | |
Davis JN, Williams BJ, Herron JT, Galiano FJ, Meyers S. ETO-2, a new member of the ETO-family of nuclear proteins. Oncogene. 1999;18(6):1375–1383. | |
Davis JN, McGhee L, Meyers S. The ETO (MTG8) gene family. Gene. 2003;303:1–10. | |
Peterson LF, Zhang DE. The 8;21 translocation in leukemogenesis. Oncogene. 2004;23(24):4255–4262. | |
Rowley JD. Identificaton of a translocation with quinacrine fluorescence in a patient with acute leukemia. Ann Genet. 1973;16(2):109–112. | |
Salomon-Nguyen F, Busson-Le Coniat M, Lafage Pochitaloff M, Mozziconacci J, Berger R, Bernard OA. AML1-MTG16 fusion gene in therapy-related acute leukemia with t(16;21)(q24;q22):two new cases. Leukemia. 2000;14(9):1704–1705. | |
Park IJ, Park JE, Kim HJ, Jung HJ, Lee WG, Cho SR. Acute myeloid leukemia with t(16;21)(q24;q22) and eosinophilia: case report and review of the literature. Cancer Genet Cytogenet. 2010;196(1):105–108. | |
Athanasiadou A, Stalika E, Sidi V, Papaioannou M, Gaitatzi M, Anagnostopoulos A. RUNX1-MTG16 fusion gene in de novo acute myeloblastic leukemia with t(16;21)(q24;q22). Leuk Lymphoma. 2011;52(1):145–147. | |
Gruber TA, Larson Gedman A, Zhang J, et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell. 2012;22(5):683–697. | |
Masetti R, Pigazzi M, Togni M, et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood. 2013;121(17):3469–3472. | |
Fischer MA, Moreno-Miralles I, Hunt A, Chyla BJ, Hiebert SW. Myeloid translocation gene 16 is required for maintenance of haematopoietic stem cell quiescence. EMBO J. 2012;31(6):1494–1505. | |
Calabi F, Pannell R, Pavloska G. Gene targeting reveals a crucial role for MTG8 in the gut. Mol Cell Biol. 2001;21(16):5658–5666. | |
Amann JM, Chyla BJ, Ellis TC, et al. Mtgr1 is a transcriptional corepressor that is required for maintenance of the secretory cell lineage in the small intestine. Mol Cell Biol. 2005;25(21):9576–9585. | |
McGhee L, Bryan J, Elliott L, et al. Gfi-1 attaches to the nuclear matrix, associates with ETO (MTG8) and histone deacetylase proteins, and represses transcription using a TSA-sensitive mechanism. J Cell Biochem. 2003;89(5):1005–1018. | |
Melnick AM, Westendorf JJ, Polinger A, et al. The ETO protein disrupted in t(8;21)-associated acute myeloid leukemia is a corepressor for the promyelocytic leukemia zinc finger protein. Mol Cell Biol. 2000;20(6):2075–2086. | |
Moore AC, Amann JM, Williams CS, et al. Myeloid translocation gene family members associate with T-cell factors (TCFs) and influence TCF-dependent transcription. Mol Cell Biol. 2008;28(3):977–987. | |
Grosveld F, Rodriguez P, Meier N, et al. Isolation and characterization of hematopoietic transcription factor complexes by in vivo biotinylation tagging and mass spectrometry. Ann N Y Acad Sci. 2005;1054: 55–67. | |
Chevallier N, Corcoran CM, Lennon C, et al. ETO protein of t(8;21) AML is a corepressor for Bcl-6 B-cell lymphoma oncoprotein. Blood. 2004;103(4):1454–1463. | |
Hamlett I, Draper J, Strouboulis J, Iborra F, Porcher C, Vyas P. Characterization of megakaryocyte GATA1-interacting proteins: the corepressor ETO2 and GATA1 interact to regulate terminal megakaryocyte maturation. Blood. 2008;112(7):2738–2749. | |
Schuh AH, Tipping AJ, Clark AJ, et al. ETO-2 associates with SCL in erythroid cells and megakaryocytes and provides repressor functions in erythropoiesis. Mol Cell Biol. 2005;25(23):10235–10250. | |
Amann JM, Nip J, Strom DK, et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21(19):6470–6483. | |
Lutterbach B, Westendorf JJ, Linggi B, et al. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol. 1998;18(12):7176–7184. | |
Liu Y, Cheney MD, Gaudet JJ, et al. The tetramer structure of the Nervy homology two domain, NHR2, is critical for AML1/ETO’s activity. Cancer Cell. 2006;9(4):249–260. | |
Zhang J, Hug BA, Huang EY, et al. Oligomerization of ETO is obligatory for corepressor interaction. Mol Cell Biol. 2001;21(1):156–163. | |
Fukuyama T, Sueoka E, Sugio Y, et al. MTG8 proto-oncoprotein interacts with the regulatory subunit of type II cyclic AMP-dependent protein kinase in lymphocytes. Oncogene. 2001;20(43):6225–6232. | |
Schillace RV, Andrews SF, Liberty GA, Davey MP, Carr DW. Identification and characterization of myeloid translocation gene 16b as a novel a kinase anchoring protein in T lymphocytes. J Immunol. 2002;168(4):1590–1599. | |
Lutterbach B, Sun D, Schuetz J, Hiebert SW. The MYND motif is required for repression of basal transcription from the multidrug resistance 1 promoter by the t(8;21) fusion protein. Mol Cell Biol. 1998;18(6):3604–3611. | |
Barseguian K, Lutterbach B, Hiebert SW, et al. Multiple subnuclear targeting signals of the leukemia-related AML1/ETO and ETO repressor proteins. Proc Natl Acad Sci U S A. 2002;99(24):15434–15439. | |
Odaka Y, Mally A, Elliott LT, Meyers S. Nuclear import and subnuclear localization of the proto-oncoprotein ETO (MTG8). Oncogene. 2000;19(32):3584–3597. | |
Sacchi N, Tamanini F, Willemsen R, Denis-Donini S, Campiglio S, Hoogeveen AT. Subcellular localization of the oncoprotein MTG8 (CDR/ETO) in neural cells. Oncogene. 1998;16(20):2609–2615. | |
Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274(52):37413–37420. | |
Schmitt J, Hess H, Stunnenberg HG. Affinity purification of histidine-tagged proteins. Mol Biol Rep. 1993;18(3):223–230. | |
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. | |
Markham NO, Cooper T, Goff M, Gribben EM, Carnahan RH, Reynolds AB. Monoclonal antibodies to DIPA: a novel binding partner of p120-catenin isoform 1. Hybridoma. 2012;31(4):246–254. | |
Akingbade D, Kingsley PJ, Shuck SC, et al. Selection of monoclonal antibodies against 6-oxo-M(1)dG and their use in an LC-MS/MS assay for the presence of 6-oxo-M(1)dG in vivo. Chem Res Toxicol. 2012;25(2):454–461. | |
Wood LD, Irvin BJ, Nucifora G, Luce KS, Hiebert SW. Small ubiquitin-like modifier conjugation regulates nuclear export of TEL, a putative tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(6):3257–3262. | |
Pan MG, Xiong Y, Chen F. NFAT gene family in inflammation and cancer. Curr Mol Med. 2013;13(4):543–554. | |
Reich NC. STATs get their move on. JAKSTAT. 2013;2(4):e27080. | |
Chen X, Xu L. Mechanism and regulation of nucleocytoplasmic trafficking of smad. Cell Biosci. 2011;1(1):40. | |
Chen LF, Greene WC. Shaping the nuclear action of NF-kappaB. Nat Rev Mol Cell Biol. 2004;5(5):392–401. | |
Pratt WB. Control of steroid receptor function and cytoplasmic-nuclear transport by heat shock proteins. Bioessays. 1992;14(12):841–848. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.