")
Back to Journals » International Journal of Nanomedicine » Volume 13
Anti-JNK2 peptide–siRNA nanostructures improve plaque endothelium and reduce thrombotic risk in atherosclerotic mice
Authors Pan H, Palekar RU, Hou KK, Bacon J, Yan H, Springer LE, Akk A, Yang L, Miller MJ, Pham CTN , Schlesinger PH, Wickline SA
Received 18 March 2018
Accepted for publication 15 June 2018
Published 6 September 2018 Volume 2018:13 Pages 5187—5205
DOI https://doi.org/10.2147/IJN.S168556
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Hua Pan,1 Rohun U Palekar,2 Kirk K Hou,3 John Bacon,2 Huimin Yan,3 Luke E Springer,3 Antonina Akk,3 Lihua Yang,3 Mark J Miller,3 Christine TN Pham,3 Paul H Schlesinger,3 Samuel A Wickline1
1Department of Cardiovascular Sciences, USF Health, Morsani College of Medicine, The USF Health Heart Institute, University of South Florida, Tampa, FL, USA; 2Department of Medicine, Washington University, St Louis, MO, USA; 3Department of Biomedical Engineering, Washington University, St Louis, MO, USA
Background: A direct and independent role of inflammation in atherothrombosis was recently highlighted by the Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) trial, showing the benefit of inhibiting signaling molecules, eg, interleukins. Accordingly, we sought to devise a flexible platform for preventing the inflammatory drivers at their source to preserve plaque endothelium and mitigate procoagulant risk.
Methods: p5RHH-siRNA nanoparticles were formulated through self-assembly processes. The therapeutic efficacy of p5RHH-JNK2 siRNA nanoparticles was evaluated both in vitro and in vivo.
Results: Because JNK2 is critical to macrophage uptake of oxidized lipids through scavenger receptors that engender expression of myriad inflammatory molecules, we designed an RNA-silencing approach based on peptide–siRNA nanoparticles (p5RHH-siRNA) that localize to atherosclerotic plaques exhibiting disrupted endothelial barriers to achieve control of JNK2 expression by macrophages. After seven doses of p5RHH-JNK2 siRNA nanoparticles over 3.5 weeks in ApoE-/- mice on a Western diet, both JNK2 mRNA and protein levels were significantly decreased by 26% (P=0.044) and 42% (P=0.042), respectively. Plaque-macrophage populations were markedly depleted and NFκB and STAT3-signaling pathways inhibited by 47% (P<0.001) and 46% (P=0.004), respectively. Endothelial barrier integrity was restored (2.6-fold reduced permeability to circulating 200 nm nanoparticles in vivo, P=0.003) and thrombotic risk attenuated (200% increased clotting times to carotid artery injury, P=0.02), despite blood-cholesterol levels persistently exceeding 1,000 mg/dL. No adaptive or innate immunoresponses toward the nanoparticles were observed, and blood tests after the completion of treatment confirmed the largely nontoxic nature of this approach.
Conclusion: The ability to formulate these nanostructures rapidly and easily interchange or multiplex their oligonucleotide content represents a promising approach for controlling deleterious signaling events locally in advanced atherosclerosis.
Keywords: siRNA, JNK2, peptide nanoparticles, perfluorocarbon nanoparticles, macrophages, atherosclerosis, plaque, ApoE-/- mice, thrombosis, endothelium, erosions, scavenger receptors
Introduction
Atherosclerotic lesions emerge from the interplay of numerous cell types, blood components, hemodynamic factors, and genetic predilections that engender a smoldering inflammatory process exacerbated by such risk factors as hypertension, diabetes, and smoking.1 Among the earliest events in lesion formation is the activation of endothelial cells lining blood vessels in atherosclerosis-prone sites by complex mechanical and fluid shear stresses,2,3 resulting in upregulation of surface receptors for adhesion molecules, such as VCAM1, that facilitate entry, proliferation, and differentiation of monocytes in the intima. The uptake of oxidized lipids (eg, oxidized low-density lipoprotein [oxLDL]) by polarized macrophages to form foam cells generates a panoply of cytokines and chemokines that accelerate endothelial injury, ultimately leading to extensive endothelial cell sloughing,4 plaque erosion, and/or plaque-cap rupture that exposes the inflamed procoagulant milieu to acute thrombosis.5–9
Recently, we reported that plaque endothelial barrier disruption and procoagulant activity in ApoE−/− mice on a Western diet was able to be mitigated rapidly within a few weeks by periodic treatment with thrombin-targeted nanoparticles.10 These particles carry PPACK (proline–phenylalanine–arginine–chloromethyl ketone), an irreversible inhibitor of thrombin around three orders of magnitude more selective for factor IIa than for Xa.11 When injected systemically, the therapeutic nanostructures passively permeate damaged endothelial barriers, but not normal vascular barriers, and are retained in the plaque intima to exert sustained thrombin surveillance without inducing systemic bleeding diathesis. Restoration of endothelial barrier function and abrogation of thrombotic risk were accompanied by reductions in thrombin–antithrombin complexes, tissue factor, and classical inflammatory drivers (ie, NFκB) in endothelium and macrophages in conjunction with reduced plaque burden, despite persistent hypercholesterolemia (>1,000 mg/dL).
Taken together with recent reports of newer anticoagulants used to forestall atherosclerosis, and in light of the recent Canakinumab Antiinflammatory Thrombosis Outcome Study (CANTOS) clinical trial reporting the benefit of inhibiting inflammatory drivers of atherothrombosis (IL1β) despite hypercholesterolemia,12 these experimental data prompted us to inquire if more selective and highly specific anti-inflammatory plaque-targeted nanoparticle formulations could similarly normalize endothelial barrier function and thrombotic risk without affecting clotting function locally or systemically. While thrombin is clearly a local target for intervention in advanced plaques, it exerts highly pleiotropic procoagulant and inflammatory signaling effects (via PARs) in advanced plaques, but also serves a physiologic function as a critical wound-healing molecule. Therefore, we sought to interdict key inflammatory signaling events at their source, the plaque macrophage, without affecting either clotting or the physiological function of thrombin itself.
Accordingly, we selected the JNK pathway as a target for testing a new class of recently developed therapeutic nanoparticles that could be used to deliver highly specific siRNA for JNK protein knockdown in procoagulant plaques. JNKs are members of the MAPK family and play a critical role as proapoptotic responders to a variety of environmental stresses, cytokines, and growth factors.13–15 JNK2, one of three isoforms in the JNK family (also termed “stress-activated protein kinases”), is a principal mediator of foam-cell formation through the scavenger-receptor-signaling pathway. JNK2 stimulates macrophage uptake and internalization of oxLDL to form resident foam cells by phosphorylating scavenger receptors.16 The scavenger-receptor-signaling pathway plays a critical role in macrophage uptake of oxidized lipids and foam-cell formation.17 Although JNK2 represents a therapeutic target for suppressing atherosclerosis,13,16 there are no selective pharmaceutical agents that might facilitate elucidation of JNK-isoform activities in vivo. The immediate goal was to develop a specific RNA-silencing approach for suppressing JNK2 locally in plaques as proof of concept for a flexible and interchangeable platform capable of targeting any mRNA of interest in inflamed plaques.
RNA interference induced by siRNA is based on complete base-pair match with the mRNA of interest and induced solely by mRNA cleavage. This interference mechanism enables multiple mRNAs to be degraded by one siRNA and achieve efficient and long-lasting knockdown effects. In fact, cell division is the only dilution limit in siRNA-induced mRNA knockdown.18 Although siRNA holds great promise as a therapeutic approach, its clinical translation has been limited, due to challenges in effective delivery to the site of interest. Besides viral-based delivery strategies, cationic lipids, polymers, and peptides are employed for siRNA delivery. Although cationic lipid is the best characterized and most efficient delivery approach, clinical translation has been limited, due to its cytotoxicity19 and systemic immunoresponses.20 The protonatable moieties of the cationic polymers, including polyethylenimine, cyclodextrin, and polysaccharide chitosan,21 provide robust endosomolysis for siRNA endosomal escape. However, increased cytotoxicity from destabilization of cell membrane and reactive oxygen-species production limits their clinical translation.22,23 Because of promoted endocytosis and lack of cytotoxicity, cationic peptides have gained popularity in siRNA delivery. Unfortunately, the application of peptide vectors is limited by endosomal entrapment and serum instability.24,25 Accordingly, we recently developed a novel peptide that overcomes the limitations of other cationic peptides, and incorporated pH-sensing residues to release siRNA and enable endosomal escape.26–28
We report herein the application of this flexible peptide-based siRNA-delivery system26–28 for selective silencing of JNK2 in atherosclerotic plaques of ApoE−/− mice on a Western diet. We describe an engineered 21-amino acid, amphipathic, cationic peptide (p5RHH) that condenses oligonucleotides into a 55 nm particle in a simple mixing procedure ready for injection. These nanostructures produce no measurable innate or adaptive immunoresponses and no apparent organ toxicity. We show that p5RHH-JNK2 siRNA nanoparticles rapidly penetrate disrupted plaque endothelial barriers to downregulate local JNK2 expression, resulting in attenuation of vessel-procoagulant activity, recovery of endothelial barrier integrity, and reduction in plaque macrophages after sequential dosing for <1 month. Additionally, we demonstrate that JNK2 silencing reduces plaque inflammation in part by inhibiting NFκB and STAT3 signaling.
Methods
p5RHH-siRNA-nanoparticle preparation
Peptide structures for condensation of siRNA into 55 nm particles have been described in prior publications.26,27 p5RHH peptide was synthesized by GenScript and siRNAs with or without fluorescence labeling were ordered from Sigma-Aldrich (St Louis, MO, USA). p5RHH peptide (NH2-VLTTGLPALISWIRRRHRRHC-COOH) was prepared at 20 mM in molecular biology-grade water (46-000-CI; Corning, New York, NY, USA), while siRNAs were prepared at 100 μM in siRNA buffer diluted from 5× siRNA buffer (B-002000-UB-100; Thermo Fisher Scientific, Waltham, MA, USA). The peptide:siRNA mole ratio was 100:1. For in vitro applications, p5RHH-siRNA nanoparticles were formulated by mixing p5RHH and siRNA in Hanks’ Balanced Salt solution (HBSS) (14025-092; Thermo Fisher Scientific) and incubated at 37°C for 40 minutes, followed by albumin stabilization, as previously reported.27 For in vivo studies, peptide and siRNA mixtures in HBSS were incubated on ice for 10 minutes prior to intravenous (IV) injection with 0.5 mg siRNA/kg. Physical characterizations of albumin-coated p5RHH-siRNA nanoparticles showed a particle size of 55 nm with polydispersity of 0.282 and ζ-potential of −33.24 mV.
Perfluorocarbon-nanoparticle formulation
Perfluorocarbon (PFC) nanoparticles were produced using previously described methods.29 Briefly, a lipid mixture of 99 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphocholine and 1 mol% 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (850355P and 850705P, respectively; Avanti Polar Lipids, Alabaster, AL, USA) was prepared in a chloroform–methanol mixture (3:1 v:v). The solvents were then evaporated to generate a lipid film that was dried overnight in a vacuum oven at 50°C. The lipid film (2.0% w:v), perfluoro-15-crown-5-ether (CE; 20% w:v; Gateway Specialty Chemicals, St Peters, MO, USA), and Milli-Q water were sonicated and emulsified for five passes at 20,000 psi (138,000 kPa) in an ice bath (LV-1; Microfluidics, Newton, MA, USA). The emulsion was modified to replace the crown ether core with perfluorooctyl bromide (PFOB) for use as a 19F magnetic resonance spectroscopy (MRS) reference standard for quantification of nanoparticle concentration in plaques.
Cell culture
Mouse monocytic RAW 264.7 cells (Washington University Tissue Culture Support Center, St Louis, MO, USA) were maintained in DMEM (30-2001; ATCC, Manassas, VA, USA) with 10% v:v heat inactivated fetal bovine serum (FBS) (F2442; Sigma-Aldrich) in a humidified environment with 5% CO2. RAW 264.7 cells were seeded in six-well plates and maintained in the aforementioned culture conditions for 24 hours before p5RHH-siRNA-nanoparticle treatment. p5RHH-siRNA nanoparticles were formulated and incubated with RAW 264.7 cells at siRNA concentration of 50 nM for 4 hours. Then, cells were washed twice with PBS (14040117; Thermo Fisher Scientific) and incubated with the aforementioned culture medium for another 24–72 hours, before reverse-transcription polymerase chain reaction (RT2-PCR) or Western blot.
Confocal imaging
RAW 264.7 cells were cultured on glass coverslips and treated with p5RHH-siRNA nanoparticles generated by using Cy3-labeled siRNA (Sigma-Aldrich). At 48 hours after treatment, cells were washed three times in PBS with Ca2+ and Mg2+ and fixed in 4% paraformaldehyde (157-4-100; Electron Microscopy Sciences, Hatfield, PA, USA) before being mounted on glass slides using mounting medium with 4′,6-diamidino-2-phenylindole (H-1200; Vector Labs, Burlingame, CA, USA). Cells were imaged with confocal microscopy (Meta 510; Carl Zeiss, Oberkochen, Germany).
Two-photon imaging
ApoE−/− mice fed a Western diet for 3.5 months received IV injections of p5RHH-Cy3-labeled siRNA nanoparticles or control nanoparticles without fluorescence labeling and were euthanized 24 hours later to collect aortas. En face imaging of aorta samples was performed on a custom-built video-rate two-photon microscope equipped with a Chameleon Vision Sapphire IITi laser (Coherent, Santa Clara, CA, USA).
IVIS fluorescence imaging
ApoE−/− mice fed a Western diet for 3.5 months received IV injections of p5RHH-Cy5.5-labeled siRNA nanoparticles or control nanoparticles without fluorescence labeling. At 24 hours after injection, aortas were collected for ex vivo IVIS imaging (Xenogen IVIS Spectrum; PerkinElmer, Waltham, MA, USA). The following settings were used for image acquisition: excitation 675 nm, emission 720 nm, binning factor 8, field of view 12.9, exposure time 0.5 seconds, and f-value 2. The pseudocolor “efficiency image” was presented to illustrate fluorescence-labeled nanoparticle distribution in the aortas. Fluorescence data were calibrated quantitatively in units of radiant efficiency or photon flux per unit excitation intensity (photons/sec/cm2/steradian)/(μW/cm2).
Real-time PCR
Total RNA from RAW 264.7 cells or aortas was isolated using an RNeasy minikit (74104; Qiagen, Venlo, the Netherlands). RNA (1 μg) was used to synthesize cDNA by reverse transcription with an RT2 first-strand kit (330401; Qiagen). Real-time PCR analysis was performed on an ABI 7300 system (Thermo Fisher Scientific) with RT2 first SYBR green/ROX PCR master mix (330530; Qiagen). Specific primers for each gene were purchased from Qiagen. Genes of interest were normalized to mouse β-actin.
Western blot
Radioimmunoprecipitation assay (RIPA) buffer (R0278-500ML; Sigma-Aldrich) with protease inhibitors (4906837001; Sigma-Aldrich) per 10 mL RIPA buffer and phenylmethylsulfonyl fluoride (8553; Cell Signaling Technology, Danvers, MA, USA) at a final concentration of 1 mM was used to extract proteins from RAW 264.7 cells or aortas. Briefly, cells were disrupted and sonicated in the aforementioned buffer and protein lysates obtained by centrifugation for 10 minutes at 12,000× g at 4°C. For the aortas, homogenization was performed with a Bullet Blender Storm (Next Advance, Troy, NY), followed by the same centrifugation. Protein concentration was quantified with BCA protein assay (23225; Thermo Fisher Scientific). Under reducing conditions, equivalent amounts of total protein were fractionated using sodium dodecyl sulfate polyacrylamide-gel electrophoresis. Membranes were probed with rabbit anti-JNK2 or anti-STAT3 (1:1,000 dilution, 4,672S or 12,640S, respectively; Cell Signaling Technology) and rabbit anti-GAPDH (1:1,000 dilution, sc-25778; Santa Cruz Biotechnology, Dallas, TX, USA). Membranes were washed and incubated with secondary antibody antirabbit HRP (1:10,000 dilution, sc-2313; Santa Cruz Biotechnology). Bands were visualized with Pierce ECL Western blotting substrate (32106; Thermo Fisher Scientific) using ChemiDoc MP (Bio-Rad Laboratories, Hercules, CA, USA). Knockdown of proteins was quantified using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Foam-cell-formation assay
At 48 hours after p5RHH-JNK2 siRNA-nanoparticle treatment, RAW 264.7 cells were treated with or without 50 μg/mL acetylated LDL (acLDL; RP-045; Intracel Resources, Frederick, MD, USA) for an additional 24 hours. To visualize foam-cell formation, RAW 264.7 cells underwent oil red O staining as previously published.27
Immunofluorescence imaging
Aortas were embedded in optimal cutting-temperature compound before sectioning and staining. Briefly, the frozen aorta sections of 8 μm were fixed in acetone at −20°C for 20 minutes before incubation with rabbit anti-JNK2 (1:200 dilution, ab76125, Abcam, Cambridge, UK), rat antimonocyte+ macrophage (Moma2; 1:200 dilution, ab33451; Abcam), or rabbit anti-NFκB p65 (D14E12) XP (1:200 dilution, 8242; Cell Signaling Technology), followed with incubation in appropriate secondary antibodies: goat pAb to rabbit IgG (DyLight 488; 1:500 dilution, ab96899; Abcam) or goat pAb to rat IgG (Alexa Fluor 594; 1:500 dilution, ab150160; Abcam). All images were acquired using an Olympus microscope at the same exposure times and analyzed with ImageJ. The percentage of acellular (anucleate) area in plaques was evaluated in blinded fashion by an experienced reviewer (HY) in these same aorta sections in mice with or without treatment based on regions of interest segmented in semiautomated objective fashion with the use of ImageJ to select all regions devoid of blue-counterstained nuclei. This method was consistent with what has been reported, and yielded comparable percentile ranges for plaque acellular compartments in ApoE−/− mice.30
Atherosclerosis induction and treatment
Male ApoE−/− mice (4–6 weeks old; Jackson Laboratory, Bar Harbor, ME, USA) were fed a Western diet (TD.88137; Envigo, Huntingdon, UK) for 14 weeks before treatment with p5RHH-JNK2 siRNA nanoparticles at 0.5 mg siRNA/kg or HBSS via tail-vein injection for 3.5 weeks, with a total of seven doses. During treatment, all mice were maintained on the Western diet. Animals were killed 24 hours after the final dose.
Carotid-artery thrombosis methodology
To define vessel-procoagulant activity, mice were anesthetized with a cocktail of xylazine and ketamine (37 and 87 mg/kg, respectively), and for carotid-artery photochemical injury, to measure the time required for vessel occlusion, as previously reported.10,31,32 Time to occlusion was recorded and used as a quantitative index of procoagulant activity and global thrombotic risk. Detailed methodology is included in Figure S1.
19F MRS quantification of nanoparticle deposition in aortic plaques
19F MRS of isolated whole aortas was measured by using a Varian 11.7 T scanner with an in-house-designed solenoid radiofrequency coil and procedures as published previously.33 Aortas were placed in centrifuge tubes and measured together with an internal standard of 0.1% PFOB nanoparticles. 19F MRS (flip angle 90°, number of averages 2,048 (aortas) using spin-echo sequence) was recorded for quantitative evaluation of PFC (crown ether) nanoparticle accumulation in the aortas. The crown ether 19F signal was compared with the distinct 19F signal of the PFOB standard containing a known concentration of 19F atoms. The calibrated crown ether nanoparticle amount was normalized to the weight of the aorta sample and expressed as picoliters per milligram (pL/mg), representing the content of PFC nanoparticles taken up by aortic plaques.
Compared to static endothelial staining methods for vascular barrier assessment, the use of semipermeating PFC nanoparticles with 250 nm diameter in vivo circulating provides a selective and functional metric for evaluating endothelial barrier damage in the atherosclerotic plaques, as these PFC nanoparticles do not penetrate vessels without missing endothelium. Moreover, these PFC nanoparticles do not register endothelial dysfunction with tight-junction weakening, and instead are specific to stages of atherosclerosis with plaque endothelial disruption, which correlates with procoagulant activity directly.32 Quantitative measurement based on 19F spectroscopy is objective, because a full aortic sample is used. Therefore, it is not subjective to selection bias.
Hematologic parameters and serum chemistry
Blood was collected from left ventricles and evaluated for blood counts and serum chemistry. All tests were done at the Department of Comparative Medicine, Washington University.
Splenic cell-subpopulation analysis
Spleens were collected and dissociated mechanically to get single-cell suspensions. R-phycoerythrin-conjugated anti-CD25 (561065, PC61; BD Biosciences, San Jose, CA, USA), R-phycoerythrin-conjugated anti-NK1.1 (12-5941-63, PK136; Thermo Fisher Scientific), fluorescein isothiocyanate- or peridinin–chlorophyll–protein complex-conjugated anti-CD4 (550280, L3T4; BD Biosciences), fluorescein isothiocyanate-conjugated anti-CD19 (550284, 1D3; BD Biosciences), peridinin–chlorophyll–protein complex- or allophycocyanin-conjugated anti-CD8 (553032, 53-6.7; BD Biosciences), and Gr1 (14-5931-81, Ly-6G; Thermo Fisher Scientific) were used. Briefly, 106 cells were used and blocked in the anti-FcR monoclonal antibody 2.4 G2, followed with staining by incubation with the indicated antibodies at 4°C for 20 minutes. Cells were then washed and resuspended before flow-cytometry analysis. A FoxP3 staining kit (71-5775; Thermo Fisher Scientific) was used to investigate FoxP3 expression. A FACSCalibur cell analyzer was used for flow-cytometry evaluations and CellQuest Pro software used for data analysis (BD Biosciences).
Purification and stimulation of CD4+ T cells
To isolate CD4+ T cells from mouse spleens, positive magnetic sorting was performed using an AutoMACS separator (130-104-454, MACS CD4+ microbeads; Miltenyi Biotech, Bergisch Gladbach, Germany). Fluorescence-labeled anti-CD4, anti-CD8, and anti-CD19 monoclonal antibodies (BD Biosciences) were used to verify isolated CD4+ population purity using flow cytometry. After purification, 2×106 cells/mL CD4+ T cells were resuspended in RPMI 1640 supplemented with 1% v:v NEAA, 10% v:v FBS, 2 mM L-glutamine, 1% v:v sodium pyruvate, 10 mM HEPES, 100 μg/mL streptomycin, 50 μM 2-mercaptoethanol, and 100 U/mL penicillin, then seeded at 2×105 cells/well in flat-bottomed 96-well microtiter plates coated with 5 μg/mL anti-CD3 monoclonal antibody (555273; BD Biosciences). Cells were cultured for 72 hours, before collection of culture supernatants for cytokine evaluations using BD cytometric bead arrays (552364 mouse inflammation kit; BD Biosciences).
Complement activation
Mice received IV injections of p5RHH-JNK2 siRNA nanoparticles at 0.5 mg siRNA/kg. At 30 minutes postinjection, blood was drawn from the inferior vena cava. To prevent further ex vivo complement activation, blood was transferred immediately into tubes with 10 mM EDTA to get fresh plasma, which was used for C3a enzyme-linked immunosorbent assay (ELISA).34 Briefly, rat antimouse C3a (4 μg/mL) monoclonal antibody (558250; BD Biosciences) was used to coat plates by overnight incubation at 4°C. After blocking (1% BSA in PBS at room temperature for 1 hour) and washing (three times with 0.05% v:v Tween 20 in PBS), plates were incubated with 100 μL freshly collected 1:100-diluted plasma samples at room temperature for 1 hour. Plates were then washed three times and incubated with biotinylated antimouse C3a at 250 ng/mL at room temperature for 1 hour. Plates were then washed and incubated with 400 ng/mL streptavidin–peroxidase for 30 minutes. After the incubation and wash, 100 μL peroxide–chromogen solution was added per well, and a SpectraMax Plus reader (Molecular Devices, Sunnyvale, CA, USA) was used to read at 450 nm. The standard curve was established by using mouse recombinant C3a (558618; BD Biosciences).
Immunoresponses to nanoparticles
To evaluate IgM- or IgG-specific response to p5RHH-JNK2 siRNA nanoparticles, 96-well plates (3855; Immulon 4 HBX; Thermo Fisher Scientific) coated with nanoparticles were formulated as previously reported.35 Plates were washed and then blocked with 1% BSA in PBS before the addition of diluted mouse sera (1:10–1:100 dilution) to wells. After incubation at room temperature for 1 hour and washing, HRP-conjugated goat antimouse IgM or goat antimouse IgG (1021-05 or 1030-05, respectively; Southern Biotechnology Associates, Birmingham, AL, USA) antibody was added. After the incubation and wash, 100 μL peroxide–chromogen solution was added to each well, and the SpectraMax Plus reader was used to read at 450 nm.
Statistics
Data are represented as means ± standard error (SE). Unpaired two-tailed Student’s t-tests were performed to assess differences between two groups. For comparison between more than two groups, one-way analysis of variance (ANOVA) followed with Scheffé post hoc test was performed. P<0.05 was considered statistically significant.
Animal study approval
Animal experiments were completed in compliance with US federal laws and in accordance with Washington University Division of Comparative Medicine guidelines. The animal protocol is reviewed annually and approved by the Washington University Animal Studies Committee.
Results
p5RHH-JNK2 siRNA decreases plaque JNK2 expression in hypercholesterolemic ApoE−/− mice
In vivo delivery of p5RHH-JNK2 siRNA was tested for efficacy by measurements of JNK2 mRNA and protein in ApoE−/− mice after seven serial doses administered over 3.5 weeks, starting at 14 weeks after initiation of a Western diet. Compared to controls, which were ApoE−/− mice treated with HBSS, JNK2 mRNA levels decreased by 26% in the whole aorta from the ApoE−/− mice receiving p5RHH-JNK2 siRNA nanoparticles (P=0.044; Figure 1A). However, p5RHH-scrambled siRNA-nanoparticle treatment over the same duration in the age-matched ApoE−/− mice receiving the same diet regimen exerted no effect on JNK2 mRNA levels (Figure 1A). Moreover, JNK1 mRNA levels were not altered by p5RHH-JNK2 siRNA-nanoparticle treatment (Figure 1B), thereby confirming isoform specificity of the p5RHH-JNK2 siRNA nanoparticles. Because nanoparticles packaged with scrambled siRNA did not alter the target JNK2 mRNA expression in vivo, ApoE−/− mice receiving HBSS treatment served as controls to evaluate in vivo inhibition of JNK2 protein expression and functional effects of p5RHH-JNK2-nanoparticle treatments. JNK2 protein levels were reduced by 42% in whole aortas from ApoE−/− mice receiving p5RHH-JNK2 siRNA nanoparticles compared to those in the control group (P=0.042; Figure 1C and D). Examples of p5RHH-siRNA nanoparticles residing in plaque are illustrated in Figure S2.
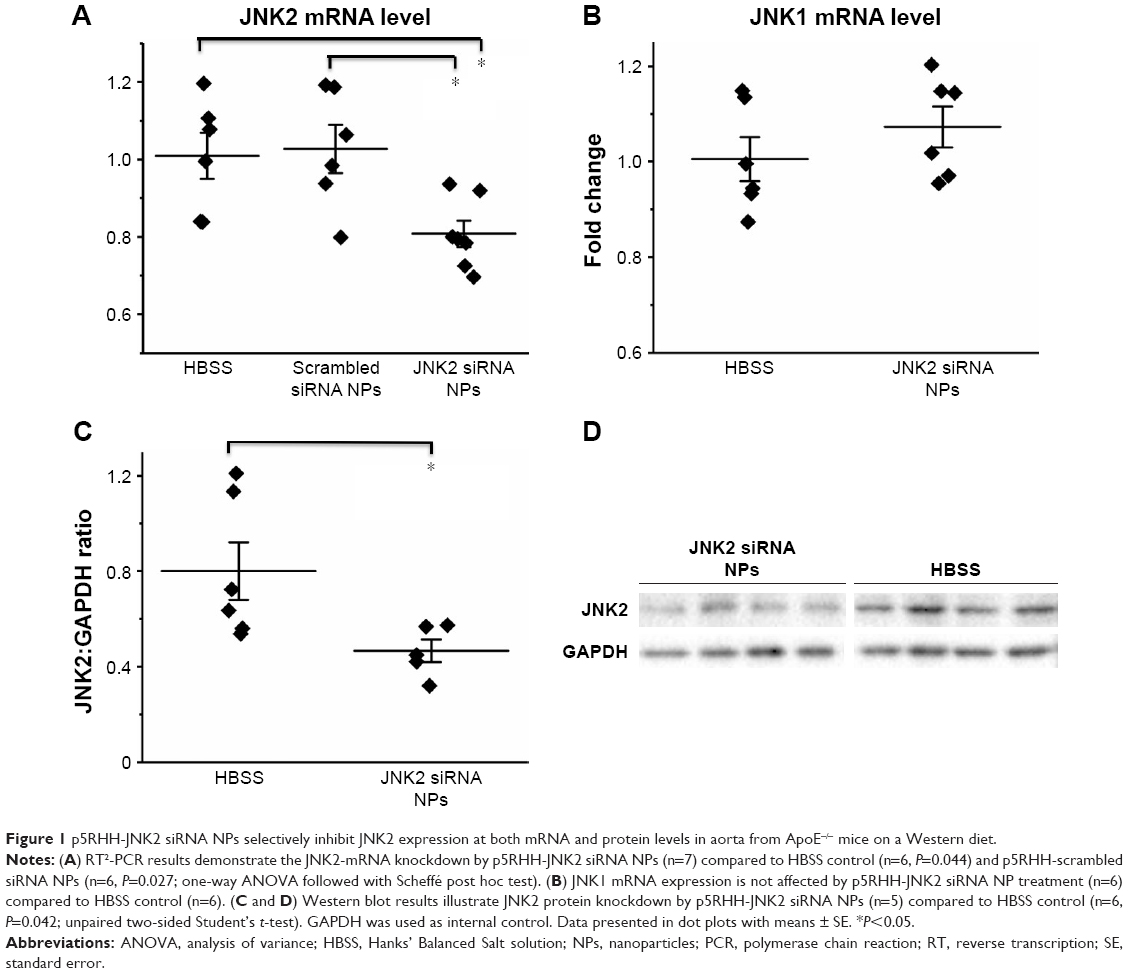 | Figure 1 p5RHH-JNK2 siRNA NPs selectively inhibit JNK2 expression at both mRNA and protein levels in aorta from ApoE−/− mice on a Western diet. |
JNK2 silencing reduces thrombotic risk
To evaluate the effect of p5RHH-JNK2 siRNA-nanoparticle treatment on vessel prothrombotic activity, ApoE−/− mice with p5RHH-JNK2 siRNA treatment or HBSS control treatment underwent a carotid-artery-injury procedure10,11,32 to elicit focal clotting, in order to quantify vascular procoagulant activity as previously reported. In this widely used procedure,36–38 shorter occlusion times indicate more aggressive clotting and heightened thrombotic risk. As illustrated in Figure 2A, the time to complete occlusion of siRNA nanoparticle-treated ApoE−/− mice increased 200% over that of the control subjects (47.2±4.4 vs 22.4±6.8 minutes, treated vs control; P=0.02), indicating that even relatively short-term therapy with p5RHH-JNK2 siRNA nanoparticles dramatically attenuated thrombotic risk.
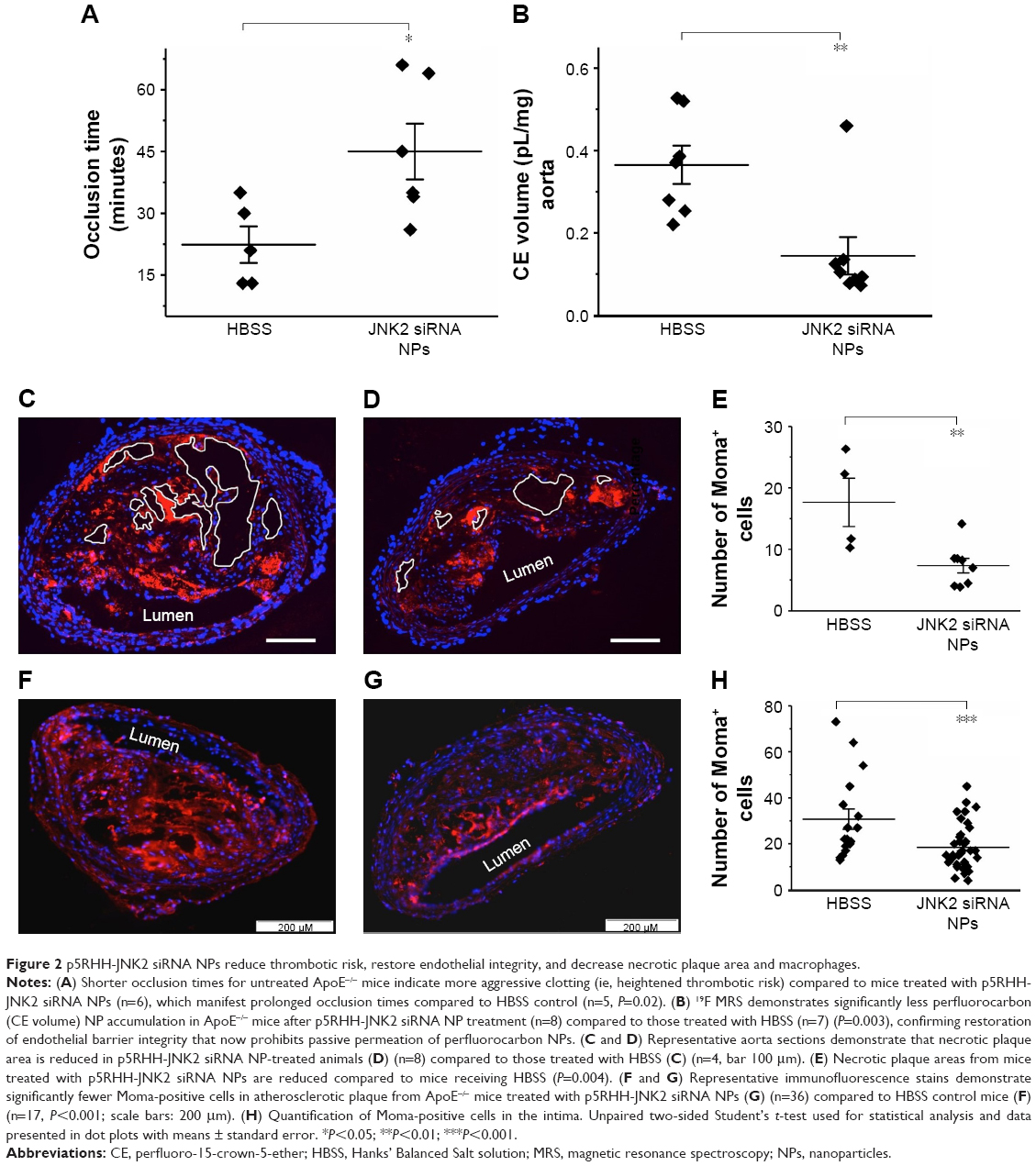 | Figure 2 p5RHH-JNK2 siRNA NPs reduce thrombotic risk, restore endothelial integrity, and decrease necrotic plaque area and macrophages. |
Restoration of endothelial barrier function by JNK2 silencing
To probe postulated mechanisms associated with reduced thrombotic risk, the status of endothelial vascular barriers in ApoE−/− mice receiving p5RHH-JNK2 siRNA-nanoparticle treatment or HBSS control treatment was elucidated by quantifying the plaque uptake of semipermeating PFC nanoparticles that diffuse passively in vivo across damaged endothelium, but not intact vasculature. As reported previously,10,32,39 PFC nanoparticles of nominal 200 nm diameter will passively penetrate deeply and rapidly into atherosclerotic plaques exhibiting endothelial disruption, but are excluded from adjacent vessel regions with intact endothelial barriers. Based on the unique 19F MR signature of the PFC nanoparticles, absolute quantification of PFC nanoparticles accumulating in the plaque components of the aorta after 2 hours of in vivo circulation can be calculated by registering the 19F MR spectrum and comparing it to a known fluorine-calibration standard. Accordingly, quantification of PFC nanoparticles in the whole aorta provides an objective index of vascular barrier disruption. Figure 2B illustrates the accumulation of PFC nanoparticles in aortas from ApoE−/− mice on a Western diet. Those mice receiving p5RHH-JNK2 siRNA nanoparticles manifested a 62% reduction in 19F signals emanating from plaque-trapped nanoparticles compared to control mice (0.150±0.046 pL/mg vs 0.365±0.047 pL/mg for treated vs controls, P=0.003), representing a 2.6-fold decrease in barrier permeability and disruption after treatment.
JNK2 silencing reduces plaque necrotic area and macrophage prevalence
As shown in Figure 2, the acellular plaque area (Figure 2C–E) was reduced by 58% (P=0.004), suggesting reduced necrosis. Macrophage staining was quantified to define the role of anti-JNK2 siRNA on plaque-macrophage burden. As demonstrated in Figure 2F–H, macrophage populations in the atherosclerotic plaques were reduced significantly in the plaques of ApoE−/− mice treated with p5RHH-JNK2 siRNA nanoparticles compared to mice receiving HBSS control treatment.
JNK2 silencing reduces inflammatory signaling
Because NFκB signaling is a pivotal driver of inflammation and a transcriptional mediator of numerous atherosclerotic gene families,34,40–42 NFκB-activation status was evaluated after p5RHH-JNK2 siRNA-nanoparticle treatment. Immunostaining of p65, a canonical NFκB protein, was performed on aorta sections from ApoE−/− mice receiving p5RHH-JNK2 siRNA treatment or HBSS control treatment. Confocal images on the plaque region revealed significantly more cells harboring both cytoplasmic and nuclear p65 in the sections from the ApoE−/− mice with HBSS control treatment (Figure 3A) than in those treated with the p5RHH-JNK2 siRNA nanoparticles (Figure 3B). Semiquantification of immunofluorescence-stained sections indicated that 42.66% of all cells in plaques from control animals exhibited activated NFκB signaling, whereas only 22.48% of cells were positive in animals undergoing treatment (Figure 3C); a 47% reduction. Given the cross talk between NFκB and STAT343–46 and the crucial role of STAT3 signaling in the progression of atherosclerosis,47–49 STAT3 protein expression in the whole aorta was evaluated (Figure 3D and E). p5RHH-JNK2 siRNA nanoparticle-treated animals exhibited a 46% reduction in whole-aorta STAT3 expression (P=0.004) compared to controls.
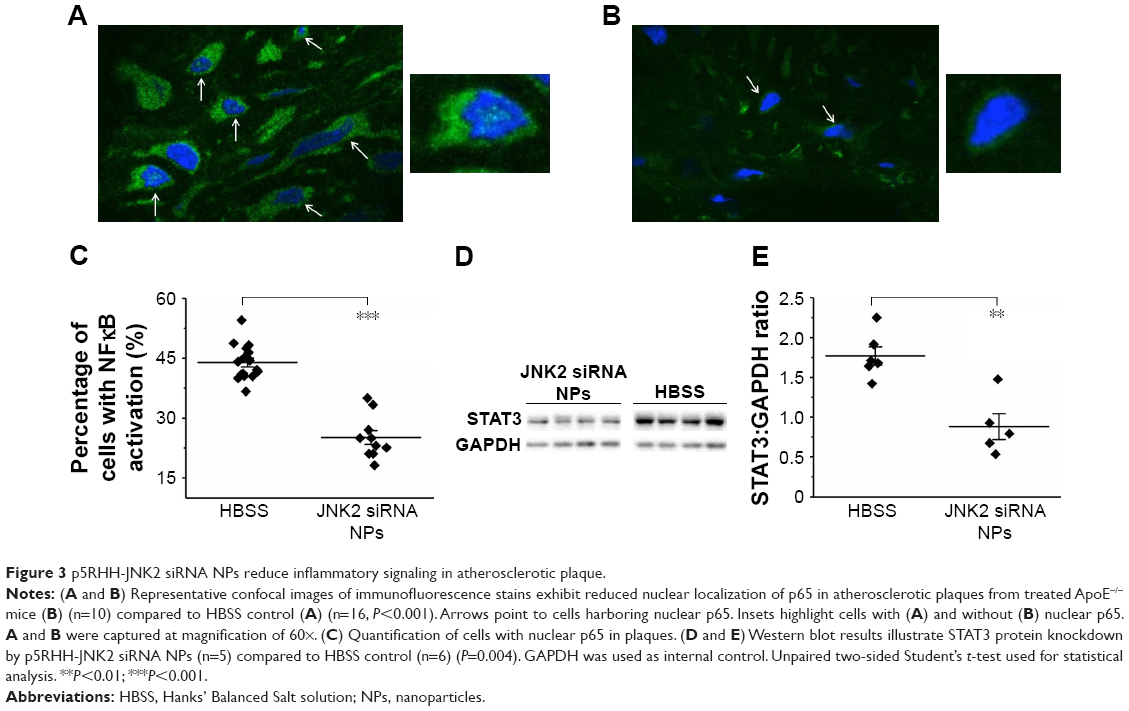 | Figure 3 p5RHH-JNK2 siRNA NPs reduce inflammatory signaling in atherosclerotic plaque. |
Effects of p5RHH-JNK2 siRNA on macrophages
JNK2 expression has been reported to be two- to threefold higher in human atherosclerotic plaques compared to normal arteries.50 To define potential cell type(s) that could be affected by the local delivery of p5RHH-JNK2 siRNA nanoparticles, the expression of JNK2 within different cell types, macrophages, smooth-muscle cells, and endothelial cells was evaluated. Costaining of JNK2 and markers for macrophages, smooth-muscle cells, and endothelial cells demonstrated that macrophages were the principal source of plaque JNK2 (Figures S3 and S4). Therefore, we evaluated consequences of p5RHH-siRNA-nanoparticle treatment of cultured RAW 264.7 macrophages. To investigate cytoplasmic delivery of siRNA to RAW 264.7 cells, Cy3-labeled eGFP siRNA nanoparticles were prepared and visualized by confocal microscopy. Macrophages were incubated with either p5RHH-Cy3-labeled eGFP siRNA nanoparticles or free Cy3-labeled eGFP siRNAs for 48 hours before microscopic imaging. Surface rendered Z-stack series demonstrated significantly more uptake of siRNAs packaged into nanoparticles (Figure 4A) than for free Cy3-labeled siRNAs (Figure 4B). Moreover, higher-magnification confocal images illustrated siRNA endosomal escape and release of siRNAs into cytoplasm directly abutting nuclear structures (Figure 4C).
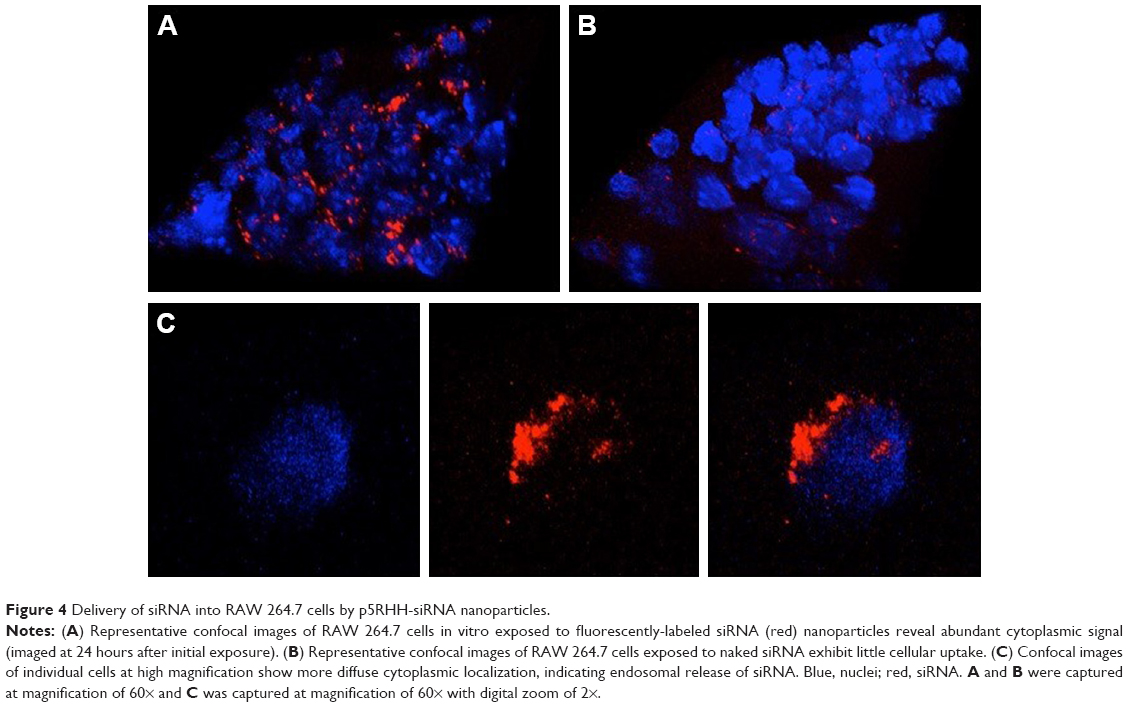 | Figure 4 Delivery of siRNA into RAW 264.7 cells by p5RHH-siRNA nanoparticles. |
To understand the effects of p5RHH-JNK2 siRNA nanoparticles on macrophage JNK2 mRNA and protein levels and their role in foam-cell formation, macrophages were challenged with acLDL. First, RAW 264.7 cells were treated with p5RHH-JNK2 siRNA nanoparticles at siRNA concentration of 50 nM and harvested at 24, 48, and 72 hours for RNA extraction. Macrophages treated with p5RHH-JNK2 siRNA nanoparticles after 24, 48, and 72 hours exhibited JNK2 mRNA diminution of 79%±3%, 80%±2%, and 75%±2%, respectively, vs cells without p5RHH-JNK2 siRNA-nanoparticle treatment (Figure 5A).
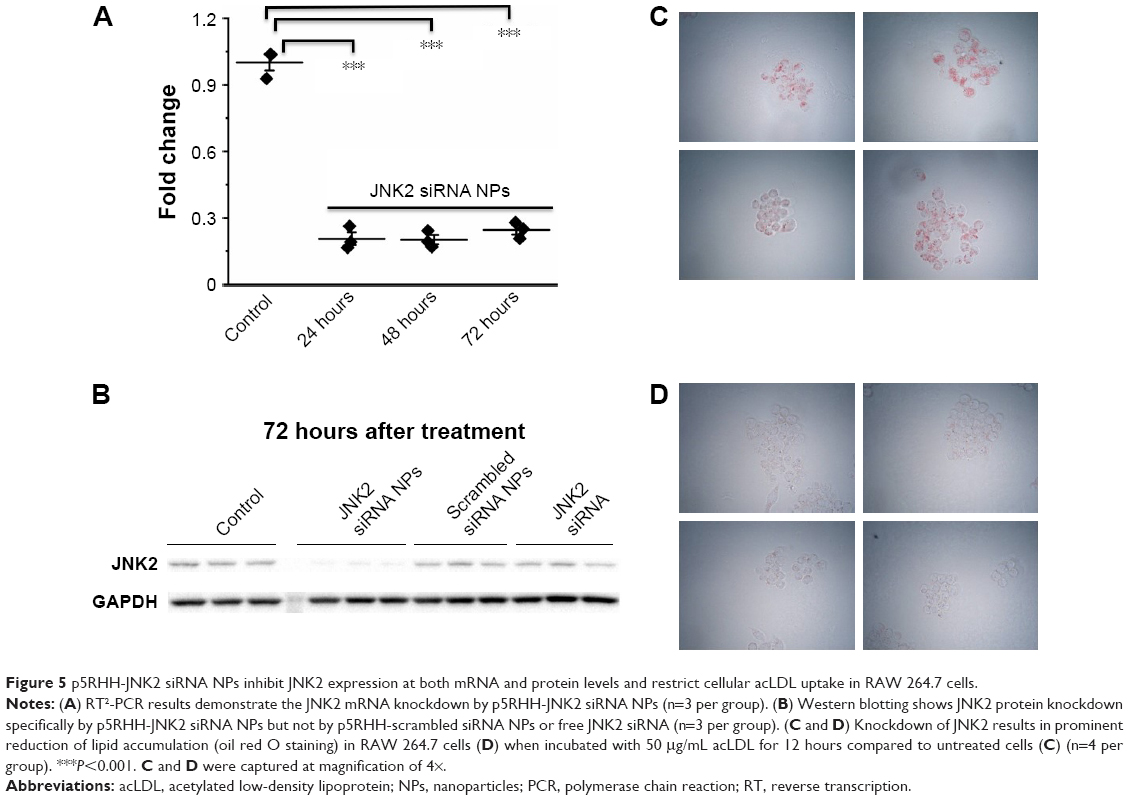 | Figure 5 p5RHH-JNK2 siRNA NPs inhibit JNK2 expression at both mRNA and protein levels and restrict cellular acLDL uptake in RAW 264.7 cells. |
JNK2 protein knockdown was determined at the 72-hour time point in macrophages receiving either p5RHH-JNK2 siRNA nanoparticles, p5RHH-scrambled siRNA nanoparticles, or free JNK2 siRNA, all at siRNA concentration of 50 nM. Macrophages without treatment served as additional controls. Only macrophages treated with p5RHH-JNK2 siRNA nanoparticles manifested significant JNK2 protein knockdown (Figure 5B).
To assess the functional consequences of JNK2 knockdown in macrophages, the effect of p5RHH-JNK2 siRNA nanoparticles on macrophage acLDL cellular uptake was evaluated. RAW 264.7 cells were treated with p5RHH-JNK2 siRNA nanoparticles at JNK2 siRNA concentration of 50 nM, and cells without treatment served as controls. After 48 hours, cells were incubated with 50 μg/mL acLDL for another 24 hours, and then oil red O staining was applied to delineate cellular uptake of acLDL. Treatment with p5RHH-JNK2 siRNA nanoparticles substantially inhibited the cellular uptake of acLDL (Figure 5C) compared to controls (Figure 5D). These results confirmed that p5RHH-JNK2 siRNA nanoparticles were capable of decreasing JNK2 expression at both mRNA and protein levels to inhibit foam-cell formation.
Safety of p5RHH-JNK2 siRNA-nanoparticle treatment
Blood chemistry, off-target immune-cell functions, and innate/adaptive immunoresponses were evaluated. Blood was collected after ApoE−/− mice had received serial IV injection of seven doses of p5RHH-JNK2 siRNA nanoparticles at 0.5 mg siRNA/kg over 3.5 weeks, which is the same regimen used for therapeutic evaluation. Blood from HBSS-treated mice served as control. Blood-chemistry results from the serum evaluation are summarized in Table 1, which indicates that multiple doses of p5RHH-JNK2 siRNA nanoparticles did not alter electrolyte profiles, liver/kidney function, or red blood cell/platelet counts.
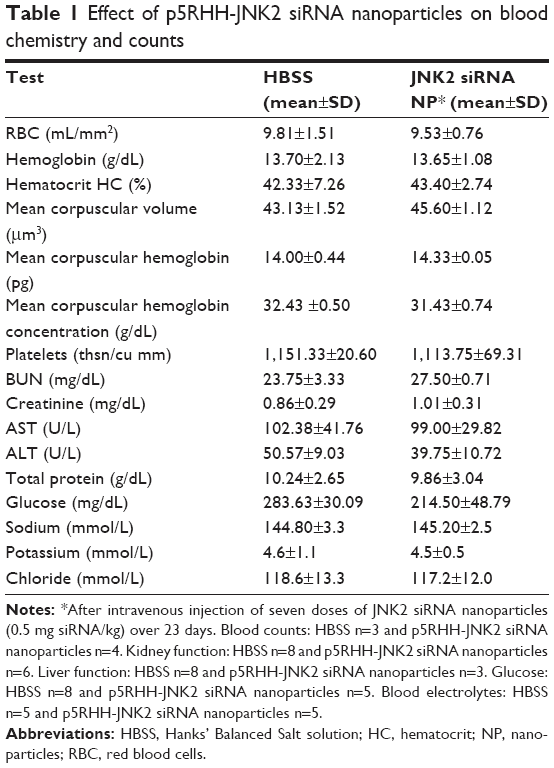 | Table 1 Effect of p5RHH-JNK2 siRNA nanoparticles on blood chemistry and counts |
To evaluate the effect of p5RHH-JNK2 siRNA nanoparticles on the functions of immune cells, 24 hours after the last dose, spleens from ApoE−/− mice that had received p5RHH-JNK2 siRNA-nanoparticle treatment and the control group were harvested for splenocyte-subpopulation analysis. As illustrated in Figure 6A, the number of splenocytes was significantly lower in ApoE−/− mice treated with p5RHH-JNK2 siRNA nanoparticles compared to those without treatment (55.00±14×106 vs 94.33±16×106, treated vs control). Figure 6B indicates that spleen weights also were 54% lower in treated vs HBSS control mice (0.13±0.07 g vs 0.28±0.17 g, treated vs control; P=0.001). Moreover, spleen weights in treated ApoE−/− mice approximated more those of C57BL/6 mice on normal chow (0.13±0.07 g vs 0.08±0.01 g, treated ApoE−/− mice vs C57BL/6; Figure 6B), and the post-hoc test suggested that spleen weights of ApoE−/− mice treated with p5RHH-JNK2 siRNA nanoparticles were not significantly different from C57BL/6 mice (P=0.511).
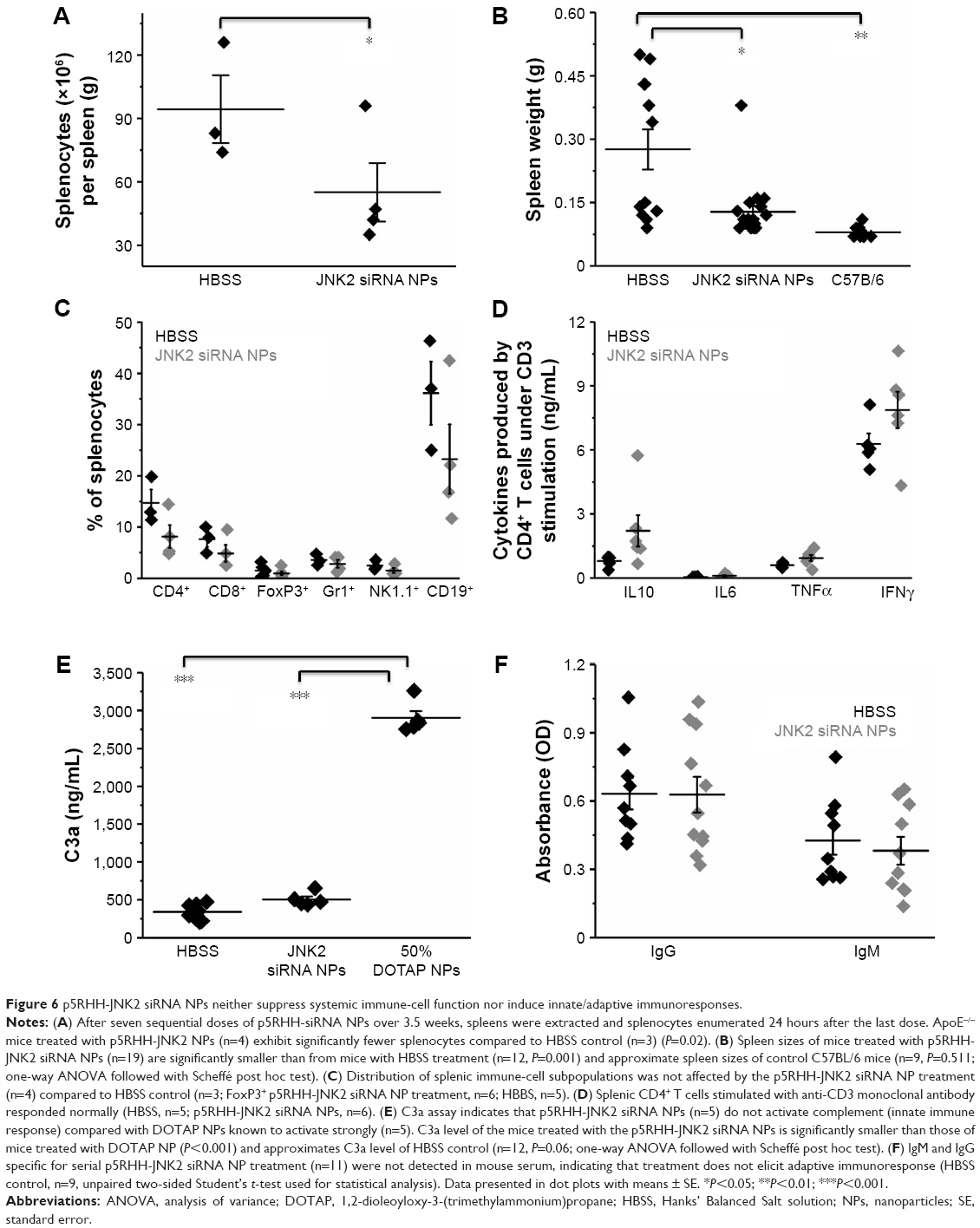 | Figure 6 p5RHH-JNK2 siRNA NPs neither suppress systemic immune-cell function nor induce innate/adaptive immunoresponses. |
Splenocyte-subpopulation differences were examined in treated vs untreated ApoE−/− mice: CD8+ and CD4+ T cells, FoxP3+ T-regulatory cells, NK1.1+ natural killer cells, and GR1+, and CD19+ B cells. Figure 6C depicts the similarity between splenocyte subpopulations in these two groups. Additionally, following treatment with p5RHH-JNK2 siRNA nanoparticles, immune-cell function was assessed by ex vivo stimulating isolated and purified splenic CD4+ T cells with anti-CD3 monoclonal antibody. The results (Figure 6D) suggested that under anti-CD3 activation, CD4+ T cells from control and treated animals released comparable amounts of cytokines, including IL10 (1.72±0.75 vs 0.80±0.12 ng/mL), IL6 (0.106±0.037 vs 0.04±0.006 ng/mL), TNFα (0.94±0.14 ng/mL vs 0.61±0.04), and IFNγ (7.87±0.86 ng/mL vs 6.28±0.50) for treated vs control groups, respectively, indicative of preserved immune-system functionality after anti-JNK2 siRNA treatment.
Host innate immunoresponses to p5RHH-JNK2 siRNA nanoparticles were assessed. Plasma from ApoE−/− mice was collected 30 minutes after IV injection of HBSS (as control) or p5RHH-JNK2 siRNA nanoparticles for quantification of C3a, a degradation product of C3, which is an in vivo indicator of complement activation. As demonstrated in Figure 6E, observed was a minimal increase in C3a level in mice that received p5RHH-JNK2 siRNA nanoparticles compared to HBBS controls (504.86±39.01 vs 339.71±24.61 ng/mL, treated vs control). In contrast, Figure 6E indicates that 1,2-dioleoyloxy-3-(trimethylammonium)propane (DOTAP), a known lipidic complement activator, enhanced C3a release and complement activation nearly sixfold (C3a 2,900.40±91.93 ng/mL), indicative of the relative safety margin against immediate immunoresponsiveness.
To evaluate the adaptive immunogenicity of p5RHH-JNK2 siRNA nanoparticles, IgG- and IgM-antibody responses against p5RHH-JNK2 siRNA nanoparticles were evaluated after the completion of treatment with seven serial total doses over 3.5 weeks (IV administration twice a week). Sera were collected from mice that were treated with either p5RHH-JNK2 siRNA nanoparticles or HBSS, and IgG and IgM antibodies specific for p5RHH-JNK2 siRNA nanoparticles were assessed by ELISA assays on plates coated with p5RHH-JNK2 siRNA nanoparticles. There was no significant difference observed between control and treatment groups (Figure 6F), demonstrating that p5RHH-JNK2 siRNA nanoparticles did not elicit appreciable adaptive immunoresponses at these time points.
Discussion
The goal of interfering with macrophage uptake of modified lipoproteins seeks to curtail atherosclerosis by removing a major stimulus for elaboration of numerous factors that perpetuate plaque inflammation. In this report, we describe plaque-homing peptide-based siRNA nanoparticles for in vivo RNA silencing of critical members of the MAPK-signaling system (JNK) that focally suppress inflammatory drivers of atherosclerotic plaques. Significant new observations arising from this study were: anti-JNK2 siRNA nanoparticles rapidly attenuated thrombotic risk, restored endothelial barrier integrity, reduced plaque necrosis, and depleted plaque-macrophage content after only a few weeks; JNK2 inhibition exerted suppression of proinflammatory molecules (eg, NFκB, STAT3); and nanoparticle treatment did not compromise critical systemic innate or adaptive immunoresponsiveness, and thus the approach exhibits a benign safety profile.
JNK2 expression has been reported to be two- to threefold higher in human atherosclerotic plaques compared to normal arteries.50 ApoE−/−JNK2−/− animals develop fewer atherosclerotic plaques, due to suppression of JNK2-mediated foam-cell formation.16 Moreover, it has been reported that enhanced local macrophage proliferation caused by scavenger-receptor A-mediated oxLDL uptake by macrophages is a key driver of plaque growth.51 In this report, focal plaque-macrophage targeting likely resulted from endothelial barrier disruption, which occurs in advanced plaques prior to neovascularization. In prior studies, we have observed no significant plaque angiogenesis at the time points studied in this ApoE−/− model,32 which accords with the observations of Moulton et al that ApoE−/− mice require at least 6 months on a Western diet to develop significant neovascularization.52,53 Nevertheless, for human atherosclerotic plaques, neovascularization accompanies chronic inflammation,54–59 which might be expected to facilitate intimal access via leaky neovasculature penetrating from adventitia. Under other experimental conditions, such as collagen antibody-induced arthritis, neovascular routes to inflammatory macrophages for p5RHH-siRNA nanoparticles may be operative.35
The origin of macrophages in atherosclerotic plaques remains an intriguing and debated question.60 Although further studies are required to elucidate fully the mechanism of the reduction of plaque-macrophage content observed in this study, it is possible that both reduced recruitment and decreased local proliferation contributed. It has been reported that proliferation of local macrophages accounts for most of the macrophage population in atherosclerotic plaques51 and oxLDL uptake promotes macrophage proliferation.61–64 In light of the fact that JNK2 inhibition is known to reduce oxLDL uptake by macrophages,16 Figure 5 (C and D) confirms that knockdown of JNK2 with peptide-siRNA nanoparticles in macrophage cell lines significantly decreased intracellular accumulation of modified LDL. Because oxLDL internalization induces proliferation,61–64 one likely avenue for the therapeutic efficacy of p5RHH-JNK2 siRNA pertains to reduced local macrophage proliferation (Figure 4A–C). The inhibition of JNK2 expression observed in the whole-aorta protein extracts (Figure 2) is consistent with decreased macrophage proliferation. Alternatively, Figure 5 (A and B) indicates that treatment reduced spleen size and number of splenocytes. It has been reported that monocytes originating from the spleen can migrate to plaques and contribute to plaque growth as a consequence of local expression of inflammatory cytokines/chemokines at atherosclerotic sites.65–67 Reduced local plaque inflammation might attenuate homing signals for splenic monocyte populations.
The anti-inflammatory benefits of this approach are suggested by reduced levels of the key STAT3- and NFκB-signaling pathways. A direct causal relationship between JNK2 expression and NFκB/STAT3 signaling is not specified by the current observations. It is possible that the JNK2 inhibition engendered an overall downregulation of local inflammation simply by inhibiting foam-cell formation. Indeed, our recent work with this siRNA-delivery system for inhibition of canonical and noncanonical NFκB in experimental models of rheumatism and osteoarthritis indicated that broad suppression of cytokine/chemokine production by macrophages and other activated cell types might be anticipated.35,68 Although it has been reported that macrophage-specific NFκB inhibition was proatherogenic in LDL-receptor-deficient mice,69 the overall modulation of intense inflammatory plaque signaling effected by inhibition of JNK2 in this study appears to prevail as a net-positive factor.
RNA silencing for suppressing JNK2-mediated foam-cell formation rapidly exerts additional benefits for plaque healing in the form of improved vascular barrier integrity and reduced vascular procoagulant activity. Because endothelial apoptosis promotes thrombosis,70,71 the rapid restoration of endothelial barrier integrity observed here lends support to mechanistic strategies aimed at preventing endothelial apoptosis, enhancing endothelial proliferation, or both. A correlation was observed between endothelial barrier recovery and reduced plaque macrophages (Figures 3 and 4).
Although the cellular actions of these siRNA nanostructures appear to predominate in the local plaque environment, there may be responses at a distance. The observation of reduced spleen weight after JNK2 inhibition was unexpected, but intriguing. The sterile inflammation in the spleen that accompanies atherosclerosis has been characterized variously as both protective72 and contributory,73 based on the activation of selected cellular compartments. We detected no specific differences in splenic immune-cell populations (Figure 6) in treated or untreated mice, although spleen size differed dramatically. Evaluation of immunoactivity toward the peptide-nanoparticle agent itself revealed no specific innate or adaptive immunoresponses (Figure 6) that might have triggered spleen expansion. Prior work on this peptide-siRNA nanoparticle has shown that these transfecting nanoparticles are not taken up in the spleen35 and exert no noticeable immunoactivation, perhaps militating against direct suppression of splenic immunoactivity to effect plaque healing. Moreover, body weights in the treated and control mice were similar, suggesting that a global effect on inflammation might not be operative. Rather, we propose that local resolution of plaque inflammation after JNK2 inhibition and the related depletion of circulating stimulatory factors within microparticles and/or exosomes that are released from plaque and traffic to distant sites, such as the spleen, could represent an alternative mechanism for future exploration. Nevertheless, current observations do raise the question of whether splenic targeting of JNK2 expression in reservoir macrophages with subsequent suppression of activated monocyte trafficking to plaques could play a supportive role in improved vascular barrier function, reduced procoagulant activity, and plaque healing that would be amenable to future study.
The present regimen could be useful for short-term intensive therapies requiring rapid control of hypercoagulable plaques under such circumstances as recent non-ST-elevation myocardial infarction, unstable angina, refractory angina, stroke, and other related pathologies associated with vascular barrier disruption and heightened thrombotic risk. Moreover, because these nanostructures can be formulated with any siRNA or even mixtures of multiplexed oligonucleotides,68 the approach could serve as a translational test bed for examining myriad signaling events of interest.
Acknowledgment
This study was supported by US National Institutes of Health grants HL073646, HL112303, DK102691, and AR067491 (to SAW) and AR067491 (to CTNP).
Disclosure
SAW has equity in Trasir Therapeutics. The authors report no other conflicts of interest in this work.
References
Libby P, Ridker PM, Hansson GK. Inflammation in atherosclerosis: from pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129–2138. | ||
Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327–387. | ||
Gimbrone MA, García-Cardeña G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22(1):9–15. | ||
Damani S, Bacconi A, Libiger O, et al. Characterization of circulating endothelial cells in acute myocardial infarction. Sci Transl Med. 2012;4(126):126ra33. | ||
Burke AP, Virmani R. Pathophysiology of acute myocardial infarction. Med Clin North Am. 2007;91(4):553–572; ix. | ||
Farb A, Burke AP, Tang AL, et al. Coronary plaque erosion without rupture into a lipid core: a frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996;93(7):1354–1363. | ||
Narula J, Nakano M, Virmani R, et al. Histopathologic characteristics of atherosclerotic coronary disease and implications of the findings for the invasive and noninvasive detection of vulnerable plaques. J Am Coll Cardiol. 2013;61(10):1041–1051. | ||
Prati F, Uemura S, Souteyrand G, et al. OCT-based diagnosis and management of STEMI associated with intact fibrous cap. JACC Cardiovasc Imaging. 2013;6(3):283–287. | ||
Yazdani SK, Vorpahl M, Ladich E, Virmani R. Pathology and vulnerability of atherosclerotic plaque: identification, treatment options, and individual patient differences for prevention of stroke. Curr Treat Options Cardiovasc Med. 2010;12(3):297–314. | ||
Palekar RU, Jallouk AP, Myerson JW, Pan H, Wickline SA. Inhibition of thrombin with PPACK-nanoparticles restores disrupted endothelial barriers and attenuates thrombotic risk in experimental atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(3):446–455. | ||
Myerson J, He L, Lanza G, Tollefsen D, Wickline S. Thrombin-inhibiting perfluorocarbon nanoparticles provide a novel strategy for the treatment and magnetic resonance imaging of acute thrombosis. J Thromb Haemost. 2011;9(7):1292–1300. | ||
Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–1131. | ||
Sumara G, Belwal M, Ricci R. “JNKing” atherosclerosis. Cell Mol Life Sci. 2005;62(21):2487–2494. | ||
Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci. 2008;115(7):203–218. | ||
Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. c-Jun N-terminal kinase (JNK) signaling: recent advances and challenges. Biochim Biophys Acta. 2010;1804(3):463–475. | ||
Ricci R, Sumara G, Sumara I, et al. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306(5701):1558–1561. | ||
Dickhout JG, Basseri S, Austin RC. Macrophage function and its impact on atherosclerotic lesion composition, progression, and stability: the good, the bad, and the ugly. Arterioscler Thromb Vasc Biol. 2008;28(8):1413–1415. | ||
Singh S, Narang AS, Mahato RI. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm Res. 2011;28(12):2996–3015. | ||
Filion MC, Phillips NC. Toxicity and immunomodulatory activity of liposomal vectors formulated with cationic lipids toward immune effector cells. Biochim Biophys Acta. 1997;1329(2):345–356. | ||
Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–530. | ||
Gary DJ, Puri N, Won YY. Polymer-based siRNA delivery: perspectives on the fundamental and phenomenological distinctions from polymer-based DNA delivery. J Control Release. 2007;121(1–2):64–73. | ||
Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114(1):100–109. | ||
Cheng J, Zeidan R, Mishra S, et al. Structure-function correlation of chloroquine and analogues as transgene expression enhancers in nonviral gene delivery. J Med Chem. 2006;49(22):6522–6531. | ||
Crombez L, Divita G. A non-covalent peptide-based strategy for siRNA delivery. Methods Mol Biol. 2011;683:349–360. | ||
Mäe M, Andaloussi SE, Lehto T, Langel U. Chemically modified cell-penetrating peptides for the delivery of nucleic acids. Expert Opin Drug Deliv. 2009;6(11):1195–1205. | ||
Hou KK, Pan H, Lanza GM, Wickline SA. Melittin derived peptides for nanoparticle based siRNA transfection. Biomaterials. 2013;34(12):3110–3119. | ||
Hou KK, Pan H, Ratner L, Schlesinger PH, Wickline SA. Mechanisms of nanoparticle-mediated siRNA transfection by melittin-derived peptides. ACS Nano. 2013;7(10):8605–8615. | ||
Hou KK, Pan H, Schlesinger PH, Wickline SA. A role for peptides in overcoming endosomal entrapment in siRNA delivery: a focus on melittin. Biotechnol Adv. 2015;33(6):931–940. | ||
Winter PM, Neubauer AM, Caruthers SD, et al. Endothelial ανβ3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2006;26(9):2103–2109. | ||
Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A. 2003;100(18):10423–10428. | ||
Myerson JW, He L, Allen JS, et al. Thrombin-inhibiting nanoparticles rapidly constitute versatile and detectable anticlotting surfaces. Nanotechnology. 2014;25(39):395101. | ||
Palekar RU, Jallouk AP, Goette MJ, et al. Quantifying progression and regression of thrombotic risk in experimental atherosclerosis. FASEB J. 2015;29(7):3100–3109. | ||
Waters EA, Chen J, Yang X, et al. Detection of targeted perfluorocarbon nanoparticle binding using 19F diffusion weighted MR spectroscopy. Magn Reson Med. 2008;60(5):1232–1236. | ||
Pham CT, Thomas DG, Beiser J, et al. Application of a hemolysis assay for analysis of complement activation by perfluorocarbon nanoparticles. Nanomedicine. 2014;10(3):651–660. | ||
Zhou HF, Yan H, Pan H, et al. Peptide-siRNA nanocomplexes targeting NF-κB subunit p65 suppress nascent experimental arthritis. J Clin Invest. 2014;124(10):4363–4374. | ||
Watts LT, Zheng W, Garling RJ, Frohlich VC, Lechleiter JD. Rose bengal photothrombosis by confocal optical imaging in vivo: a model of single vessel stroke. J Vis Exp. 2015;(100):e52794. | ||
Vicente CP, He L, Pavão MS, Tollefsen DM. Antithrombotic activity of dermatan sulfate in heparin cofactor II-deficient mice. Blood. 2004;104(13):3965–3970. | ||
Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis. Arterioscler Thromb Vasc Biol. 2007;27(10):2079–2093. | ||
Zhang H, Zhang L, Myerson J, et al. Quantifying the evolution of vascular barrier disruption in advanced atherosclerosis with semipermeant nanoparticle contrast agents. PLoS One. 2011;6(10):e26385. | ||
Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170(2):191–203. | ||
Cuaz-Pérolin C, Billiet L, Baugé E, et al. Antiinflammatory and antiatherogenic effects of the NF-κB inhibitor acetyl-11-keto-β-boswellic acid in LPS-challenged ApoE-/- mice. Arterioscler Thromb Vasc Biol. 2008;28(2):272–277. | ||
Wolfrum S, Teupser D, Tan M, Chen KY, Breslow JL. The protective effect of A20 on atherosclerosis in apolipoprotein E-deficient mice is associated with reduced expression of NF-κB target genes. Proc Natl Acad Sci U S A. 2007;104(47):18601–18606. | ||
Fan Y, Mao R, Yang J. NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell. 2013;4(3):176–185. | ||
Grivennikov SI, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010;21(1):11–19. | ||
He G, Karin M. NF-κB and STAT3: key players in liver inflammation and cancer. Cell Res. 2011;21(1):159–168. | ||
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. | ||
Wang R, Zhang Y, Xu L, et al. Protein inhibitor of activated STAT3 suppresses oxidized LDL-induced cell responses during atherosclerosis in apolipoprotein E-deficient mice. Sci Rep. 2016;6:36790. | ||
Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res. 2015;106(3):365–374. | ||
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(5):969–979. | ||
Slevin M, Elasbali AB, Turu M, Krupinski J, Badimon L, Gaffney J. Identification of differential protein expression associated with development of unstable human carotid plaques. Am J Pathol. 2006;168(3):1004–1021. | ||
Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19(9):1166–1172. | ||
Moulton KS, Vakili K, Zurakowski D, et al. Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc Natl Acad Sci U S A. 2003;100(8):4736–4741. | ||
Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation. 1999;99(13):1726–1732. | ||
Barger AC, Beeuwkes R, Lainey LL, Silverman KJ. Hypothesis: vasa vasorum and neovascularization of human coronary arteries – a possible role in the pathophysiology of atherosclerosis. N Engl J Med. 1984;310(3):175–177. | ||
Kamat BR, Galli SJ, Barger AC, Lainey LL, Silverman KJ. Neovascularization and coronary atherosclerotic plaque: cinematographic localization and quantitative histologic analysis. Hum Pathol. 1987;18(10):1036–1042. | ||
Zamir M, Silver MD. Vasculature in the walls of human coronary arteries. Arch Pathol Lab Med. 1985;109(7):659–662. | ||
Zhang Y, Cliff WJ, Schoefl GI, Higgins G. Immunohistochemical study of intimal microvessels in coronary atherosclerosis. Am J Pathol. 1993;143(1):164–172. | ||
Sueishi K, Yonemitsu Y, Nakagawa K, Kaneda Y, Kumamoto M, Nakashima Y. Atherosclerosis and angiogenesis: its pathophysiological significance in humans as well as in an animal model induced by the gene transfer of vascular endothelial growth factor. Ann N Y Acad Sci. 1997;811(1):311–324. | ||
Kumamoto M, Nakashima Y, Sueishi K. Intimal neovascularization in human coronary atherosclerosis: its origin and pathophysiological significance. Hum Pathol. 1995;26(4):450–456. | ||
Potteaux S, Ait-Oufella H, Mallat Z. Role of splenic monocytes in atherosclerosis. Curr Opin Lipidol. 2015;26(5):457–463. | ||
Hundal RS, Salh BS, Schrader JW, Gómez-Muñoz A, Duronio V, Steinbrecher UP. Oxidized low density lipoprotein inhibits macrophage apoptosis through activation of the PI 3-kinase/PKB pathway. J Lipid Res. 2001;42(9):1483–1491. | ||
Matsumura T, Sakai M, Kobori S, et al. Two intracellular signaling pathways for activation of protein kinase C are involved in oxidized low-density lipoprotein-induced macrophage growth. Arterioscler Thromb Vasc Biol. 1997;17(11):3013–3020. | ||
Biwa T, Hakamata H, Sakai M, et al. Induction of murine macrophage growth by oxidized low density lipoprotein is mediated by granulocyte macrophage colony-stimulating factor. J Biol Chem. 1998;273(43):28305–28313. | ||
Sakai M, Miyazaki A, Hakamata H, et al. Lysophosphatidylcholine potentiates the mitogenic activity of modified LDL for human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. 1996;16(4):600–605. | ||
Wang M, Subramanian M, Abramowicz S, et al. Interleukin-3/granulocyte macrophage colony-stimulating factor receptor promotes stem cell expansion, monocytosis, and atheroma macrophage burden in mice with hematopoietic ApoE deficiency. Arterioscler Thromb Vasc Biol. 2014;34(5):976–984. | ||
Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. | ||
Li J, Ley K. Lymphocyte migration into atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2015;35(1):40–49. | ||
Yan H, Duan X, Pan H, et al. Suppression of NF-κB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc Natl Acad Sci U S A. 2016;113(41):E6199–E6208. | ||
Kanters E, Pasparakis M, Gijbels MJ, et al. Inhibition of NF-κB activation in macrophages increases atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2003;112(8):1176–1185. | ||
Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95. | ||
Durand E, Scoazec A, Lafont A, et al. In vivo induction of endothelial apoptosis leads to vessel thrombosis and endothelial denudation: a clue to the understanding of the mechanisms of thrombotic plaque erosion. Circulation. 2004;109(21):2503–2506. | ||
Grasset EK, Duhlin A, Agardh HE, et al. Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc Natl Acad Sci U S A. 2015;112(16):E2030–E2038. | ||
Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339(6116):161–166. |
Supplementary materials
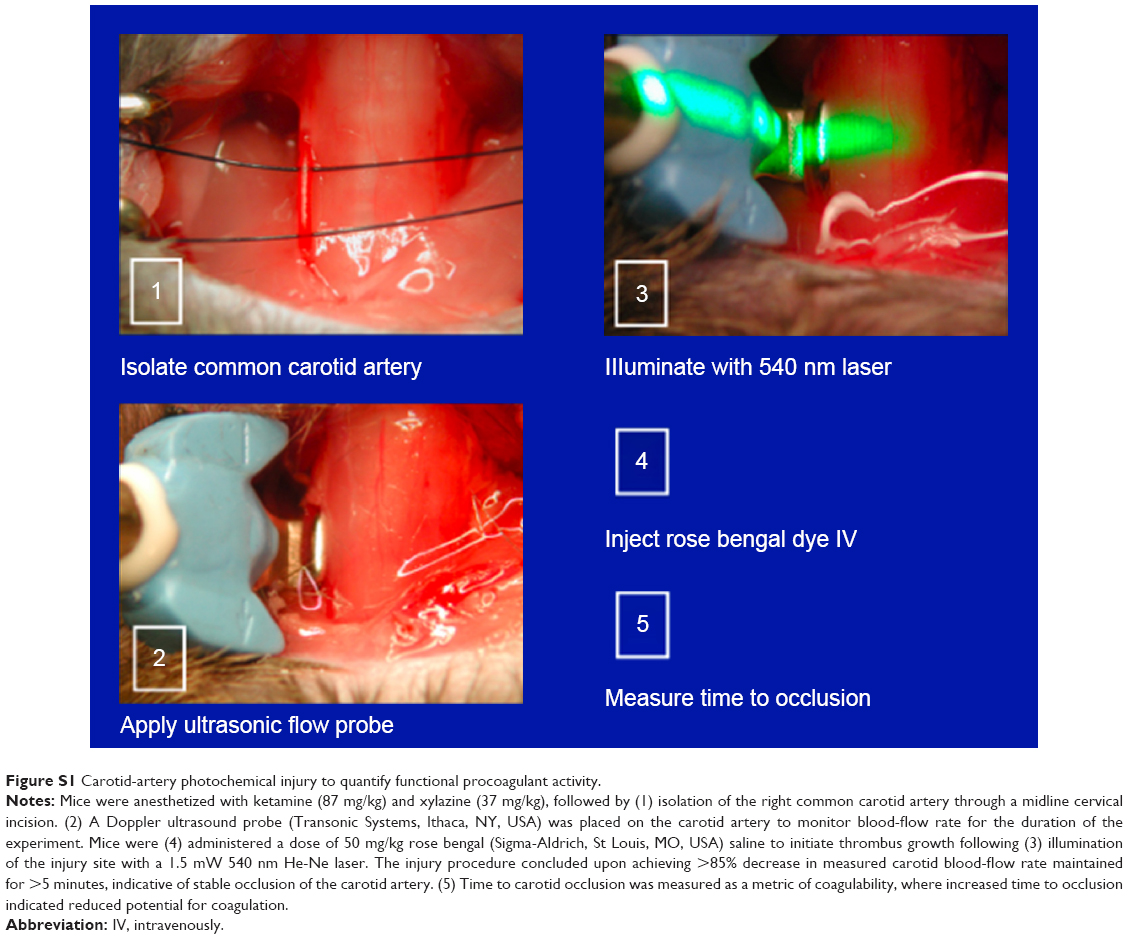 | Figure S1 Carotid-artery photochemical injury to quantify functional procoagulant activity. |
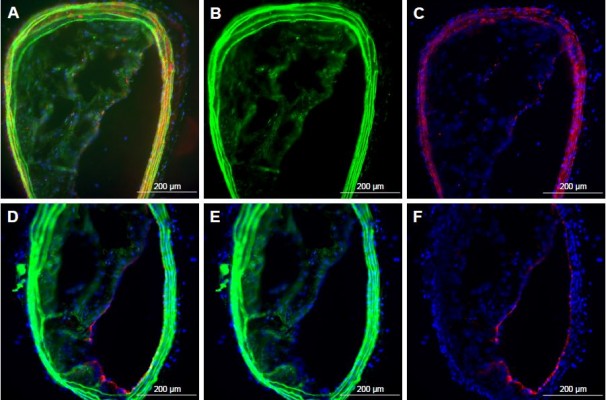
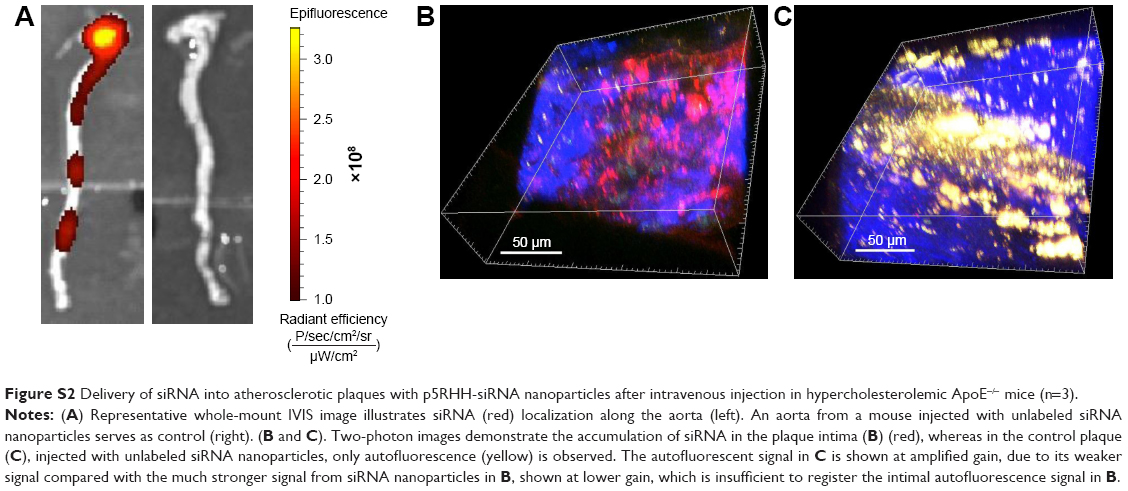 | Figure S2 Delivery of siRNA into atherosclerotic plaques with p5RHH-siRNA nanoparticles after intravenous injection in hypercholesterolemic ApoE−/− mice (n=3). |
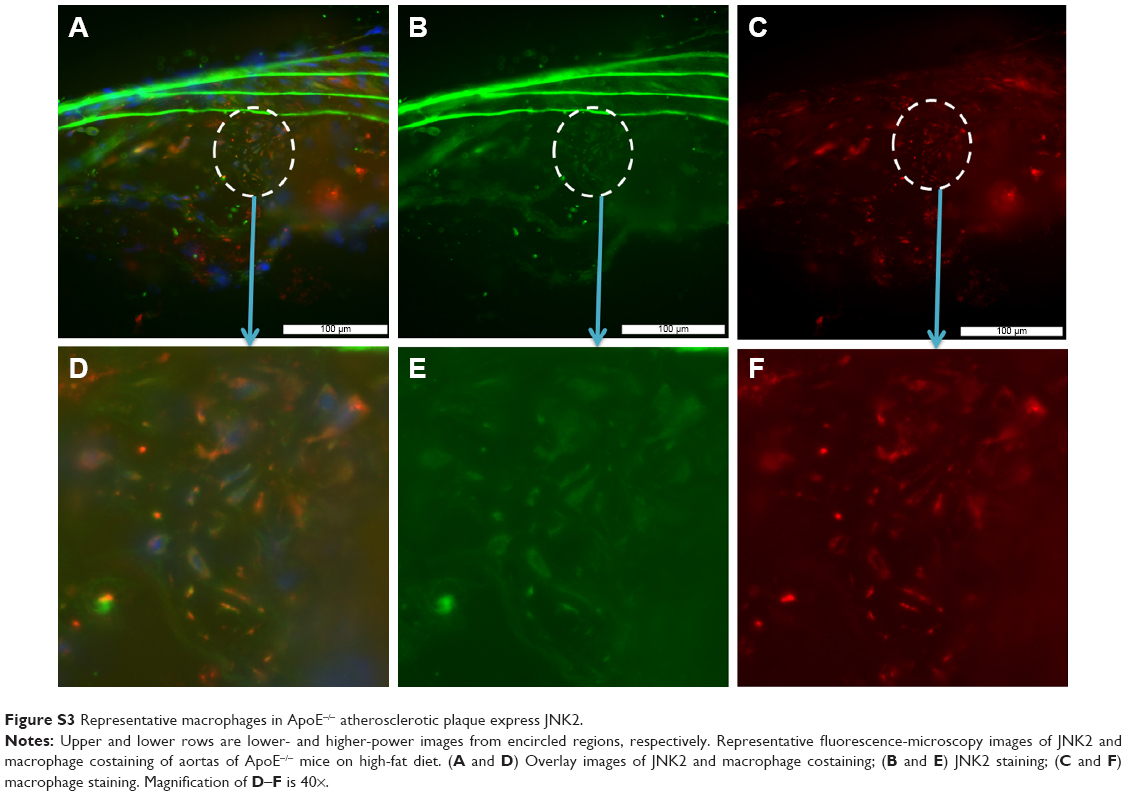 | Figure S3 Representative macrophages in ApoE−/− atherosclerotic plaque express JNK2. |
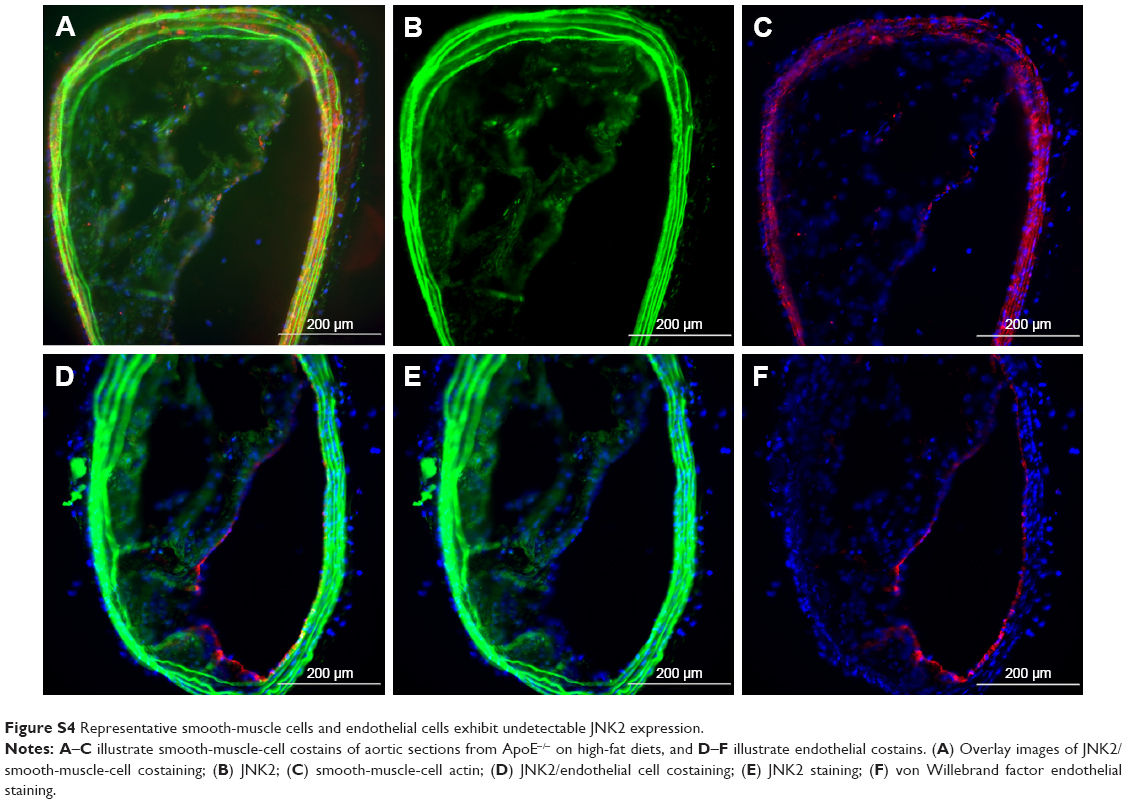 | Figure S4 Representative smooth-muscle cells and endothelial cells exhibit undetectable JNK2 expression. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.