")
Back to Journals » OncoTargets and Therapy » Volume 13
Anti-Cancer Activity of Acriflavine as Metabolic Inhibitor of OXPHOS in Pancreas Cancer Xenografts
Authors Bulle A , Dekervel J , Deschuttere L, Nittner D , Van Cutsem E, Verslype C , van Pelt J
Received 7 January 2020
Accepted for publication 13 May 2020
Published 15 July 2020 Volume 2020:13 Pages 6907—6916
DOI https://doi.org/10.2147/OTT.S245134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Takuya Aoki
Ashenafi Bulle,1 Jeroen Dekervel,1 Lise Deschuttere,1 David Nittner,2,3 Eric Van Cutsem,1 Chris Verslype,1 Jos van Pelt1
1Laboratory of Clinical Digestive Oncology, Department of Oncology, KU Leuven & University Hospitals Leuven and Leuven Cancer Institute (LKI), Leuven, Belgium; 2Histopathology Expertise Center, VIB-KU Leuven Center for Cancer Biology, VIB, Leuven, Belgium; 3Department of Oncology, KU Leuven, Leuven, Belgium
Correspondence: Jos van Pelt
Laboratory of Clinical Digestive Oncology, Department of Oncology, KU Leuven & University Hospitals Leuven and Leuven Cancer Institute (LKI), Leuven, Belgium
Tel +32-16-330 694
Fax +32-16-330 718
Email [email protected]
Purpose: All currently available therapies for the treatment of pancreatic ductal adenocarcinoma (PDAC) show limited success. PDACs fast progression depends on the tumor characteristics and can be influenced by the microenvironment. The antibacterial drug acriflavine (ACF) has been shown to have anti-cancer activities in cell lines.
Materials and Methods: To understand the working mechanism of ACF on cancer progression and tumor-stromal interplay, we evaluated pancreatic cancer in cell culture (Panc-1) (morphology, cell invasion and RNA expression) and the macrophage cell line THP1 (representing innate immune stromal cells). In the translational arm, the activity of ACF on xenograft models of human PDAC tumors representing different clinical subclasses was investigated (tumor growth, morphology and immunofluorescence, RNA expression and pathway analysis).
Results: In vitro, ACF reduces epithelial-to-mesenchymal transition (EMT) and invasion of Panc-1 cells and shifts macrophage polarization to a M1-like anti-tumoral phenotype. At non-toxic concentrations, ACF downregulates cell metabolism. In xenografts, effect on tumor growth and metabolism could be confirmed; however, the innate immune stromal cells did not respond. Importantly, only in the moderately differentiated PDAC model, ACF could significantly suppress tumor growth and not in the fast-growing EMT-high model. Pathway analysis shows that ACF highly significantly downregulates metabolic pathways, especially OXPHOS and MYC/cell proliferation pathways in xenografts.
Conclusion: ACF, with known pleiotropic effects on cancer cells, is in this study shown to be an attractive therapeutic based on its novel observed metabolic activity. Repurposing this compound for cancer treatment should be in the setting with other targeting agents, which offers reduced chance of resistance development in PDAC. Further evaluation should best be done in biological complex models such as human xenografts or syngeneic cancer models.
Keywords: pancreatic ductal adenocarcinoma, PDAC, tumor microenvironment, Hif1α, gene set enrichment analysis, GSEA, metabolic reprogramming, drug repurposing
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer (>90%)1 and the incidence is rising.2 Pancreatic cancer has a bad prognosis and little improvement is to be expected in the near future.3 Surgery, radiation therapy and chemotherapy are treatment approaches for PDAC that may prolong survival and/or relieve symptoms in many patients, but they seldom result in a real cure.4 Gemcitabine monotherapy has long been used as the standard chemotherapy for pancreatic cancer patients, but most patients do not respond well and end up with resistance and disease progression. Several chemotherapeutics drug combinations are also being used but none is very successful in larger groups of patients. The most important recent breakthrough is found in the adjuvant setting, where FOLFIRINOX has been shown to reduce the recurrence rate and to improve the survival of patients operated for pancreatic cancer.5 There are also several targeted therapies under pre-clinical investigation, but these have not generally been accepted for use.6,7 Immunotherapy trials in pancreatic cancer have shown no clinical benefit.8 Improvement in therapeutic strategies for this form of cancer are therefore still needed.
PDAC is different from most other solid tumors for its high stromal content that can effectively shield drugs from reaching the tumor9 and its high degree of hypoxia.10 Furthermore, the interplay between the cancer and the stroma (cells and a-cellular constituents) is important for tumor progression. Epithelial-to-mesenchymal transition (EMT) of tumor cells is considered a driver of drug-resistance, migratory capacity, of invasion, development of fibrosis, metabolism and cancer progression.11 Various biochemical and biophysical factors in the tumor microenvironment, including hypoxia, growth factors and cytokines are capable of inducing EMT.12 During the process of EMT, the cancer cells lose epithelial markers such as E-cadherin, certain cytokeratins, occludin, and claudin and gain mesenchymal markers such as N-cadherin, vimentin, and fibronectin.13 These changes are mediated by transcription factors SNAI1, SNAI2, TWIST, and ZEB.
Acriflavine (ACF) is a heteroaromatic dye with antibacterial and antiviral effects.14 More recently, its potential as an anti-cancer agent emerged as acriflavine has topo-isomerase inhibitor activity.15 ACF also blocks the HIF pathway by preventing the dimerization of the HIF-1 subunits, HIF-1α and HIF-1β.16 Furthermore, there are reports that show a close link between HIF signaling, EMT and cancer aggressiveness.17 HIF-1 has been linked to glucose uptake and drug resistance in pancreatic cancer.18,19 And, in a recent study, we show that acriflavine inhibits the ATF4 transcriptional program and re-sensitizes drug-resistant cells.20 These characteristics encourage further studies to repurpose ACF for systemic cancer therapy.
In this study, we investigated the working of ACF in vitro on pancreatic tumor cells and macrophage cell lines (representing innate immune cells as found in PDTX); components for tumor-stroma crosstalk. In addition, we tested ACF in two human PDAC xenografts (PDTX): classical type (moderately differentiated and relative slow progression: PAC006) and quasi-mesenchymal type (poorly differentiated, EMT characteristics and faster growth: PAC010).21,22 Importantly, in PDTX models the human stroma is replaced by murine stroma and we use this to dissect the molecular characteristics of the stroma (mouse) and tumor cells (human).22 Further, in PDTX models, we can only investigate the tumor-infiltrating cells from the innate immune system of which a large fraction are the tumor-associated macrophages (TAMs). Conventionally, these TAMs are classified as classical activated (M1, proinflammatory and anti-tumoral function) and nonclassical activated (M2, anti-inflammatory/pro-tumoral) that reflect their distinct cellular function and metabolism. Shift in the levels of M1 and M2 macrophages can influence cancer progression.
Materials and Methods
Panc-1 Cell Culture, EMT Induction and Acriflavine Treatment
Panc-1, human pancreatic cancer cells were obtained from ATCC (CRL-1469, Rockville, MD, USA). EMT was induced by transforming growth factor-β1 (TGF-β1: 5 ng/mL for 48 hours) or cobalt chloride (CoCl2: 200 μM for 48 hours). To inhibit EMT, a non-toxic concentration of acriflavine (2.5 μM, determined previously20) was added to the cell culture together with TGF-β1 or CoCl2.
THP1 Cell Culture, Macrophage Polarization and Acriflavine Treatment
THP-1 monocytic cells were purchased from ATCC (TIB-202), cultured in RPMI supplemented with 10% FCS. To generate M0 macrophages, the THP-1 cells were treated with 120 ng/mL phorbol 12-myristate 13-acetate (PMA) for 24 hours. Cells were washed and medium replaced. We differentiated M0 cells with 50 ng/mL of LPS to M1 phenotype or with 20 ng/mL of hIL-4 and 20 ng/mL of hIL-13 to induce M2 phenotype for 48 hours.23,24 Dose response curves in M0 cells were determined (XTT assay, Roche Applied Science, Penzberg, Germany, see Supplementary data) that gave IC50 for acriflavine (ACF, 4 µM), metformin (MET, 20 mM), Curcumin (CUR, 15 µM) and Chloroquine (CQ, 40 µM). These compounds (at a concentration resulting in maximal 15% cell death) were added simultaneously to the M0 cells with either LPS or IL-4/IL-13. After 48 hours, cells were harvested for RNA expression. For each experiment, conditions were analyzed in triplicate.
Tumor Cell Invasion Assay
Invasion studies were performed using BioCoat Matrigel Invasion Chambers (Corning, NY, USA)20,25 (see Supplementary data).
Compounds
Acriflavine (Sigma-Aldrich, MO, USA) was dissolved in 100% dimethyl sulfoxide (DMSO) and stored at room temperature. TGF-β1 (R & D Systems Europe Ltd) was dissolved in 4mM HCL containing 0.1% BSA and stored at −20°C. Cobalt chloride (CoCl2) (Sigma) was dissolved in distilled water (100 mg/mL) and stored at room temperature. PMA, LPS, Curcumin, Metformin and Chloroquine were obtained from Sigma, hIL4 and hIL13 from R&D Systems.
PDTX, Tumor Implantation, Treatment Procedure, Growth and Sample Collection
We recently reported in detail the development of the PDTX models and their characterization.21,22,26 In short: tumor tissue was implanted subcutaneously into the interscapular fat pad of mice. After the tumor reached a volume of 100–200 mm3, the mice were randomly divided into groups of 8 mice and treated intraperitoneal: A) the control group that was treated with vehicle (0.9% NaCl) and B) acriflavine: schedule four doses of acriflavine injected daily with a fixed dose of 5 mg/kg Monday to Thursday and single dose on Friday for the weekend of 10 mg/kg. The body weight and tumor size were measured thrice a week. Tumor tissues were harvested as soon as the volume reached 1000 –1500 mm3, weighed, photographed and stored for histological analysis and molecular profiling.
Animal care and all research procedures were executed in accordance with the applicable legal guidelines (Belgian legislation Royal Decree regarding the protection of laboratory animals of 29 May, 2013 and European directive (2010/63/EU)) and under approval of the medical ethical committee for laboratory animals of the KU Leuven (approval numbers P147/2012, P038/2015).21,22,26
Histology and Immunohistochemistry
Immunohistochemistry (IHC) using human-specific anti-Cytokeratin-pan antibody and immunofluorescence double staining for TAMs was performed on formalin-fixed paraffin-embedded (FFPE) tumor tissue sections (see Supplementary data).
qRT-PCR
RNA was isolated with the RNeasy Kit (Qiagen, Chatsworth, CA) and reverse transcribed into cDNA using SuperScript II reverse transcriptase. The PCR reaction was carried using TaqMan MGB probe (Applied Biosystems, Foster City, USA), beta-2-microglobulin was used as housekeeping gene using the Lightcycler 96 (Roche). Each sample was assayed in duplicate.
Transcriptome Analysis of Cell Culture Experiments
Panc-1 cell culture samples (TGF-β1 experiment) were analyzed using the Affymetrix Human PrimeView Array. For the Panc-1 CoCl2 experiment, RNA gene expression was determined on an Illumina NextSeq 500 half-flow cell. Reads were aligned to the reference genome of Homo sapiens (GRCh37.73) and a generalized linear model (GLM) was fitted against the normalized counts. The resulting p-values were corrected for multiple testing to control false discovery rate (FDR q). A gene was considered differentially expressed if a 2log fold change >+1 or <-1 and a FDR q < 0.05. All samples were analyzed in triplicate (biological controls). Microarray and RNA sequencing data are available at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE82299.
Gene Expression of PDTX Samples
RNA sequencing and processing was performed by VIB Nucleomics Core (www.nucleomics.be). In short, the poly-A containing mRNA molecules were purified using poly-T oligo-attached magnetic beads, converted into cDNA and sequenced on an Illumina HiSeq 4000 full flow-cell. After preprocessing, reads were mapped to the reference human and mouse genome using ICRG method.22 The data were analyzed using Ingenuity Pathway Analysis (IPA) (www.ingenuity.com) and Gene Set Enrichment Analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp). Hierarchic clustering of the individual samples was done using PermutMatrix program (Linkage: McQuitty’s criteria and Dissimilarity: Euclidean distance).
RNA sequencing data of PDTX study have been submitted to the Gene Expression Omnibus (NCBI) under number GSE118197.
Statistical Analysis
All statistics on RT-qPCR were performed using SPSS v23 (IBM). Statistical differences between groups were assessed with a Student’s t-test or the Mann–Whitney Rank Sum Test when appropriate. For differences in gene expression assessed by RT-qPCR, ANOVA test with post hoc Tukey’s procedure was used. A P value below 0.05 was considered statistically significant.
Results
Observations in vitro
Effect of ACF on Pancreatic Cancer Cells in vitro
The in vitro section of this manuscript reanalyzes and expands on our previous work.20 TGF-β1 induces EMT-characteristic morphological changes in Panc-1 cells (reduced cell contact, spindle-shaped) (Figure 1A–C). Analysis of the gene expression confirms EMT (Figure 1D, hierarchical clustering of 55 EMT-related genes, Figure 1E, 8-gene signature, for details see Supplementary data), showing separation of TGF-β1-treated Panc-1 cells from untreated cells. TGF-β1 affects processes like cell adhesion, cellular motion and invasion (Figure 1F, for details, see Supplementary data). In short, Panc-1 cells acquire all of the characteristics of an aggressive form of pancreatic cancer. When ACF was given to the Panc-1 cells in combination with TGF-β1, it blocked most of these effects. Cobalt chloride (used to simulate hypoxia) also induced increased invasion and morphological EMT in Panc-1 cells that was reversed by ACF (shown previously). Findings in both models suggest that ACF overwrites stress signals that had shifted the tumor cell to a more invasive-EMT phenotype.
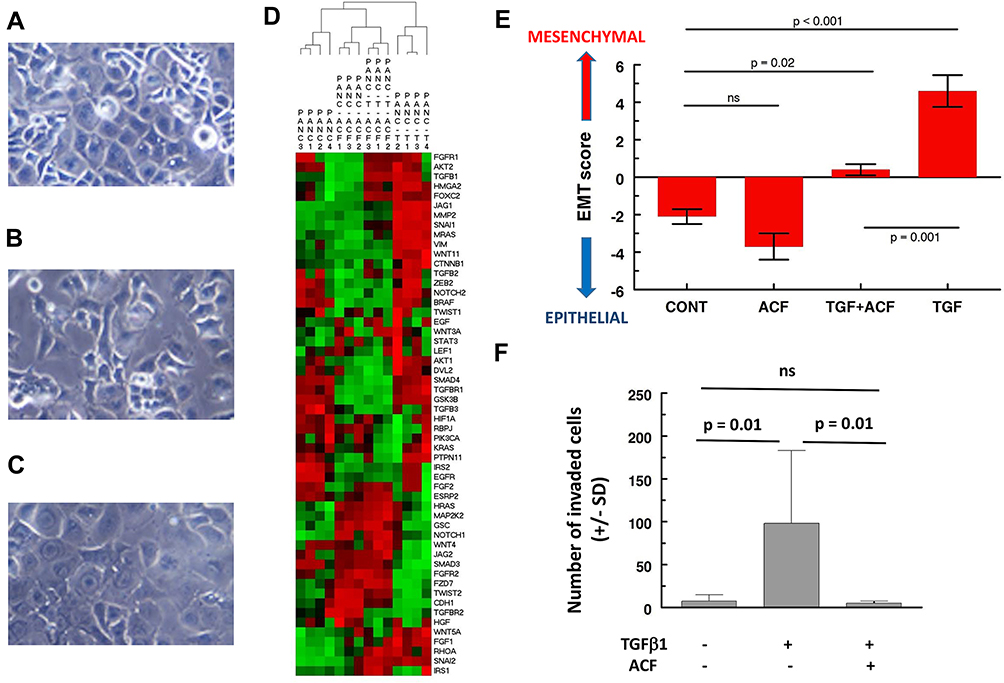 |
Figure 1 Effect of acriflavine (ACF) on epithelial-to-mesenchymal (EMT) induction in Panc-1 cells and invasion in vitro. (A–C) Morphological appearance of Panc-1 cells: (A) untreated, (B) after treatment with TGF-β1 (5 ng/mL) or (C) when treated with the combination of TGF-β1 (5 ng/mL) and ACF (2.5 µM). Treatment for 48 hours, original magnification is 20×, adapted from Dekervel J, Bulle A, Windmolders P, et al. Acriflavine inhibits acquired drug resistance by blocking the epithelial-to-mesenchymal transition and the unfolded protein response. Transl Oncol. 2017;10(1):59–69., Copyright (2017), with permission from Elsevier.20 (D) Two-way hierarchical clustering of gene expression of Panc-1 cells treated with ACF or TGF-β1 (=T) or a combination (T-ACF) (GSE82299). A panel of 55 EMT-related genes was used (from IPA, Supplementary Table 1). The color scale illustrates the relative gene expression level: red, above the mean Z-score, green below the mean. (E) To quantify epithelial vs mesenchymal phenotype of the cells we used a representative 8-gene relative EMT score (EMT-score = ∑ SNAI1+SNAI2+TGFB1+VIM+ZEB2-CDH1-CTNNB1-AKT2, using normalized, 2log expression values, see also Supplementary File). (F) Invasion assay studies. Cells were added on top of a matrigel insert. After 48 hours the number of invaded cells was scored under a microscope on five random fields per insert (± SD). Abbreviations: CONT, untreated control; ns, not significant. |
Stress induced by TGF-β1 or CoCl2 resulted in an upregulation of metabolism of the cancer cells (GSEA: KEGG hsa01100). We found a suppressive effect of ACF on the metabolism with a negative enrichment of this pathway opposite from what was seen after stress (Figure 2), this will be further investigated in xenografts (see Supplementary Table 2).
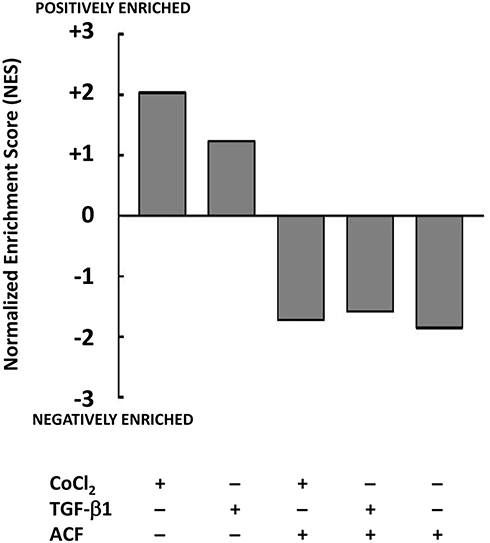 |
Figure 2 Effect of acriflavine (ACF) on Panc-1 metabolism in cell culture. Relative gene expression was determined for the different conditions relative to untreated Panc-1 cells (GSE82299). Gene Set Enrichment Analysis (GSEA) identified the normalized enrichment score (NES) of the hallmark gene set for Metabolism (KEGG: hsa01100) as significantly positively enriched when cells were treated with TGF-β1 or CoCl2, this was reversed by ACF or when Panc-1 cells were treated with ACF only (2.5 µM). |
Effect of ACF on the Human Macrophage Cell Line (THP1) in vitro
In a recent PDAC study, we have shown that the drug gemcitabine shifts the innate immune cells in xenografts to a pro-tumoral phenotype.22 We used the THP-1 cells as an in vitro model for TAMs. THP-1 cells can be differentiated by PMA to a so-called M0-like phenotype. The M0-cells can subsequently be polarized to macrophages, either to a M1-like phenotype (anti-tumoral) or to M2-like phenotype (pro-tumoral).24 We studied how ACF influences the polarization of M0 cells. We determined experimentally mRNA markers by qRT-PCR that can classify the THP-1 derived cells, four markers were selected and the differential gene expression relative to the M0 cells was used. M1 type cells are characterized by mRNA CD14up, CD163up, IL10up and MRC1down, M2-type cells as mRNA CD14down, CD163up, IL10up and MRC1up (Figure 3). Hierarchical clustering results in three main groups (M1, M0 and M2). The expression markers we selected do not fully overlap with the cell surface protein markers that are described for macrophages isolated from blood but are practical to separate the differentiated THP1 cells. As already described, stimulation of M0 cells with LPS yields M1 cells.24 However, LPS in the presence of the drug curcumin, chloroquine or metformin resulted in pro-tumoral M2-like cells. In contrast, when M0 cells are stimulated with IL4 and IL13 this gives rise to M2 cells. When IL4/IL13 was used in combination with curcumin, chloroquine or metformin the cells clustered intermediate between M0 and M2. A different effect is seen when M0 cells (induced to M2) are co-stimulated with acriflavine, this drug shifts the expression of the macrophage markers CD163 and MRC1 resulting in a clustering of these cells to M1 anti-tumoral phenotype.
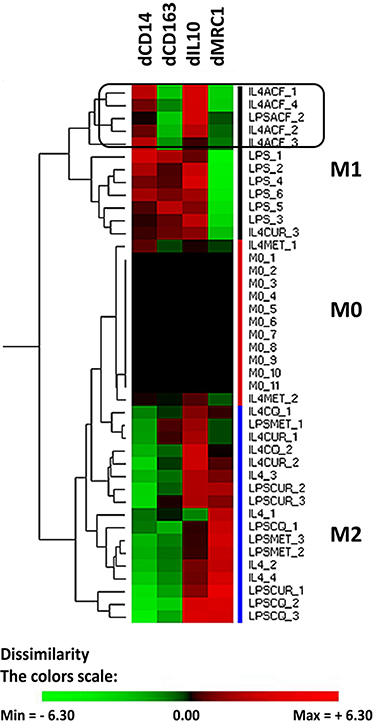 |
Figure 3 Effect of acriflavine (ACF) on phenotype of THP1 cells in vitro. Human THP1 cells were differentiated to M0 cells and subsequently incubated for 48 hours with Lipopolysaccharides (LPS) (resulting in M1-like cells) or IL4/IL13 (resulting in M2-like cells), with or without drugs. The differential gene expression (d) of selected marker genes (CD14, CD163, IL10 and MRC1) was computed relative to that in M0 cells as cycle number by qRT-PCR in the corresponding experiment. Hierarchical clustering was performed to group the cell’s phenotype. Drug concentrations were chosen to have no more than 15% cell death. Drugs used: acriflavine (ACF, 2.5 µM), metformin (MET, 5 mM), Curcumin (CUR, 10 µM) and Chloroquine (CQ, 25 µM). The color scale illustrates the relative gene expression level compared to M0 cells in the corresponding experiment (2log values, ΔΔCq-method): red, increase of expression, green reduced expression. |
Effects of ACF in Xenografts Models of Human PDAC
The Effect of ACF on Tumor Growth
As described above, from in vitro experiments we have indications that treatment with ACF might be beneficial for a patient with pancreatic cancer. Treatment of the mouse xenograft models PAC006 (well differentiated PDAC) and PAC010 (poorly differentiated PDAC) with ACF for a period of up to 24 days, showed different responses. Tumor growth of PAC010 was fast that required the termination of the animals after 10 days. Treatment of PAC010 with ACF did not alter the progression of tumor growth. The PAC006 has a slower cancer growth rate, when these animals were treated with ACF a significant reduction of the tumor volume was observed at day 9 compared to control. With continued treatment, the tumor volume stabilized at day 12 and remained stable until the end of the experiment (24 days) (Figure 4).
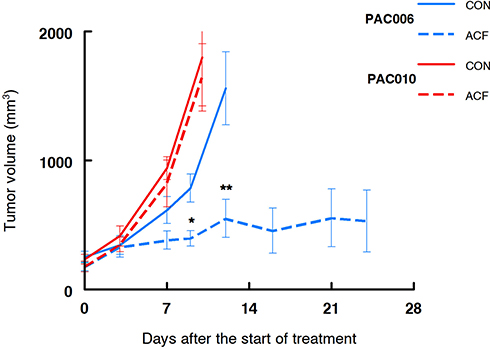 |
Figure 4 Tumor growth in human xenograft pancreatic cancer models treated with acriflavine. Growth curve of PAC010 (red, EMT-high, poorly differentiated and metastatic patient) and PAC006 (blue, EMT-low, moderately differentiated tumor model). Solid lines are control animals (CON), broken line are ACF-treated animals. Each of the groups consisted of eight animals; results are given as mean ± SEM (*p = 0.01, **p < 0.01). |
Molecular Analysis of ACF in Responsive PAC006 Model
For the rest of our analysis, we concentrated on the PAC006 model, the model where we see a growth inhibition by ACF. When PAC006 tumors were treated with ACF, the H&E staining pattern is not different from that seen in the untreated PAC006 (Figure 5A). We did not observe an effect on the ratio of tumor cells to stroma cells based on PAN-CK staining (Figure 5B). Influx of TAM was further assessed by immunofluorescence double staining that identified M1-like TAMs as MHCIIposCD206low and M2-like TAM’s as F4/80posCD206pos (see Supplementary data). ACF did not change the number of TAMs in the stroma or the ratio between the macrophage subsets with or without ACF treatment (Figure 5C).
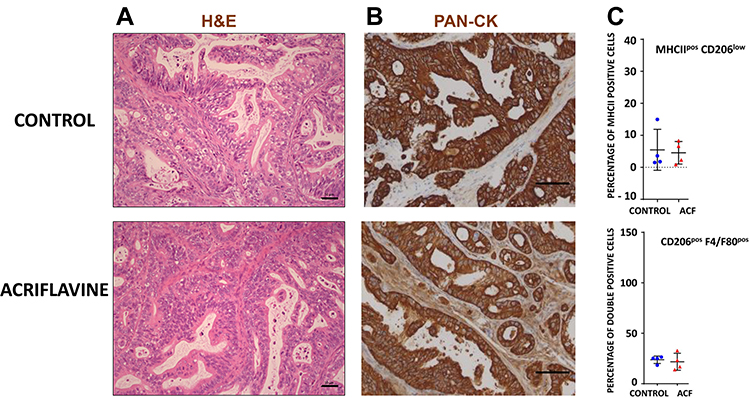 |
Figure 5 Composition of stroma and tumor of PAC006. (A) Representative Hematoxylin and eosin (H&E) staining of tumor tissues at the end of treatment (Scale bar 50 µm). (B) Tumor sections were stained with antibody for human Cytokeratin-pan antigen (PAN-CK) to visualize the area occupied by the human tumor (Scale bar 50 µm). (C) Quantification of macrophage phenotype by immunofluorescence (M1: combination of MHCIIhigh and CD206low, M2: mouse-specific macrophage marker F4/80 together with CD206high). Tumor sections of 4 animals per group were analysis per condition (see also Supplementary File). |
The morphological evaluation was followed by an analysis of the gene expression of the tumor and the stroma. The number of highly differentially expressed genes in the tumors treated with ACF versus untreated animals (log2-ratio < −1 or > +1 and p-value <0.001) was small: up = 21, down = 3 and for the stroma: up = 107 and down = 197.
For GSEA we used the lists of all genes identified by RNA sequencing ranked according to their differential expression. This analysis reveals that both tumor and stroma react to ACF treatment in a nearly similar way (see Supplementary Table 3). In the tumor, the significantly upregulated Hallmark gene sets ranked according to normalized enrichment score (NES) with FDR q-value <0.01 include: - Hedgehog signaling (NES 1.99), - interferon α (NES 1.93) and - interferon γ response (NES 1.80). The downregulated gene sets include: - oxidative phosphorylation (NES −2.85), - myc targets V1 (NES −2.58), - fatty acid metabolism (NES −1.84) and - E2F targets (NES −2.04) (Figure 6A–C). Important to note, the NES for the downregulated gene sets were high and highly significant. The GSEA therefor suggests as potential working mechanism for ACF an overall suppression of the oxidative phosphorylation and energy generation. The differentially expressed genes are found in Complexes I, III, IV and V and all are downregulated (see Supplementary Table 4). In addition, all genes in the myc targets V1 genes (96 detected) are also downregulated.
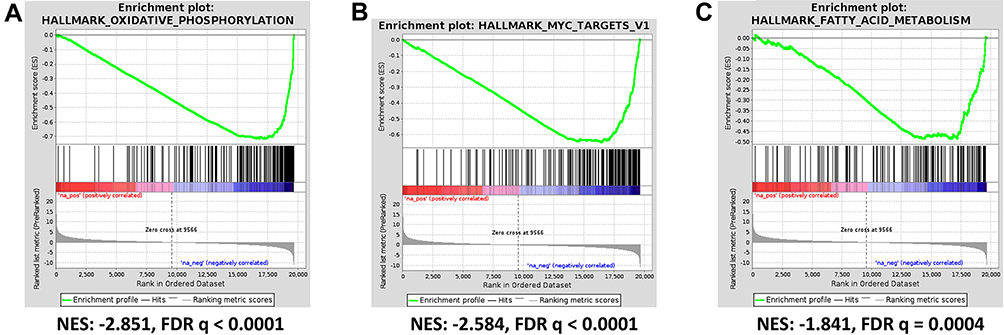 |
Figure 6 Acriflavine inhibits oxidative phosphorylation and other pathways in human xenograft pancreatic cancer model PAC006. Differentially expressed genes between ACF-treated samples (n=3) and controls (n=4) was determined and GSEA was performed (see Supplementary Table 3). The statistically most significant hallmark gene sets include (A) OXIDATIVE PHOSPHORYLATION, this GSEA Hallmark gene set includes 195 genes, 97 were detected and all were downregulated (see Supplementary Table 4). (B) MYC TARGETS V1 and (C) FATTY ACID METABOLISM. Abbreviations: NES, normalized enrichment score; FDR, false discovery rate. |
Discussion
For many solid cancers, growth and metastasis are linked to EMT and hypoxia. ACF has been suggested as a promising candidate anti-cancer drug based on its interference in these mechanisms.15,16,20 For this study, we complemented in vitro observations with detailed transcriptome analysis. In cell line models, we found a downregulation of EMT and of metabolic pathways. Together with a reduced migration when ACF was given to cells further suggests a beneficial effect of ACF in cancer treatment. It has been established that the tumor microenvironment can modulate cancer progression, with recruitment of immunological cells from the circulation and through stroma-cancer cell signaling.27 We used THP1 cells as a model for tumor-associated macrophages (TAM), cells that we had found to be recruited in PDAC xenografts under drug treatment.22 ACF in vitro could shift macrophages to M1-(anti-tumoral)-phenotype in contrast to other similar drugs. This promises to be an additional therapeutic benefit of ACF in PDAC treatment.
However, these in vitro observations were not fully translated when ACF was applied in more biological complex PDTX models. In a fast-growing PDTX model (PAC010), ACF could not impact on the tumor progression. In contrast, in the PAC006 model derived from a patient with a well-moderately differentiated and relatively slow growing tumor, ACF did show significant reduction of tumor growth and size stabilization. Gene expression, morphology and immunohistochemical staining did not indicate a reduction of EMT of the cancer cells or a shift in TAM-subpopulations as seen in vitro (ACF 24 days vs control 10 days). We speculate that the molecular mechanisms that were observed in vitro do not work in PDTX or are compensated, hence unlikely that they will contribute substantial when ACF is applied in patients. Other mechanisms should explain the anti-cancer effect of ACF in xenograft.
What was found when analyzing the gene expression, both in Panc-1 cells and in the PAC006 model, was a downregulation of metabolism and in xenografts also processes related to energy production (in particular oxidative phosphorylation (OXPHOS) and fatty acid metabolism). Furthermore, in PDTX we found a down regulation of proliferation (MYC targets and E2F targets). A high percentage of genes in mitochondrial Complex I, III, IV and V of OXPHOS are affected; at RNA expression level almost all OXPHOS genes are downregulated by 25% to 35% in the tumor (Supplementary Table 4). Such a general downregulation in a single pathway can explain in part the observed effect of ACF in xenografts. This blocking by ACF is also found in the stroma (Supplementary Table 3). On this mechanism, there is one publication from 1978 that reported inhibition of mitochondrial energy production by ACF28 other information is lacking. A subgroup of cancers including PDAC and lymphomas rely on mitochondrial metabolism.29,31 Therefore, in the last 5 years, the metabolisms in pancreatic cancer has become a field of interest for research.32 For instance, OXPHOS inhibition to treat cancers has gained more attention.31 A number of clinical studies have been conducted especially with metformin mostly in combination with radiation therapy.33 There are concerns for metformin and other drugs that target oxidative metabolism with regard to their pharmacological limitations (inadequate potency, transport-mediated accumulation and off-target pharmacology).34 New molecules have recently been reported and tested in vitro and in xenografts.34,35 We find that inhibition by ACF of tumor growth requires a period before it is observed. After 10 days of treatment, ACF starts to have an effect on tumor growth probably through a metabolic reprogramming and reduced cell division. The mechanism likely involves myc transcription factor that was also identified by GSEA and is known to play a central role in metabolic reprogramming and reduced proliferation.36
Can these observations find their way into the clinic? The single-target therapy approach, although it has great potential, has taught us that aiming for one process frequently results in the development of resistance; combination treatment looks the way forward.37 Secondly, OXPHOS is a very general process; strong interference with this process will also affect healthy cells and tissues (working not restricted to tumor, see Supplementary Table 3). ACF appears to be moderate in its action and this opens a window for combination with other tumor-targeting agents, this should increase the applicability for PDAC treatment. Our observation supports the need for further evaluation of ACF in animal models as part of such a combination treatment specifically for the subgroup of relative slow growing PDAC. Whether tumors in animals (and patients) that have an intact immune system are responsive to ACF treatment should be determined. Furthermore, PDAC is a type of tumor with a dense stroma that is highly hypoxic,9,10 whether our observations also hold for other types of tumors with a different microenvironment we are not sure.
Conclusion
We conclude that cell culture models are not sufficient for evaluation of ACF in cancer treatment. This xenograft study provides arguments to further investigate ACF for use in PDAC. Improvement for PDAC patients should be expected with an early diagnosis, patient selection based on histological and molecular characteristics and through combination treatment targeting different pathways that can include ACF.
Acknowledgments
The authors wish to thank Prof L Libbrecht (Hospital Saint-Luc, Sint-Lambrechts-Woluwe, Belgium) and Prof D Lambrechts (VIB Center for Cancer Biology, Leuven, Belgium) for support, members of TRACE (TRACE: a patient-derived tumor xenograft platform (PDX) in translational cancer research at KU Leuven, Belgium) for their excellent work in the development of the EUS-PDTX models. We also want to thank the VIB Nucleomics Core facility, Leuven Belgium (Dr R Janky, Dr S Plaisance) for bioinformatics support.
Disclosure
Prof. Dr. Eric Van Cutsem reports grants from Bayer, Boehringer Ingelheim, Celgene, Ipsen, Lilly, Merck, Merck KgA, Novartis, Roche, and Servier, and has served on advisory boards for Array, AstraZeneca, Bayer, Biocartis, Bristol Myers Squib, Celgene, Daiichi, Halozyme, GSK, Incyte, Ipsen, Lilly, Merck, Merck KgA, Novartis, Roche, and Servier, outside the submitted work. Prof. Dr. Chris Verslype reports grants from Ipsen, Bayer, and Novartis, outside the submitted work. The authors report no other potential conflicts of interest in this work.
References
1. Karandish F, Mallik S. Biomarkers and targeted therapy in pancreatic cancer. Biomark Cancer. 2016;8(S1):27–35. doi:10.4137/BiC.s34414
2. American Cancer Society. Cancer facts & figures. 2019. Available from: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf.
3. Ferlay J, Partensky C, Bray F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016;55(9–10):1158–1160. doi:10.1080/0284186X.2016.1197419
4. Chandana S, Mahadevan D. Translational advances and novel therapies for pancreatic ductal adenocarcinoma: hope or hype? Expert Rev Mol Med. 2009;11:1–31. doi:10.1017/S1462399409001240
5. Conroy T, Hammel P, Hebbar M, et al. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N Engl J Med. 2018;379(25):239. doi:10.1056/NEJMoa1809775
6. Ducreux M, Cuhna AS, Caramella C, et al. Cancer of the pancreas: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:56–68. doi:10.1093/annonc/mdv295
7. Hingorani SR, Harris WP, Beck JT, et al. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res. 2016;22(12):2848–2854. doi:10.1158/1078-0432.CCR-15-2010
8. Kunk PR, Bauer TW, Slingluff CL, et al. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J ImmunoTherapy Cancer. 2016;4(1):14. doi:10.1186/s40425-016-0119-z
9. Mei L, Du W, Ma WW. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: controversies and promises. J Gastrointest Oncol. 2016;7(3):487–494. doi:10.21037/jgo.2016.03.03
10. Span PN, Bussink J. Biology of hypoxia. Semin Nucl Med. 2015;45(2):101–109. doi:10.1053/j.semnuclmed.2014.10.002
11. Persa O-D, Niessen CM. Epithelial polarity limits EMT. Nat Cell Biol. 2019;21(3):297–304. doi:10.1038/s41556-019-0284-7
12. Maier HJ, Wirth T, Beug H. Epithelial-mesenchymal transition in pancreatic carcinoma. Cancers. 2010;2(4):2058–2083. doi:10.3390/cancers2042058
13. Barriere G, Fici P, Gallerani G, et al. Epithelial mesenchymal transition: a double-edged sword. Clin Transl Med. 2015;4(1):14. doi:10.1186/s40169-015-0055-4
14. Wainwright M. Acridine – a neglected antibacterial chromophore. J Antimicrob Chemother. 2001;47:1–13. doi:10.1093/jac/47.1.1
15. Hassan S, Laryea D, Mahteme H, et al. Novel activity of acriflavine against colorectal cancer tumor cells. Cancer Sci. 2011;102:2206–2213. doi:10.1111/j.1349-7006.2011.02097.x
16. Lee K, Zhang H, Qiana DZ, et al. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. PNAS. 2009;106(42):7910–7915. doi:10.1073/pnas.0909353106
17. Ye L-Y, Chen W, Bai X-L, et al. Hypoxia-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res. 2016;76(4):818–830. doi:10.1158/0008-5472.CAN-15-0977
18. Shukla SK, Purohit V, Mehla K, et al. MUC1 and HIF-1alpha signaling crosstalk induces anabolic glucose metabolism to impart gemcitabine resistance to pancreatic cancer. Cancer Cell. 2017;32(1):71–87. doi:10.1016/j.ccell.2017.06.004
19. Chaika NV, Gebregiworgis T, Lewallen ME, et al. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1 alpha to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci U S A. 2012;109(34):13787–13792. doi:10.1073/pnas.1203339109
20. Dekervel J, Bulle A, Windmolders P, et al. Acriflavine inhibits acquired drug resistance by blocking the epithelial-to-mesenchymal transition and the unfolded protein response. Transl Oncol. 2017;10(1):59–69. doi:10.1016/j.tranon.2016.11.008
21. Bulle A, Dekervel J, Libbrecht L, et al. Gemcitabine induces epithelial-to-mesenchymal transition in patient-derived pancreatic ductal adenocarcinoma xenografts. Am J Transl Res. 2019;11(2):765–779.
22. Bulle A, Dekervel J, Deschuttere L, et al. Gemcitabine recruits M2-type tumor-associated macrophages into the stroma of pancreatic cancer. Transl Oncol. 2020;13(3):100743. doi:10.1016/j.tranon.2020.01.004
23. Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014;23(1):37–45. doi:10.1016/j.intimp.2014.08.002
24. Genin M, Clement F, Fattaccioli A, et al. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15(1):577. doi:10.1186/s12885-015-1546-9
25. van Malenstein H, Dekervel J, Verslype C, et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013;329(1):74–83. doi:10.1016/j.canlet.2012.10.021
26. Hermans E, Van der Merwe SW, Depreeuw J, et al. Successful application of endoscopic ultrasound-guided fine needle biopsy to establish pancreatic patient-derived tumor xenografts: a pilot study. Endoscopy. 2016;48(11):1016–1022. doi:10.1055/s-0042-113597
27. Luo G, Long J, Zhang B, et al. Stroma and pancreatic ductal adenocarcinoma: an interaction loop. Biochim Biophys Acta. 2012;1826(1):170–178. doi:10.1016/j.bbcan.2012.04.002
28. Higuti T, Arakaki N, Yokota M, et al. Acriflavine: anisotropic inhibitor of energy transduction in oxidative phosphorylation of rat liver mitochondria. FEBS Lett. 1978;87:87–91.
29. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11(1):9–15. doi:10.1038/nchembio.1712
30. Lonardo E, Cioffi M, Sancho P, et al. Metformin targets the metabolic achilles heel of human pancreatic cancer stem cells. PLoS One. 2013;8(10):e76518. doi:10.1371/journal.pone.0076518
31. Ashton TM, McKenna WG, Kunz-Schughart LA, Higgins GS. Oxidative phosphorylation as an emerging target in cancer therapy. Clin Cancer Res. 2018;24(11):2482–2490. doi:10.1158/1078-0432.CCR-17-3070
32. Qin C, Yang G, Yang J, et al. Metabolism of pancreatic cancer: paving the way to better anticancer strategies. Mol Cancer. 2020;19(1):50. doi:10.1186/s12943-020-01169-7
33. Koritzinsky M. Metformin: a novel biological modifier of tumor response to radiation therapy. Int J Radiat Oncol Biol Phys. 2015;93(2):454–464. doi:10.1016/j.ijrobp.2015.06.003
34. Molina JR, Sun Y, Protopopova M, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–1046. doi:10.1038/s41591-018-0052-4
35. Shi Y, Lim SK, Liang Q, et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature. 2019;567(7748):341–346. doi:10.1038/s41586-019-0993-x
36. Chen H, Liu H, Qing G. Targeting oncogenic mica as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. doi:10.1038/s41392-018-0008-7
37. Chiorean EG, Coveler AL. Pancreatic cancer: optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–3545. doi:10.2147/DDDT.S60328
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.