")
Back to Journals » OncoTargets and Therapy » Volume 12
Animal models of well-differentiated/dedifferentiated liposarcoma: utility and limitations
Authors Codenotti S, Mansoury W, Pinardi L, Monti E , Marampon F, Fanzani A
Received 16 February 2019
Accepted for publication 4 June 2019
Published 3 July 2019 Volume 2019:12 Pages 5257—5268
DOI https://doi.org/10.2147/OTT.S175710
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Sanjay Singh
Silvia Codenotti,1 Walaa Mansoury,1 Luca Pinardi,1 Eugenio Monti,1 Francesco Marampon,2 Alessandro Fanzani1
1Department of Molecular and Translational Medicine, University of Brescia, Brescia, Italy; 2Department of Radiotherapy, Policlinico Umberto I, “Sapienza” University of Rome, Rome, Italy
Abstract: Liposarcoma is a malignant neoplasm of fat tissue. Well-differentiated and dedifferentiated liposarcoma (WDL/DDL) represent the two most clinically observed histotypes occurring in middle-aged to older adults, particularly within the retroperitoneum or extremities. WDL/DDL are thought to represent the broad spectrum of one disease, as they are both associated with the amplification in the chromosomal 12q13-15 region that causes MDM2 and CDK4 overexpression, the most useful predictor for liposarcoma diagnosis. In comparison to WDL, DDL contains additional genetic abnormalities, principally coamplifications of 1p32 and 6q23, that increase recurrence and metastatic rate. In this review, we discuss the xenograft and transgenic animal models generated for studying progression of WDL/DDL, highlighting utilities and pitfalls in such approaches that can facilitate or impede the development of new therapies.
Keywords: liposarcoma, transgenic animal model, xenograft
Introduction
Liposarcoma is an often fatal cancer of adipose tissue that accounts for approximately 20% of all adult soft tissue sarcomas.1 It can arise in almost any body district, although the most frequent sites are the extremities (24%) and the retroperitoneal region (45%), with a peak occurrence around the 5th and 6th decade and a slight predominance in males.1 Liposarcoma presents in 2 largest groups, indicated as well-differentiated liposarcoma (WDL) and dedifferentiated liposarcoma (DDL), in addition to the less frequent myxoid and pleomorphic subtypes. The diagnosis of each subtype is based on anatomical location, clinical behavior, histology appearance, and cytogenetic features.2 Only WDL has no tendency to metastasize (unless it contains a dedifferentiated component) and may be therefore considered as a low-grade tumor, whilst the other subtypes show significant metastatic rates, ranging from 15% up to 50%.2 WDL/DDL histotypes share similar genetics despite a different prognosis and embody the most common cases observed clinically.3 WDL represents the largest group of malignant adipocytic neoplasms, accounting for approximately 40–45% of all cases.2 WDL is a slowly growing tumor distinguished by the presence of malignant adipocytes and spindle cells showing fibroblastic/myofibroblastic differentiation and giving rise to four subtypes, namely, adipocytic (or lipoma‐like), sclerosing, inflammatory, and spindle cell variants. The most important prognostic factor for this tumor is anatomical location, which is also the main predictor of relapse. WDL arising at somatic soft tissue sites, such as limbs or the trunk wall, is alternatively termed atypical lipomatous tumor (ALT) since its complete surgical resection is usually curative. Instead, the term WDL is preferable used for tumors occurring in the retroperitoneum or other visceral sites, since the risk of local recurrences is about 50% and associated to a dedifferentiation process, causing an even higher mortality rate than that associated with metastasis.2 The term DDL was first introduced by Evans in 1979 to define the morphological progression from ALT/WDL to a non‐lipogenic sarcoma.4 DDL indeed is considered a biphasic tumor consisting of a WDL component juxtaposed to either a high-grade undifferentiated sarcoma with malignant fibrous histiocytoma or fibrosarcoma-like features or with a lower-grade sarcoma having the appearance of myxofibrosarcoma. DDL is more often recurrent, requires multi-organ resection more frequently and presents a shorter disease-free interval when compared to WDL.5 DDL variants are more predisposed to metastasize, while ALT/WDL subtypes do not metastasize without dedifferentiation.6 DDL behavior is peculiar showing also the tendency to develop heterologous myogenic (rhabdomyosarcomatous or leiomyosarcomatous), osteo/chondrosarcomatous, and rarely angiosarcomatous differentiation in approximately 5–10% of the cases.7,8 Both WDL and DDL are poorly responsive to conventional chemotherapy, and surgical resection represents the best management for operable disease.9 Adjuvant radiation is employed to reduce risk of local recurrence in case of high-grade DDL, whereas a first-line chemotherapy consisting of single-agent doxorubicin treatment is generally reserved for unresectable or metastatic diseases.9
Genomic landscape in liposarcoma
Each liposarcoma subtype is characterized by a distinctive set of genetic signatures.10–13 Myxoid tumor type harbors the recurrent translocation t(12;16)(q13;p11) associated to the FUS-DDIT3 gene fusion product,14 whereas pleomorphic tumor is a complex-karyotype sarcoma frequently characterized by loss of TP53, RB1, and NF1.15 Nearby all WDL/DDL tumors are associated with the presence of one or more supernumerary circular (“ring”) and/or giant rod chromosomes.16 This leads to high-level amplifications in the chromosomal 12q13-15 region that causes overexpression of MDM2, the most observed amplification in WDL/DDL (close to 100%), and CDK4 (over 90% of the cases).17,18 The diagnostic detection of MDM2 and CDK4 by fluorescence in situ hybridization represents a reliable tool to discriminate WDL/DDL from other adipocytic tumors.19 Unsurprisingly, WDL shows high expression of genes associated with lipid metabolism and adipocytic differentiation, while DDL is characterized by upregulation of genes involved in proliferation and DNA repair as a result of additional genetic abnormalities, including losses, fusion transcripts, and amplifications.20–22 Unlike WDL, DDL frequently contains 1p32, 6q23, and 12q amplifications causing oncogenic overexpression of AP-1, HMGA2, GLI1, MAP3K12, CDK2, ALX1, and TBX5.23–27 Over the last years, novel gene amplifications (UAP1, MIR557, LAMA4, CPM, IGF2, ERBB3, IGF1R), deletions at chromosome 1p (RUNX3, ARID1A), chromosome 11q (ATM, CHEK1), and chromosome 13q14.2 (MIR15A, MIR16-1),28 and recurrent mutations in members of PI3KCA, PTEN, WNT, ERBB, MAPK, and JAK-STAT pathways have been detected in DDL.28,29 Finally, an important role for epigenetic mechanisms in the dedifferentiation process is emerging, since CEBPα methylation was found in 24% of the DDL30 and CDKN2A gene promoter hypermethylation was observed in DDL but not in recurrent WDL.31
The development of animal models recapitulating tumor progression, resistance, recurrence, and metastasis is vital for drug screening and biomarker analysis. Such approach includes two strategies, ie, the use of xenograft models and the development of transgenic models. Here we review the main liposarcoma models generated so far and discuss the advantages and limitations of such approaches.
Xenograft models of liposarcoma
The engraftment of human tumor cells into immunocompromised hosts (xenograft), despite being a limitation for studying the role of the immune system on tumor progression, is widely used to study cancer.32 For liposarcoma, this strategy represents the best option, given the difficulty to develop transgenic animal models (as highlighted in the next paragraph). As depicted in Figure 1, tumor samples obtained from surgical specimens are dissociated to single-cell suspensions for in vitro study or for cell-derived xenograft (CDX) injection into immunocompromised hosts. This can be done either under the skin (ectopic xenograft) or into the organ type in which the tumor originated (orthotopic xenograft). Orthotopic models ensure a more appropriate microenvironment but are more technically complex compared to ectopic models. A more personalized solution for patients with cancer is the use of tumor tissue fragments engrafted into immunocompromised mice. These patient-derived xenograft (PDX) tumors preserve the characteristics of the live tumor and better recapitulate tumor biology and intratumor heterogeneity of patient tumors.33 The process of PDX generation for individualized care in advanced sarcoma has been set up: it takes 1 to 6 months, and approximately 75% of the implanted tumors grow successfully in mice.34 In this manner, while patients are receiving surgery and treatment with first-line therapy, the tumor is expanded across more generations of mice to test more appropriate treatments. The PDX models generated from surgical specimens maintain the tumor microenvironment present in the human host and the genetic features associated to intratumor heterogeneity, including gene expression profile, copy number variants, and treatment susceptibility. However, it should be advised that one recent study has reported that PDX models of varied tumor types develop mutations with serial passages that diverge from those observed in the patients.35 Recently, 5 PDX models were successfully established from surgical specimens and biopsies of 31 DDL patients.36 The tumors fragments were implanted bilaterally into the subcutaneous space of immunodeficient mice. Fragments from collected tumors were bilaterally re-implanted and passaged over multiple generations. Bilateral tumor engraftment and cryopreservation approaches of PDX models were used to reduce the number of mice required over time. Cryopreserved PDX tumors were successfully re-engrafted in mice.36 Such strategies are not avoid of limitations, including the need of a sufficient amount of fresh tumor tissue, the time of propagation and the failure rate of about 20% of the tumor implantation that, however, may predict lower aggressiveness. As reported in Table 1, several xenograft DDL models have been generated, while the engraftment of WDL lines is more difficult to obtain.37,38 In this regard, it has been proposed to keep the tumors inside a vascularized chamber during their growth into host mice to improve the engraftment success.39
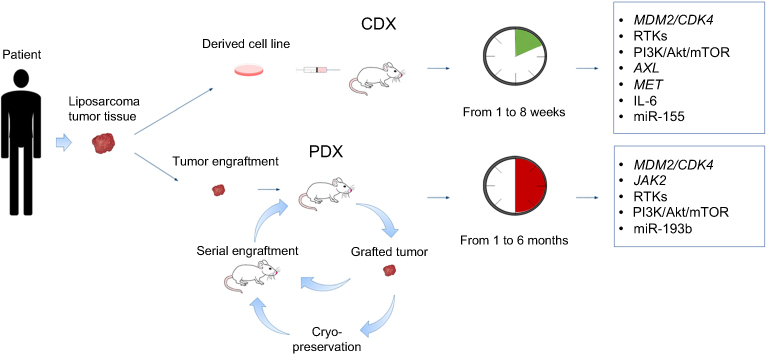 |
Figure 1 Xenograft animal models of liposarcoma. Surgically resected tumors are dissociated to obtain cell suspensions that are injected into immunocompromised mice to generate CDX tumors. This technique is simple, the success of tumor engraftment is relatively high and the time of growth ranges from 1 to 8 weeks. However, the tumor dissociation into cell line disrupts tumor microenvironment and alters intratumor heterogeneity. Alternatively, small fragments (~2–3 mm in diameter) of resected tumor are entirely xenotransplanted into immunocompromised mice to generate PDX tumors that can be further used for serial engraftments. Alternatively, grafted tumors are cryopreserved prior to further utilization. The PDX technique usually requires more time, but it preserves the tumor characteristics, allowing a preclinical drug testing for personalized therapies. The genetic abnormalities of CDX and PDX liposarcoma tumors are highlighted in the boxes.Abbreviations: CDX, cell-derived xenograft; PDX, patient-derived xenograft. |
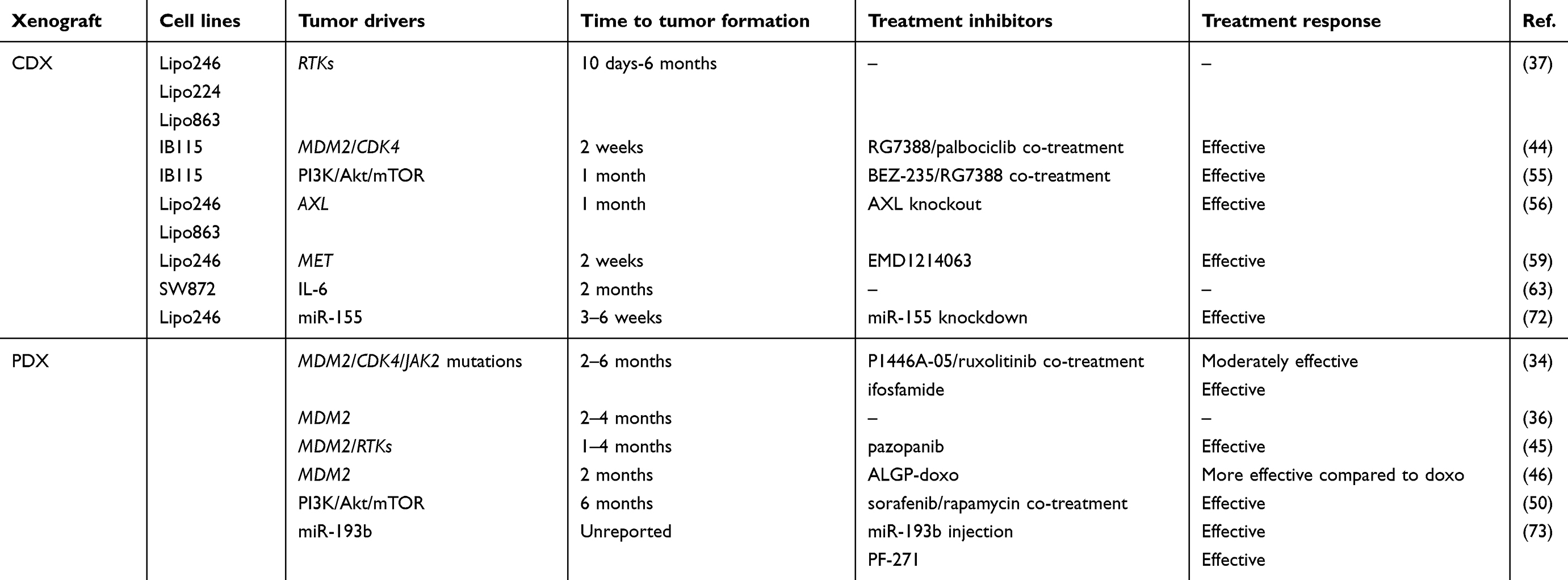 |
Table 1 Mouse xenograft models for DDL |
MDM2/CDK4
As stated before, MDM2/CDK4 coamplification is the most observed genetic signature featuring WDL/DDL. The E3 ubiquitin ligase Mdm2 is a negative regulator of p53 tumor suppressor,40–43 whereas Cdk4 promotes cell cycle G1 phase progression through Rb protein phosphorylation. A CDX model was established to test a dual inhibitors strategy based on RG7388 and palbociclib compounds, inhibiting the p53-Mdm2 complex and Cdk4 activity, respectively.44 Over a 3-week treatment, the tumor volume was decreased and the progression-free survival was increased without evident toxic effects.44 In a PDX model established from a tumor specimen of a man presenting a high-grade DDL of the mesentery,34 genome sequencing showed MDM2/CDK4 coamplification and mutation on JAK2. Though, a limited clinical benefit was observed for the patient receiving Cdk4 inhibitor (P1446A-05) and further supplementation with Jak2 inhibitor (ruxolitinib). Since prospective drug sensitivity in the PDX model revealed the efficacy of ifosfamide treatment, this revealed a good efficacy in the patient.34 In 5 PDX models generated from surgical specimens of 31 DDL patients,36 FISH analysis revealed MDM2 amplification throughout passages. Some of these PDX models have already been successfully used for in vivo testing of a tyrosine kinase inhibitor (pazopanib)45 and a cytotoxic prodrug PhAC-ALGP-doxorubicin (ALGP-doxo).46 In two bilaterally transplanted PDX models, treatment with ALGP-doxo, that is converted to doxorubicin by peptidases present in tumor cells and/or tumor microenvironment, showed a significantly higher antiproliferative effect compared to doxorubicin.46
RTKs and downstream pathways
WDL/DDL show high expression of several RTKs, including DDR2, ERBB3, NTRK1, FGFR3, ROS1, MET, AXL, KIT, and IGFR.37,47 This leads to high activation of downstream signaling primarily through the Mapk and PI3K/Akt/mTOR pathways.48 The overactivation of Akt pathway in WDL/DDL frequently occurs because of the loss of PTEN49,50 or the presence of activating mutations in the PI3K gene (E542K and H1047R amino acid substitutions).51 This pathway, eliciting protein synthesis via mTOR,52 supports many cellular functions, including growth, metabolism, and survival.53 Moreover, oncogenic signal transduction through the PI3K-Akt pathway can enhance Mdm2-mediated p53 suppression.54 In PDX models, treatment with a tyrosine kinase inhibitor (pazopanib) has been reported to delay tumor growth primarily through angiogenesis inhibition.45 In addition, dual combination with a multikinase inhibitor (sorafenib) and an mTOR inhibitor (rapamycin) yielded a reduction of tumor growth that was more consistent compared to rapamycin treatment alone.50 In CDX tumors, concomitant inhibition of the PI3K/Akt/mTOR and Mdm2 pathway, mediated by BEZ-235 and RG7388 compounds, promoted a significant reduction of tumor growth.55 Reduced tumor growth and metastatic rate of CDX tumors were also reported upon knockdown of AXL,56 a member of the TAM family that signals through PI3K/Akt/mTOR and Mapk pathways and whose inhibition represents a promising avenue for the treatment of a wide number of cancers.57 Another emerging therapeutic target is the Met receptor, highly expressed in liposarcoma.37 After binding to Hgf ligand, Met receptor transactivates Stat3, SFKs, and Mapk pathways.58 Consistent with this, treatment with a novel Met inhibitor (EMD1214063) was reported to abrogate tumor growth in CDX models.59
IL-6
IL-6 is a cytokine often overexpressed in cancer and associated to a poor prognosis and chemoresistance.60 IL-6 expression is under the control of a number of transcription factors including NF-κB activator, AP-1, and CEBPs.61 Especially in adipose tissue, CEBPs play a pivotal role as they regulate several biological responses like proliferation, differentiation, adipocytes maturation, and cytokines production.62 To test the potential role of IL-6 on liposarcoma pathogenesis, DDL cells were intramuscularly injected into nude mice.63 Tumor growth was then monitored in both voluntary-active or inactive mice with open or restricted access to activity-wheels to test the potential effects of physical activity on tumor progression. The authors found a greater amount of the circulating IL-6 (6-fold increase) in tumor-bearing mice that was correlated with CEBP-α/β and Ppar-γ activities in comparison to controls.63 Of note, in mice subjected to a physical activity the levels of IL-6 were enhanced, inducing tumor growth, body weight loss, and lung metastasis dissemination through the activation of the autophagy program.63 The negative effect of regular physical activity was then confirmed using an orthotopic tumor model characterized by intramuscular tumor growth,64 indicating that patients with lower-extremities liposarcoma could be advised to minimize the physical activity during the preoperative period.
MicroRNA signatures
MicroRNAs (miRs) are evolutionarily conserved noncoding small RNAs of 18- to 24-nucleotides involved in post-transcriptional gene expression regulation through mRNA degradation, translational inhibition, or chromatin-based silencing mechanisms.65 The miR expression profiles are markedly altered in cancer and their signatures in human tumors are associated with diagnosis, staging, progression, prognosis, and response to treatment.66 Early detection of recurrent or metastatic disease through miR predictors could improve patient prognosis. MiR signatures that are unique to liposarcoma subtypes have been proposed.67,68 For example, miR-155 upregulation was detected in all liposarcoma tumors69 and its plasma levels have been reported as a diagnostic marker for DDL.70 On the other side, miR-25-3p and miR-92a-3p are secreted by liposarcoma cells through extracellular vesicles and may be useful as potential disease biomarkers.71 Using a miR array platform, an expression signature consisting of 4 overexpressed and 31 downregulated miRs was found to differentiate WDL/DDL from normal fat.72 The most statistically significant upregulated miR in DDL was confirmed to be miR-155, which promoted tumor cell growth by targeting CK1α that led to increased β-catenin signaling and cyclin D1 expression.72 Consistent with these results, miR-155 knockdown in CDX tumors delayed tumor growth.72 MiR-193b has lower expression in WDL/DDL compared to adipose tissue samples73 and its injection into PDX tumors significantly delayed tumor volume only after 3 weeks of treatment.73 Since miR-193b was found to target the FAK proteins, the tumor treatment with a FAK inhibitor (PF-271) reduced tumor growth via increased cell apoptosis.73
Transgenic animal models of liposarcoma
Transgenic animal models are mainly originated through genetic knock-in or knock-out approaches to express and/or inactivate specific genes in a tissue-specific and time-dependent manner. The procedures for their generation are expensive and time consuming; however, the availability of animal models developing tumor in response to genomic alterations is of great help for scientists and clinicians. Given the rarity of liposarcoma, the number of transgenic animal models developed so far is limited. Perhaps, one of the major difficulties in such approach is represented by the choice of the cell precursor in which the genetic lesions have to be introduced. In this context, it has been proposed that WDL/DDL may share a common ancestral cell of origin and that the gradual accumulation of genetic lesions could drive the progression of WDL to DDL.74 Alternatively, both WDL/DDL could arise from the same cell precursor but at different time points along the multistep process of adipogenic differentiation.75 Since approximately 5% cases of DDL show heterologous cellular composition of myogenic cells (leiomyosarcoma or rhabdomyosarcoma),8 potential candidate cells are the multipotent stem cells that can differentiate into different mesenchymal precursors. Furthermore, it must be mentioned that cell transdifferentiation processes are possible, as it has been observed that the aberrant activation of the Shh signaling in mature adipocytes is sufficient to convert them into myogenic tumor cells.76 As depicted in Figure 2, four transgenic animal models developing liposarcoma have been generated through gene manipulation in mesenchymal cell progenitors or adipocytes, as described below.
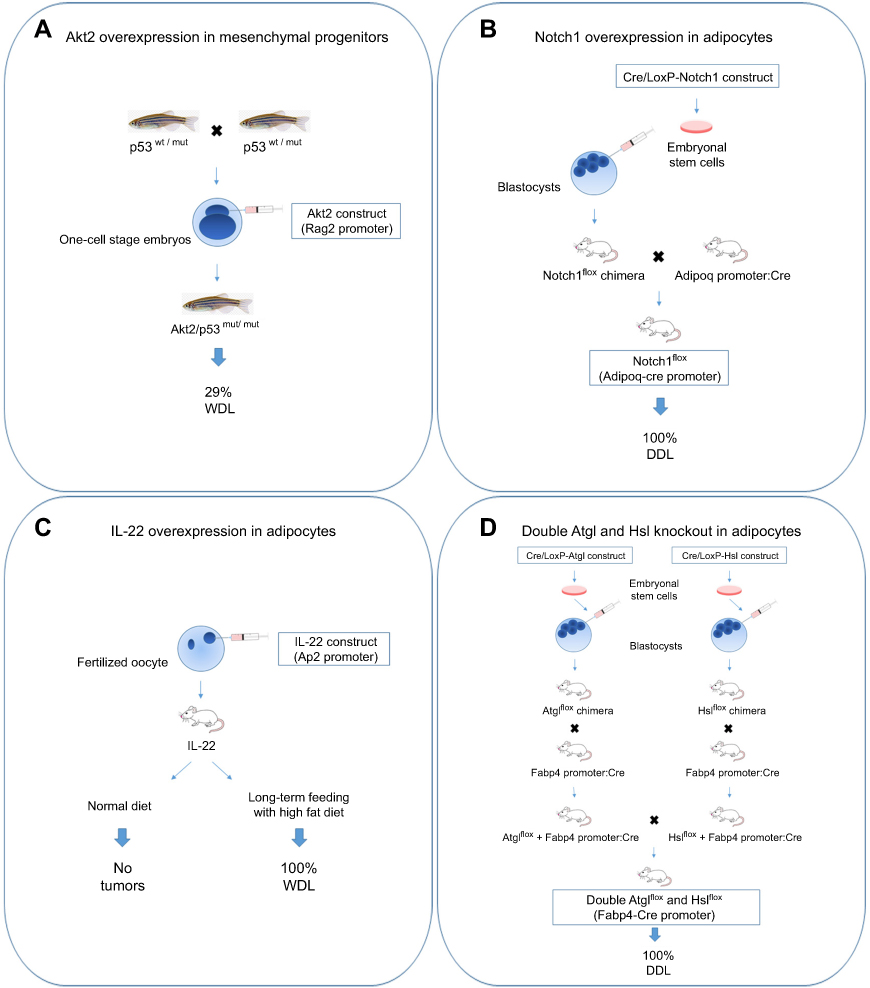 |
Figure 2 Transgenic animal models of liposarcoma. (A) The fertilized oocytes derived from zebrafish with heterozygous background for p53 mutation (M214K substitution) were microinjected with Akt2 construct. Akt2 activation in mesenchymal progenitors drove WDL development. (B) Adipocyte-restricted Notch1 overexpression in mice was obtained through an inducible Cre/LoxP approach. Embryonal mesenchymal stem cells carrying a construct in which Notch1 is flanked with two loxP sites (Notch1flox allele) were injected into mouse blastocysts. The arisen Notch1flox mice were breaded with mice expressing Cre recombinase under the Adipoq promoter, resulting in high Notch1 signaling in fat cells that caused DDL formation. (C) IL-22 construct was microinjected in mouse fertilized oocytes. Only transgenic mice fed with a long-term high fat diet developed WDL. (D) Mice double knockout for Atgl and Hsl were obtained through an inducible Cre/LoxP approach. Embryonal mesenchymal stem cells carrying Atglflox or Hslflox cassettes were injected into mouse blastocysts. The arisen Atglflox and Hslflox mice were breaded with mice expressing Cre recombinase under the Fabp4 promoter to knockout Atgl or Hsl in fat cells. Double knockout mice generated from their breeding developed DDL.Abbreviations: WDL, well-differentiated liposarcoma; DDL, dedifferentiated liposarcoma. |
Akt signaling
Oncogenic gene mutations in receptors (FGFR, EGFR) or transducers (KRAS, PI3K)77 commonly elicit deregulation of PI3K/Akt/mTOR pathway, as analogously observed in liposarcoma.28,29 A transgenic model of WDL has been generated via targeted expression of an active myristoylated Akt2 form in mesenchymal cell progenitors of zebrafish carrying p53 homozygous mutation. Following embryo microinjection with a DNA construct carrying Akt2,49 solid tumors classified as WDL (91%) and osteosarcomas (9%) developed between 1 and 4 months of age, with the highest tumor incidence rate observed in p53-homozygous mutants (about 29%) (Figure 2A). The treatment of transgenic zebrafish with a dual PI3K/mTOR inhibitor (BEZ235) was efficacious to counteract tumor growth via increased apoptosis,49 therefore confirming that this pathway synergizes with p53 loss to drive liposarcoma genesis.
Notch signaling
The evolutionarily conserved Notch signaling pathway plays a pivotal role in cell commitment, tissue development, and tumorigenesis.78,79 The Notch cascade is initiated when one of the five ligands belonging to DSL family (Jag1, −2 and Dll1, −3, −4) binds to one of the four Notch receptors.80 This interaction induces sequential cleavages in Notch receptor mediated by different proteases (Adam metalloproteases, γ-secretase protease complex) that cause the release of the Notch intracellular domain, which then translocates into the nucleus to activate the transcription of several target genes.80 The activation of Notch signaling in mature adipocytes, obtained through the Cre/LoxP technology,81 has been shown to elicit DDL formation by shaping gene expression and promoting dedifferentiation via suppression of fatty acid metabolism and Ppar-γ function (Figure 2B).82 Mice with activated Notch1 exhibited a lipodystrophy phenotype in the pre-neoplastic state, characterized by hepatic steatosis, hyperglycemia, and severe insulin resistance.82 The authors further demonstrated that the treatment of a human LPS246 xenograft model with a Notch inhibitor (dibenzazepine) was effective to reduce tumor growth.82 A selective Notch inhibitor (LY3039478) was recently tested in a phase 1a/b trial showing a modest clinical activity and a safety profile towards several sarcomas, including liposarcoma.83 Interestingly, it has been shown that Mdm2 can synergize with Notch1 to inhibit apoptosis and promote proliferation,84,85 indicating that targeting the Notch pathway may be helpful for overcoming WDL/DDL development and progression.
IL-22 overexpression
WDL formation has been observed in transgenic mice overexpressing IL-22 in adipocytes (Figure 2C).86 Interestingly, these mice had neither apparent phenotype nor metabolic alteration, but developed spontaneous tumors in adipose tissue after long-term feeding with high fat diet. IL-22 is a T-cell secreted cytokine that modulates inflammatory response in nonhematopoietic tissues such as epithelium and liver87 via primary activation of Jak1 and Tyk2 and Stat3 pathway but also of Mapk and PI3K/Akt/mTOR pathways.88 The oncogenic role of IL-22/Stat3 signaling axis has been reported in a number of tumors, such as hepatocarcinoma.89 In response to high IL-22 levels, the adipose tissue of transgenic mice became inflamed and characterized by higher levels of IL-1β, IL-6, IL-10, TNF-α, and Erk pathway activation. This work confirms the important role of diet and inflammation in tumorigenesis, since the increased secretion of steroid hormones and insulin resistance in the presence of a persistent inflammatory state, as observed in obesity, may increase tumorigenesis risk.90–92 It has been estimated that obesity in children from 2 to 14 years resulted associated with increased cancer risk in adulthood by 40% and also with a worse survival with respect to control patients.93
Epistatic gene interaction between the adipose triglyceride lipase and hormone-sensitive lipase
Epistasis refers to the observation of an unexpected phenotypic outcome when combining two genetic alleles.94 A synergistic epistatic interaction determinant for liposarcoma development has been observed between two genes of the lipolysis pathway, the adipose triglyceride lipase (Atgl) and the hormone-sensitive lipase (Hsl) (Figure 2D).95 Lipolysis has been implicated in cancer since it provides a source of fatty acids for tumor growth.96 Atgl enzyme, encoded by the PNPLA2 gene, catalyzes the hydrolysis of triglycerides to diglycerides, 97 whereas Hsl, encoded by the LIPE gene, catalyzes the second step of lipolysis, the cleavage of diglycerides to monoglycerides. Of note, deletion of PNPLA2 is reported in WDL and sarcoma,22,98 and deletions of the chromosome 19p13 region containing LIPE are frequent in DDL and correlate with poor prognosis.22 Mice lacking both Atgl and Hsl showed near-complete deficiency of lipolysis and were unable to maintain their blood glucose values over a normal postprandial fasting due to rapid depletion of carbohydrates reserves in the absence of lipid stores in adipose tissue.95 While no malignant tumors were found in white adipose tissue of transgenic mice, the brown adipose tissue was characterized by hypertrophic brown adipocytes with formation of liposarcoma tumors between 11 and 14 months of age. Expression profiling analysis in premalignant brown adipose tissue of transgenic mice revealed downregulation for the gene sets of fatty acid, triacylglycerol and ketone body metabolism, the tricarboxylic acid cycle and respiratory chain and genes of lipid metabolism. In contrast, genes involved in the immune response were upregulated. Among the differentially expressed genes, liposarcoma tumors showed highest expression of GPNMB, which encodes a circulating glycoprotein identified in several cancers,99 and lowest expression of G0S2, the endogenous inhibitor of Atgl.100 By comparing the gene expression profiles between transgenic animals and a number of 58 DDL patients,101 the authors found that LIPE, PNPLA2, and G0S2 are also among the five most downregulated genes in human liposarcoma.95
Conclusions
The generation of animal models of liposarcoma is crucial for identification of early markers, diagnosis, and development of new therapies. To date, a major obstacle in this process is the limited number of appropriate animal models recapitulating the complexity and heterogeneity of liposarcoma malignancies, resulting in poor efficiency in translating the findings of basic research to clinical applications. Surely, the generation of animal models is complicated by the choice of the potential cell of origin to be used as a recipient for the genomic editing phase. In this context, the use of PDX models allows to personalize the treatment options for patients and therefore represents an important alternative. Hopefully, the establishment of novel clinically relevant disease transgenic models will be vital for identification of the molecular mechanisms underlying liposarcoma genesis and progression and for validation of new therapies.
Abbreviation list
ADAM, A disintegrin and metalloprotease; AKT, Ak strain transforming murine thymoma viral oncogene; ALX1, Aristaless-like homeobox protein 1; AP-1, Activator protein 1; ARID1A, AT-rich interactive domain 1A; ATM, Ataxia-telangiectasia-mutated gene; CDK, Cyclin-dependent kinase; CDKN2A, Cyclin-dependent kinase inhibitor 2A; CEBPs, CCAAT/enhancer-binding proteins ; CHEK1, Checkpoint kinase 1; CK1α, Casein kinase 1 alpha; CPM, Carboxypeptidase M; DDR2, Discoidin domain receptor 2; DLL, Distal-less; DSL, Delta/serrate/lag-2; EGFR, Epidermal growth factor receptor;ERbB, Erb-b2 receptor tyrosine kinase; ERK, Extracellular signal-regulated kinase; FAK, Focal adhesion kinase; FGFR, Fibroblast growth factor receptor; FUS-DDIT3, Fusion-DNA damage-inducible transcript 3; G0S2, G0/G1 switch gene 2; GLI1, Glioma-associated oncogene homolog 1; GPNMB, Glycoprotein nonmetastatic melanoma protein B; HGF, Hepatocyte growth factor; HMGA2, High-mobility group AT-hook protein 2; IL, Interleukin; IGF2, Insulin-like growth factor 2; IGFR, Insulin-like growth factor receptor; JAG1, Jagged-1; JAK, Janus kinase; KIT, receptor tyrosine kinase; KRAS, Kirsten rat sarcoma viral oncogene homolog; LAMA4, Laminin subunit alpha 4; LIPE, Lipase E; MAPK, Mitogen-activated protein kinase; MDM2, Mouse double minute 2 homolog; MET, Mesenchymal-epithelial transition factor; mTOR, Mammalian target of rapamycin; NF1, Neurofibromin 1; NF-κB, Nuclear factor kappa B; NOTCH1, Neurogenic locus notch homolog protein 1 ; NTRK1, Neurotrophic tyrosine receptor kinase type 1 ; PI3KCA, Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha; PNPLA2, Patatin-like phospholipase domain containing 2; PPAR-γ, Peroxisome proliferator-activated receptor gamma; PTEN, Phosphatase and tensin homolog; RB1, Retinoblastoma 1; ROS1, C-ros oncogene 1; RTKs, Tyrosine kinase receptors; RUNX3, Runt-related transcription factor 3; SFKs, Src family protein kinases; SHH, Sonic hedgehog; STAT, Signal transducer and activator of transcription; TAM, Tyro3, Axl, Mertk; TBX5, T-box transcription factor 5; TNF-α, Tumor necrosis factor-alpha; TP53, Tumor suppressor protein p53; TYK2, Tyrosine kinase 2; UAP1, UDP-N-acetylglucosamine pyrophosphorylase 1; WNT, Wingless-integrated.
Acknowledgments
This work was supported by the University of Brescia (ex 60%) and Siderurgica Leonessa research funds to AF. We are grateful to Umberto Veronesi Foundation for granting FM with Post-doctoral Fellowship year-2018 Award.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Fletcher CD. The evolving classification of soft tissue tumours - an update based on the new 2013 WHO classification. Histopathology. 2014;64:2–11. doi:10.1111/his.12267
2. Dei Tos AP. Liposarcomas: diagnostic pitfalls and new insights. Histopathology. 2014;64(1):38–52. doi:10.1111/his.12311
3. Rubin BP, Fletcher CD. The cytogenetics of lipomatous tumours. Histopathology. 1997;30:507–511.
4. Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979;3:507–523.
5. Lahat G, Anaya DA, Wang X, Tuvin D, Lev D, Pollock RE. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol. 2008;15:1585–1593. doi:10.1245/s10434-007-9805-x
6. Rieker RJ, Weitz J, Lehner B, et al. Genomic profiling reveals subsets of dedifferentiated liposarcoma to follow separate molecular pathways. Virchows Arch. 2010;456:277–285. doi:10.1007/s00428-009-0869-9
7. Evans HL, Khurana KK, Kemp BL, Ayala AG. Heterologous elements in the dedifferentiated component of dedifferentiated liposarcoma. Am J Surg Pathol. 1994;18:1150–1157.
8. Binh MB, Guillou L, Hostein I, et al. Dedifferentiated liposarcomas with divergent myosarcomatous differentiation developed in the internal trunk: a study of 27 cases and comparison to conventional dedifferentiated liposarcomas and leiomyosarcomas. Am J Surg Pathol. 2007;31:1557–1566. doi:10.1097/PAS.0b013e31804b4109
9. Kollár A, Benson C. Current management options for liposarcoma and challenges for the future. Expert Rev Anticancer Ther. 2014;14:297–306. doi:10.1586/14737140.2014.869173
10. Conyers R, Young S, Thomas DM. Liposarcoma: molecular genetics and therapeutics. Sarcoma. 2011;2011:483154. doi:10.1155/2011/483154
11. Bill KL, Casadei L, Prudner BC, Iwenofu H, Strohecker AM, Pollock RE. Liposarcoma: molecular targets and therapeutic implications. Cell Mol Life Sci. 2016;73:3711–3718. doi:10.1007/s00018-016-2266-2
12. Demicco EG. Molecular updates in adipocytic neoplasms. Semin Diagn Pathol. 2019;36:85–94. doi:10.1053/j.semdp.2019.02.003
13. Thway K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: an updated review. Semin Diagn Pathol. 2019;36:112–121. doi:10.1053/j.semdp.2019.02.006
14. Crozat A, Aman P, Mandahl N, Ron D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature. 1993;363:640–644. doi:10.1038/363640a0
15. Guillou L, Aurias A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2010;456:201–217. doi:10.1007/s00428-009-0853-4
16. Pedeutour, F., Forus A, Coindre JM, et al. Structure of the supernumerary ring and giant rod chromosomes in adipose tissue tumors. Genes Chromosomes Cancer. 1999;24:30–41. doi:10.1002/(SICI)1098-2264(199901)24:1<30::AID-GCC5>3.0.CO;2-P
17. Binh, MB, Sastre-Garau X, Guillou L, et al. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol. 2005;29:1340–1347. doi:10.1097/01.pas.0000170343.09562.39
18. Sirvent N, Coindre JM, Make G, et al. Detection of MDM2-CDK4 amplification by fluorescence in situ hybridization in 200 paraffin-embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real-time PCR. Am J Surg Pathol. 2007;31:1476–1489. doi:10.1097/PAS.0b013e3180581fff
19. Thway K, Flora R, Shah C, Olmos D, Fisher C. Diagnostic utility of p16, CDK4, and MDM2 as an immunohistochemical panel in distinguishing well-differentiated and dedifferentiated liposarcomas from other adipocytic tumors. Am J Surg Pathol. 2012;36:462–469. doi:10.1097/PAS.0b013e3182417330
20. Beird HC, Wu CC, Ingram DR, et al. Genomic profiling of dedifferentiated liposarcoma compared to matched well-differentiated liposarcoma reveals higher genomic complexity and a common origin. Cold Spring Harb Mol Case Stud. 2018;4:a002386. doi:10.1101/mcs.a002386
21. Horvai AE, DeVries S, Roy R, O’Donnell RJ, Waldman F. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol. 2009;22:1477–1488. doi:10.1038/modpathol.2009.119
22. Crago AM, Socci ND, DeCarolis P, et al. Copy number losses define subgroups of dedifferentiated liposarcoma with poor prognosis and genomic instability. Clin Cancer Res. 2012;18:1334–1340. doi:10.1158/1078-0432.CCR-11-2820
23. Mariani O, Brennetot C, Coindre JM, et al. JUN oncogene amplification and overexpression block adipocytic differentiation in highly aggressive sarcomas. Cancer Cell. 2007;11:361–374. doi:10.1016/j.ccr.2007.02.007
24. Snyder EL, Sandstrom DJ, Law K, et al. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol. 2009;218:292–300. doi:10.1002/path.2564
25. Tap WD, Eilber FC, Ginther C, et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2011;50:95–112. doi:10.1002/gcc.20835
26. Creytens D, Van Gorp J, Speel EJ, Ferdinande L. Characterization of the 12q amplicons in lipomatous soft tissue tumors by multiplex ligation-dependent probe amplification-based copy number analysis. Anticancer Res. 2015;35:1835–1842.
27. Saâda-Bouzid E, Burel-Vandenbos F, Ranchère-Vince D, et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod Pathol. 2015;28:1404–1414. doi:10.1038/modpathol.2015.96
28. Kanojia D, Nagata Y, Garg M, et al. Genomic landscape of liposarcoma. Oncotarget. 2015;6:42429–42444. doi:10.18632/oncotarget.6464
29. Barretina J, Taylor BS, Banerji S, et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat Genet. 2010;42:715–721. doi:10.1038/ng.619
30. Taylor BS, DeCarolis PL, Angeles CV, et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 2011;1:587–597. doi:10.1158/2159-8290.CD-11-0181
31. He M, Aisner S, Benevenia J, Patterson F, Harrison LE, Hameed M. Epigenetic alteration of p16INK4a gene in dedifferentiation of liposarcoma. Pathol Res Pract. 2009;205:386–394. doi:10.1016/j.prp.2008.12.009
32. Morton CL, Houghton PJ. Establishment of human tumor xenografts in immunodeficient mice. Nat Protoc. 2007;2:247–250. doi:10.1038/nprot.2007.25
33. Tentler JJ, Tan AC, Weekes CD, et al. Patient-derived tumor xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi:10.1038/nrclinonc.2012.61
34. Stebbing J, Paz K, Schwartz GK, et al. Patient-derived xenografts for individualized care in advanced sarcoma. Cancer. 2014;120:2006–2015. doi:10.1002/cncr.28696
35. Ben-David U, Ha G, Tseng YY, et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat Genet. 2017;49:1567–1575. doi:10.1038/ng.3967
36. Cornillie J, Wozniak A, Li H, et al. Establishment and characterization of histologically and molecularly stable soft tissue sarcoma xenograft models for biological studies and preclinical drug testing. Mol Cancer Ther. 2019;18:1168–1178. doi:10.1158/1535-7163.MCT-18-1045
37. Peng T, Zhang P, Liu J, et al. An experimental model for the study of well-differentiated and dedifferentiated liposarcoma; deregulation of targetable tyrosine kinase receptors. Lab Invest. 2011;91:392–403. doi:10.1038/labinvest.2010.185
38. Jo EB, Hong D, Lee YS, Lee H, Park JB, Kim SJ. Establishment of a novel PDX mouse model and evaluation of the tumor suppression efficacy of bortezomib against liposarcoma. Transl Oncol. 2018;12(2):269–281. doi:10.1016/j.tranon.2018.09.015
39. Tilkorn D, Daigeler A, Stricker I, et al. Establishing efficient xenograft models with intrinsic vascularisation for growing primary human low-grade sarcomas. Anticancer Res. 2011;31:4061–4066.
40. Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245.
41. Kussie PH, Gorina S, Marechal V, et al. Structure of MDM2 oncoprotein bound to p53 tumor soppressor transactivation domain. Science. 1996;274:948–953. doi:10.1126/science.274.5289.948
42. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296–299. doi:10.1038/387296a0
43. Oliner JD, Pietenpol JA, Thiagalingam S, Gyuris J, Kinzler KW, Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature. 1993;362:857–860. doi:10.1038/362857a0
44. Laroche-Clary A, Chaire V, Algeo MP, Derieppe MA, Loarer FL, Italiano A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J Hematol Oncol. 2017;10:123. doi:10.1186/s13045-017-0482-3
45. Li H, Wozniak A, Sciot R, et al. Pazopanib, a receptor tyrosine kinase inhibitor, suppresses tumor growth through angiogenesis in dedifferentiated liposarcoma xenograft models. Transl Oncol. 2014;7(6):665–671. doi:10.1016/j.tranon.2014.09.007
46. Cornillie J, Wozniak A, Pokreisz P, et al. Antitumoral efficacy of PhAc-ALGP-doxorubicin, an enzyme-activated doxorubicin prodrug, in patient-derived soft tissue sarcoma xenograft models. Mol Cancer Ther. 2017;16:1566–1575. doi:10.1158/1535-7163.MCT-16-0832
47. Asano N, Yoshida A, Mitani S, et al. Frequent amplification of receptor tyrosine kinase genes in welldifferentiated/dedifferentiated liposarcoma. Oncotarget. 2017;8:12941–12952. doi:10.18632/oncotarget.14652
48. Ishii T, Kohashi K, Iura K, et al. Activation of the Akt-mTOR and MAPK pathways in dedifferentiated liposarcomas. Tumour Biol. 2016;37:4767–4776. doi:10.1007/s13277-015-4232-2
49. Gutierrez A, Snyder EL, Marino-Enriquez A, et al. Aberrant AKT activation drives well-differentiated liposarcoma. Proc Natl Acad Sci USA. 2011;108(39):16386–16391. doi:10.1073/pnas.1106127108
50. Smith, KB, Tran LM, Tam BM, et al. Novel dedifferentiated liposarcoma xenograft models reveal PTEN down-regulation as a malignant signature and response to PI3K pathway inhibition. Am J Pathol. 2013;182:1400–1411. doi:10.1016/j.ajpath.2013.01.002
51. Guo S, Lopez-Marquez H, Fan KC, et al. Synergistic effects of targeted PI3K signaling inhibition and chemotherapy in liposarcoma. PlosOne. 2014; 9(4):e93996. doi: 10.1371/journal.pone.0093996.
52. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;169:361–371. doi:10.1016/j.cell.2017.03.035
53. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi:10.1038/nrg1879
54. Abraham AG, O’Neill E. PI3K/Akt-mediated regulation of p53 in cancer. Biochem Soc Trans. 2014;42:798–803. doi:10.1042/BST20140070
55. Laroche A, Chaire V, Algeo MP, Karanian M, Fourneaux B, Italiano A. MDM2 antagonists synergize with PI3K/mTOR inhibition in well-differentiated/dedifferentiated liposarcomas. Oncotarget. 2017;8(33):53968–53977. doi:10.18632/oncotarget.16345
56. May CD, Garnett J, Ma XY, et al. AXL is a potential therapeutic target in dedifferentiated and pleomorphic liposarcomas. BMC Cancer. 2015;15:901. doi:10.1186/s12885-015-1584-3
57. X W, Liu X, Koul S, Lee CY, Zhang Z, Halmos B. AXL kinase as a novel target for cancer therapy. Oncotarget. 2014;5:9546–9563.
58. Peters S, Adjei AA. MET: a promising anticancer therapeutic target. Nat Rev Clin Oncol. 2012;9:314–326. doi:10.1038/nrclinonc.2012.154
59. Bill KL, Garnett J, Ma X, et al. The hepatocyte growth factor receptor as a potential therapeutic target for dedifferentiated liposarcoma. Lab Invest. 2016;95(8):951–961. doi:10.1038/labinvest.2015.62
60. Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. 2016;37:11553e72. doi:10.1007/s13277-016-5098-7
61. Hershko DD, Robb BW, Luo G, Hasselgren PO. Multiple transcription factors regulating the IL-6 gene are activated by cAMP in cultured Caco-2 cells. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1140eR8. doi:10.1152/ajpregu.00161.2002
62. Lefterova MI, Lazar MA. New developments in adipogenesis. Trends Endocrinol Metab. 2009;20:107–114. doi:10.1016/j.tem.2008.11.005
63. Assi M, Kenawi M, Ropars M, Rébillard A. Interleukin-6, C/EBP-β and PPAR-γ expression correlates with intramuscular liposarcoma growth in mice: the impact of voluntary physical activity levels. Biochem Biophys Res Commun. 2017;490(3):1026–1032. doi:10.1016/j.bbrc.2017.06.158
64. Assi M, Derbré F, Lefeuvre-Orfila L, et al. Maintaining a regular physical activity aggravates intramuscular tumor growth in an orthotopic liposarcoma model. Am J Cancer Res. 2017;7:1037–1053.
65. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi:10.1038/nature02871
66. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi:10.1038/nrc1997
67. Zhou Y, Zhang Y, Huang Y, et al. Liposarcoma miRNA signatures identified from genome-wide miRNA expression profiling. Future Oncol. 2014;10:1373–1386. doi:10.2217/fon.14.90
68. Sun R, Shen JK, Choy E, Yu Z, Hornicek FJ, Duan Z. The emerging roles and therapeutic potential of microRNAs (miRs) in liposarcoma. Discov Med. 2015;20:311–324.
69. Vincenzi B, Iuliani M, Zoccoli A, et al. Deregulation of dicer and mir-155 expression in liposarcoma. Oncotarget. 2015;6:10586–10591. doi:10.18632/oncotarget.3201
70. Boro A, Bauer D, Born W, Fuchs B. Plasma levels of miRNA-155 as a powerful diagnostic marker for dedifferentiated liposarcoma. Am J Cancer Res. 2016;6:544–552.
71. Casadei L, Calore F, Creighton CJ, et al. Exosome-derived miR-25-3p and miR-92a-3p stimulate liposarcoma progression. Cancer Res. 2017;77:3846–3856. doi:10.1158/0008-5472.CAN-16-2984
72. Zhang P, Bill K, Liu J, et al. MiR-155 is a liposarcoma oncogene that targets casein kinase-1α and enhances β-catenin signaling. Cancer Res. 2012;72(7):1751–1762. doi:10.1158/0008-5472.CAN-11-3027
73. Mazzu YZ, Hu Y, Soni RK, et al. miR-193b-regulated signaling networks serve as tumor suppressors in liposarcoma and promote adipogenesis in adipose-derived stem cells. Cancer Res. 2017;77(21):5728–5740. doi:10.1158/0008-5472.CAN-16-2253
74. Shimoji T, Kanda H, Kitagawa T, et al. Clinico-molecular study of dedifferentiation in well-differentiated liposarcoma. Biochem Biophys Res Commun. 2004;314:1133–1140.
75. Matushansky I, Hernando E, Socci ND, et al. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. Am J Pathol. 2008;172:1069–1080. doi:10.2353/ajpath.2008.070284
76. Hatley ME, Tang W, Garcia MR, et al. A mouse model of rhabdomyosarcoma originating from the adipocyte lineage. Cancer Cell. 2012;22:536–546. doi:10.1016/j.ccr.2012.09.004
77. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi:10.1016/j.cell.2011.02.013
78. Koch U, Radtke F. Notch and cancer: a double-edged sword. Cell Mol Life Sci. 2007;64:2746–2762. doi:10.1007/s00018-007-7164-1
79. Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409.
80. Andersson ER, Sandberg R, Lendahl U. Notch signaling: simplicity in design, versatility in function. Development. 2011;138:3593–3612. doi:10.1242/dev.063610
81. Le Y, Sauer B. Conditional gene knockout using cre recombinase. Methods Mol Biol. 2000;136:477–485.
82. Bi P, Yue F, Karki A, et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J Exp Med. 2016;213:2019–2037. doi:10.1084/jem.20160157
83. Mir O, Azaro A, Merchan J, et al. Notch pathway inhibition with LY3039478 in soft tissue sarcoma and gastrointestinal stromal tumours. Eur J Cancer. 2018;103:88–97. doi:10.1016/j.ejca.2018.08.012
84. Sczaniecka M, Gladstone K, Pettersson S, McLaren L, Huart AS, Wallace M. MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J Biol Chem. 2012;287:14052–14068. doi:10.1074/jbc.M111.303875
85. Pettersson S, Sczaniecka M, McLaren L, et al. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem J. 2013;450:523–536. doi:10.1042/BJ20121249
86. Wang Z, Yang L, Jiang Y, et al. High fat diet induces formation of spontaneous liposarcoma in mouse adipose tissue with overexpression of interleukin 22. PlosOne. 2011; 6(8):e23737. doi: 10.1371/journal.pone.0023737.
87. Witte E, Witte K, Warszawska K, Sabat R, Wolk K. Interleukin-22: a cytokine produced by T, NK and NKT cell subsets, with importance in the innate immune defense and tissue protection. Cytokine Growth Factor Rev. 2010;21:365–379. doi:10.1016/j.cytogfr.2010.08.002
88. Lejeune D, Dumoutier L, Constantinescu S, Kruijer W, Schuringa JJ, Renauld JC. Interleukin-22 (IL-22) activates the JAK/STAT, ERK, JNK, and p38 MAP kinase pathways in a rat hepatoma cell line. Pathways that are shared with and distinct from IL-10. J Biol Chem. 2002;277:33676–33682. doi:10.1074/jbc.M204204200
89. Jiang R, Tan Z, Deng L, et al. Interleukin-22 promotes human hepatocellular carcinoma by activation of STAT3. Hepatology. 2011;54:900–909. doi:10.1002/hep.24486
90. Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat Rev Cancer. 2004;4(8):579–591. doi:10.1038/nrc1408
91. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi:10.1016/j.cell.2010.01.025
92. De Pergola G, Silvestris F. Obesity as a major risk factor for cancer. J Obes. 2013;2013:291546. doi:10.1155/2013/291546
93. Park MH, Falconer C, Viner RM, Kinra S. The impact of childhood obesity on morbidity and mortality in adulthood: a systematic review. Obes Rev. 2012;13(1):985–1000. doi:10.1111/j.1467-789X.2012.01015.x
94. Wang X, Fu AQ, McNerney ME, White KP. Widespread genetic epistasis among cancer genes. Nat Commun. 2014;5:4828. doi:10.1038/ncomms5972
95. Wu JW, Preuss C, Wang SP, et al. Epistatic interaction between the lipaseencoding genes Pnpla2 and Lipe causes liposarcoma in mice. PLoS Genet. 2017;13:e1006716. doi:10.1371/journal.pgen.1006716
96. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013;52:585–589. doi:10.1016/j.plipres.2013.08.005
97. Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi:10.1126/science.306.5698.956a
98. Al-Zoughbi W, Pichler M, Gorkiewicz G, et al. Loss of adipose triglyceride lipase is associated with human cancer and induces mouse pulmonary neoplasia. Oncotarget. 2016;7(23):
99. Maric G, Rose AA, Annis MG, Siegel PM. Glycoprotein non-metastatic b (GPNMB): a metastatic mediator and emerging therapeutic target in cancer. Onco Targets Ther. 2013;6:839–852. doi:10.2147/OTT.S44906
100. Yang X, Lu X, Lombès M, et al. The G(0)/G(1) switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010;11(3):
101. Weinstein JN, Collisson EA, Mills GB, et al.; Network CGAR. The cancer genome atlas pan-cancer analysis project. Nat Genet. 2013;45:1113–1120. doi:10.1038/ng.2764
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.