")
Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 6
Animal models of pancreatic cancer and their application in clinical research
Authors Weidenhofer J, Colvin E, Bond D, Scarlett C
Received 6 May 2016
Accepted for publication 29 July 2016
Published 25 August 2016 Volume 2016:6 Pages 31—39
DOI https://doi.org/10.2147/GICTT.S84531
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Eileen O'Reilly
Judith Weidenhofer,1,2 Emily K Colvin,3 Danielle R Bond,1,2,4 Christopher J Scarlett2,4
1School of Biomedical Sciences and Pharmacy, The University of Newcastle, Ourimbah, 2Cancer Program, Hunter Medical Research Institute, New Lambton, 3Bill Walsh Translational Cancer Research Laboratory, Kolling Institute of Medical Research, Royal North Shore Hospital, University of Sydney, St Leonards, 4Pancreatic Cancer Research Group, School of Environmental and Life Sciences, The University of Newcastle, Ourimbah, NSW, Australia
Abstract: Pancreatic cancer has one of the highest cancer-related mortality rates of all cancers, and despite worldwide efforts to identify new curative treatments, little improvement has been made toward disease-free survival rates. Due to the effect of a heterogeneous disease phenotype in an organ where desmoplastic effects modify tumor behavior and capacity to deliver chemotherapeutics, it is clear that accurate in vivo models are imperative for the understanding of this disease, to identify and test novel therapeutics, and to assist in identifying biomarkers. This review addresses the currently available mouse models of pancreatic ductal adenocarcinoma, in particular genetically engineered and patient-derived xenograft models, focusing on their utility in the drug discovery pipeline.
Keywords: pancreatic cancer, mouse models, genetically engineered, patient-derived, orthotopic, xenograft
Introduction
Pancreatic cancer has one of the lowest 5-year survival rates (8%), meaning that despite a moderate worldwide incidence, it is the fourth leading cause of cancer-related mortality.1 Further, the survival rate has not changed over the past decades, indicating that greater research is required to understand how this cancer differs from others, where survival has improved steadily since the introduction of new treatments and screening processes.1 Pancreatic malignancies occur primarily in the form of pancreatic ductal adenocarcinoma (PDAC) in >85% of all new pancreatic cancer cases, with the remaining classified as acinar cell carcinoma, pancreatic neuroendocrine tumors, and undifferentiated carcinoma.2,3
PDAC develops initially from precursor lesions, the most common of which is pancreatic intraepithelial neoplasia (PanIN),2 with intraductal papillary mucinous neoplasms3 and mucinous cystic neoplasms,4 alternative precursors, each possessing varying tumorigenic potential. Precursor lesions are generally well differentiated and could be characterized based on the degree of cytological and architectural aberrations, with those mutations commonly seen in PDAC increasingly found in higher degree PanIN.3
Many mutations have been discovered to be involved in pancreatic cancer, with the latest large-scale sequencing study identifying commonly found mutations clustered in ten molecular mechanisms across the cohorts.5 Depending on the study design, the average number of nonsynonymous mutations per tumor is reported to be between 26 and 67.6 To date, mouse models of pancreatic cancer have utilized those commonly occurring mutations, particularly KRAS with increasing complexity derived from generating transgenic mice harboring mutations in more than one gene. Nonetheless, these mice develop tumors where these mutations are present in all cells, which is not necessarily the case in pancreatic cancer, with heterogeneity in mutation observed in the various cells of the tumor.7,8 This presents a limitation in the research of cancer, that is an accurate in vivo model that allows for the assessment of systemic involvement in the development and progression of cancer. This is particularly important for pancreatic cancer, which is known to be highly heterogeneous and also significantly affected by the tumor microenvironment.9 Thus, many model systems have been used to assess the contribution of the various genetic drivers of pancreatic cancer to disease progression and therefore clinical utility as biomarkers and therapeutic targets. This review outlines the main mouse models in terms of genetically engineered mouse models (GEMMs) and patient-derived xenografts (PDXs), their advantages and disadvantages (Table 1), and their purpose in preclinical research.
 | Table 1 Advantages and disadvantages of pancreatic cancer models Abbreviations: PanIN, pancreatic intraepithelial neoplasia; FNB, fine needle biopsies; PCac, pancreatic cancer. |
Genetically engineered mouse models
Initial attempts to model pancreatic cancer with transgenic mice utilized pancreatic-specific gene promoters, either the elastase promoter or the rat insulin promoter, to drive the expression of oncogenes (eg, SV40, H-ras) in acinar and beta cells, respectively.10–12 In general, these studies failed to accurately mimic what was known about PDAC, either not progressing to tumors or not developing PanIN prior to tumorigenesis.13 Notably, the LSL-KrasG12D inducible knock-in mouse (KC model) was the first to show the expected progression of PanIN to metastatic tumors, with the progression linked to the same disruptions to signaling that had been observed in human PDAC.14 Further studies attempted to determine which additional genetic aberrations were the drivers of progression in PDAC, as GEMMs utilizing only activating Kras mutations showed uncharacteristically long latency and relatively low metastatic involvement. Incorporation of mutations in known or suspected tumor suppressor genes, such as TP53 (KPC model), resulted in a phenotype closer to human PDAC (accelerated progression and greater proportion of anaplastic foci in the resultant tumors),15 revealing the complexity of the PDAC genetic landscape. Further, these models show resistance to gemcitabine and low median survival rates, representing an autochthonous, immunocompetent model that has been used for preclinical assessment of inhibition of progression,16,17 immunotherapy,18 and enhancing chemotherapy.19,20 To date, there are in excess of 30 different GEMMs of pancreatic cancer with varying phenotypes, and yet their clinical utility in identifying and then testing novel therapeutic targets has not resulted in the anticipated translation to increased patient survival.
GEMMs have been developed that cover the most commonly found mutations on the background of the KC model. The SMAD4 models provide a means to examine the molecular mechanisms involved in well-differentiated tumors with accelerated tumor formation.21 Modifications to this model in terms of cell type or timing of induction of the transgene also allow for the development of mucinous cystic lesions.22 Inclusion of mutations in key genes of signaling cascades also found to be mutated in cancer to the KC model has shed light on the pathways involved in metastasis and the involvement of the pancreatic tumor stroma. For instance, the involvement of paracrine hedgehog signaling from the tumor stroma in resistance to gemcitabine was shown through investigation of LSL-KrasG12D; Ink4a/Arffl/fl model.23
Models of inherited pancreatic cancer syndromes
A small proportion of pancreatic cancer cases, like most cancers, is attributed to the inheritance of susceptibilizing mutations in one or more genes including BRCA2, CDKN2A/PI16INK4A, ORSS1, STK11/LKB1, and PALB2.24 Not surprisingly, mutation of these genes is linked to familial forms of many cancers, and thus, the examination of the role these genes play in pancreatic cancer initiation and progression is vital to understand what type of mutations and cooperating gene mutations are required to drive one particular cancer over another. Furthermore, Brca2 mutant mice show phenotypic differences in the context of pancreatic cancer GEMMs, dependent on the precise mutation induced and which other genetic events are involved.25 While it is not fully understood why such large differences in latency and PanIN development have been observed for models involving Brca2 mutation, it is clear that utilizing genetic susceptibility to plan screening regimens needs to take into account many factors.
Pancreatic cancer GEMMs for preclinical testing of new treatments: immunotherapies
As described in this review, pancreatic cancer has numerous mouse models that recapitulate the initiation, progression, pathology, and molecular aberrations seen in human PDAC. These models provide a valuable resource for preclinical testing of novel therapies and have several advantages over the use of syngeneic or xenograft models, such as a similar manner of progression to human pancreatic cancer from preinvasive lesions to completely invasive and metastatic disease. In addition, several GEMMs replicate the intense desmoplastic and inflammatory stroma that is a defining feature of human pancreatic cancer and a major contributor to chemoresistance. This, combined with the fact that GEMMs arise in an immunocompetent background, makes them particularly useful in testing new therapies designed to target the immune system.
Immunotherapies that inhibit the immune checkpoint molecules programmed cell death protein 1 (PD-1), programmed death ligand 1 (PD-L1), and cytotoxic T-lymphocyte-associated antigen 4 (CTLA4) in order to boost antitumor T-cell responses have had remarkable success in several cancers, particularly melanoma. However, to date, these therapies have been largely disappointing in human pancreatic cancer.26,27 This is largely due to the immunosuppressive microenvironment present in pancreatic cancer, which has proven to be a major obstacle to improving outcomes for pancreatic cancer patients. Therefore, GEMMs that display the same immunosuppressive microenvironment are proving useful in designing novel therapies aiming to increase sensitivity to immunotherapies in pancreatic cancer.
Most studies testing novel immunotherapies in pancreatic cancer have relied on the KC/KPC model of pancreatic cancer, which mimics the immunosuppressive microenvironment seen in the human disease. Similar to human pancreatic cancer, these mice also do not respond to CTLA4 and PD-L1 antibodies.28 In this model, leukocytic infiltration is present from early PanIN lesions and increases throughout disease progression to invasive adenocarcinoma.29 These immune cells mostly comprise immunosuppressive cells such as regulatory T-cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), with CD8+ T-cells rare.29
The lack of an effector T-cell response even at the earliest stages of disease makes the KC/KPC model particularly useful as it can be used to explore why pancreatic cancer is refractory to current immunotherapies as well as identify strategies to restore antitumor immunity and/or increase sensitivity to immune checkpoint inhibitors. MDSCs in particular have been found to inversely correlate with CD8+ T-cells and suppress T-cell proliferation. Depletion of MDSCs in the KPC model leads to a significant increase in CD8+ T-cells as well as a decrease in extracellular matrix, an increase in vessel patency, and increased apoptotic tumor cells.30 Tumor-derived granulocyte macrophage colony-stimulating factor (GM-CSF) has been identified as a potent inducer of MDSCs in the pancreatic tumor microenvironment, and inhibiting GM-CSF in the KPC model leads to tumor necrosis and a decrease in infiltrating MDSCs.31 Furthermore, mutant Kras has been shown to drive production of GM-CSF from pancreatic tumor cells, leading to an increase in MDSCs and a reduction in T-cell immunity.32 These studies using the KPC mouse model suggest that targeting MDSCs either directly or by inhibiting GM-CSF in pancreatic cancer patients may be a useful approach for targeting the immunosuppressive microenvironment.
Modulating the immune microenvironment by using a CD40 agonist has met with promising results. Beatty et al18 found that combining an agonistic antibody to the costimulatory molecule CD40 with gemcitabine led to a partial response rate of 24% and stable disease in a further 52% of patients with metastatic pancreatic cancer. Importantly, when this treatment was tested in the KPC mouse model to identify the underlying mechanism of tumor regression, the response rate was similar. Unexpectedly, the antitumor effect was mediated by TAMs, rather than T-cells, and importantly, stromal degradation was observed. A follow-on study in the KPC model showed that this treatment also improved the response to CTLA4 and PD1/PD-L1 checkpoint inhibitors.33 While complete responses to the combination treatment were seen in mice engrafted subcutaneously with tumor cells from KPC mice, the same responses were not seen in the spontaneous KPC model, emphasizing the complexity of the tumor microenvironment and its influence on response to immune therapy. In addition, this study also suggests that the spontaneous model more accurately predicts patient response rates compared to using cell line models.
Another study also supports the importance of TAMs in inhibiting antitumor immune responses in pancreatic cancer. Zhu et al34 identified that inhibition of colony-stimulating factor 1 receptor was able to decrease the number of TAMs as well as reprogram the remaining macrophages to promote T-cell activation and an increase in sensitivity to immune checkpoint blockade.
The pancreatic cancer stroma has been identified to play an important part in modulating the local immune response. The presence of FAP (fibroblast activation protein)-positive cancer-associated fibroblasts has been identified as a major contributor to the immunosuppression present in the pancreatic cancer tumor microenvironment and the resistance to checkpoint inhibitors.28 This was shown to be due to FAP+ cells producing the chemokine CXCL12. Targeting CXCL12 by inhibiting its receptor CXCR4 slowed tumor growth and, when this treatment was combined with a PD-L1 inhibitor, resulted in a decrease in tumor volume.
Characterization of the KC/KPC mouse models has shown many similarities to human pancreatic cancer and is arguably the strongest model available for examining mechanisms of resistance to currently used immunotherapies as well as identifying new targets to overcome immunosuppression and restore antitumor immunity. A major limitation is that these models are labor intensive and expensive to establish and use. Syngeneic cell lines may represent a good initial starting point to test new therapies; however, as seen in the study of Winograd et al,33 these models are usually more sensitive than the spontaneous model due to differences in the microenvironment, and any results from cell line studies need to be validated in the spontaneous model. However, a recent paper published by Majumder et al proposes that orthotopic implantation of KPC tumor fragments may represent another alternative to using cell lines. In this study, implanted tumor fragments showed similar metastatic spread, desmoplasia, and leukocyte infiltration to the KPC model.35 These similarities suggest that using implanted tumor fragments may be a more time and cost-effective alternative for preclinical testing of new immunotherapies; however, this remains to be validated.
Preclinical application of mouse models
Increasingly, cancer research has focused on identifying tumor subtypes related to molecular signatures to assist in predicting outcome and in the future directing targeted treatment. Recently, Bailey et al5 conducted a comprehensive integrated genomic analysis of >450 PDAC tumors and patient-derived cell lines, from which four definitive molecular subtypes were identified. This work has highlighted the need for preclinical mouse models to not only recapitulate the commonly mutated genes but also represent one of these molecular subtypes. Intriguingly, this study also examined cell lines generated from some of the KPC mouse models, relating their gene programs to the aggressive squamous subtype. This helps to identify the important mutational events driving specific gene programs enriched in the KPC cell lines.5 Future work of this nature will assist in aligning GEMMs of pancreatic cancer with these molecular subtypes to assist in preclinical studies.
Until then the importance of selecting the most appropriate mouse model for preclinical work remains a hurdle in the advancement of novel anticancer compounds. High-throughput screening is primarily conducted in vitro in cell culture utilizing either established or patient-derived cell lines; the general lack of organized tissue structure including stroma and 3D signaling as well as clonal selection that occurs in culture predisposes this technique to the identification of false positives. Understanding the importance of a molecular subtype in terms of phenotype for identification of the appropriate mouse model for testing may lead to a greater hit rate in terms of positive preclinical testing of novel compounds. Utilizing xenografts or tumor tissue chips may assist in providing the appropriate human genome and potentially the human tumor stroma; however, the requirement for immunocompromised mouse hosts limits the assessment of novel compounds. Further, the pancreatic tumor microenvironment, as outlined earlier, is known for being immunosuppressive, which further complicates the translation from preclinical mouse model to human disease.
Success in the use of PDXs to identify the most appropriate treatment regime has been promising; however, to date, the greatest success in translating treatment efficacy has been shown for tumors arising with germline mutations36 and thus in this sense of a more homogenous nature for the driving mutation. Additionally, PDX models are not easy to expand to allow study of biological mechanisms; in this sense, GEMMs offer a distinct advantage in preclinical testing where multiple manipulations of the genome can be performed to determine the importance of particular signaling cascades to treatment efficacy. This can then be translated back into designing combined therapeutic regimes targeted toward the pathways involved to improve preclinical testing as was the case for work investigating hedgehog signaling in the LSL-Trp53R127H/− model.37
Pancreatic cancer is particularly noted for its propensity to metastasize, even following surgical resection of localized tumors. GEMMs were integral in the identification of circulating tumor cells in KPC models prior to the development of invasive tumors,37 although further work is still required to confirm that these cells are able to generate metastases in vivo. However, this still demonstrates the capacity to investigate the utility of novel or existing compounds to combat the metastatic process in a preclinical setting.
Preclinical applications of xenografts for precision medicine
The identification of the complex mutational spectrum involved in pancreatic cancer has led to the suggestion that, like other cancers showing intratumoral heterogeneity, the failure of treatment might be attributed to the specific targeting of only one driver mutation/pathway by that treatment and the generation of multiple mechanisms of drug resistance.38 Therefore, it follows that concurrent treatment with a range of therapeutics targeting multiple mutations and pathways should have greater efficacy (if tolerable) due to the limited capacity for untargeted tumor cells to receive a competitive advantage in the absence of the effectively treated tumor bulk.
While GEMMs are particularly advantageous for identifying the role of individual mutations, the necessity to modify all cells in the same way is a limitation in that it does not reflect the normal pancreatic tumor heterogeneity. Xenograft models are either created from injecting patient-derived cell lines into immunocompromised mice or from implanting a fragment of the tumor (PDX) into these animals. The use of patient-derived cell lines has clear advantage in the capacity to generate hundreds of animals from the same cell line; however, it lacks the heterogeneity of a patient tumor as well as the interactions with the stroma of the tissue of origin. Utilizing tumor fragments provides a more representative model, thereby overcoming the aforementioned disadvantages, however is severely limited by the requirement for the host animal to be immunosuppressed and the site of engraftment.39 Despite this, recent studies have shown the power of utilizing PDX models in a preclinical setting as a means to identify the best therapy for an individual whose tumor is refractory to gemcitabine.40 Further recent work using exome sequencing has shown that along with the tumor architecture, the genetic signature is maintained for at least three passages, giving scope for greater investigation of prognostic biomarkers to therapeutic response.41 This would ultimately reduce the requirement to perform PDX model testing for every individual and instead only require analysis of the primary tumor to direct therapy.
PDX models can be further enhanced through inserting the tumor fragment into the subrenal capsule (SRC) rather than subcutaneous. This has the advantage of greater perfusion providing more rapid tumor growth, however still maintains the disadvantage of requiring immunocompromised animals as well as adding the requirement for advanced monitoring systems as well as added complications from a more invasive procedure.42,43 Nonetheless, the SRC model has been effectively used in the development of trastuzumab for the treatment of Her2+ breast cancer as well as in preclinical studies of pancreatic cancer.44 The rapid tumor growth and enhanced perfusion of SRC implantation increase the rate of metastasis; however, this is still dependent on the inherent characteristics of the tumor. For example, Xue et al45 found that SRC xenografts that responded to gemcitabine were derived from patients who did not experience recurrence, while those that were refractory to gemcitabine were from patients who developed metastasis during the study period.
PDX models are being increasingly used in cancer research not only for the assessment of therapeutic efficacy but also to develop targeted delivery vehicles. PDX models are clearly showing promise as a valuable tool in evaluating the efficacy of novel treatments and have the advantage of recapitulating the natural genetic makeup of the original human patient tumor, even after passaging in vitro and in vivo, while allowing novel drug testing in an in vivo setting.41,46 This provides valuable information that can go toward the design of more effective human clinical trials to further test novel treatments for cancers such as pancreatic cancer.41 Although the use of PDX models in pancreatic cancer research is still relatively in its infancy, there are a number of studies that have utilized these models in testing the efficacy of novel drug leads against pancreatic cancer.
PDX models to assess therapeutic efficacy
Recently, using a combination of structural variation, mutational signatures, and gene mutations to define putative biomarkers of therapeutic responsiveness for platinum-based chemotherapy, Waddell et al identified that genomic instability cosegregated with inactivation of DNA maintenance genes (BRCA1, BRCA2, or PALB2) and a mutational signature of DNA damage repair deficiency. Using a PDX model assessment of mutations in BRCA pathway component genes and surrogate measures of defects in DNA maintenance (genomic instability and the BRCA mutational signature) have potential implications for therapeutic selection for pancreatic cancer. This approach enables the identification of those that may significantly respond in subgroups that are not well defined, with improved survival expected in patients with germline BRCA1 and BRCA2 mutations who receive platinum-based therapies and would significantly alter current treatment approaches to pancreatic cancer and improve overall outcomes.7 Diagnostic-therapeutic genomic-PDX approaches continue to evolve, rapidly becoming more translational to provide new opportunities in the clinic.
Kim et al also used a molecular phenotype-guided PDX model to investigate the efficacy of nab-paclitaxel delivery and SPARC (secreted protein acidic and rich in cysteine) expression and identified that specific tumor delivery of nab-paclitaxel is not directly related to SPARC expression, and nab-paclitaxel does not deplete tumor stroma, which is of particular importance as pancreatic cancer is highly desmoplastic and reliant on the stroma to drive its progression, as well as creating a barrier of fibrous tissue preventing transport of chemotherapeutics into the tumor.47
Further to this, Roy Chaudhuri et al investigated the effect of smoothened inhibitors of hedgehog signaling (sHHI) to overcome the stromal barrier and promote neovascularization and enhance tumor permeability to low-molecular weight compounds in a PDX model of pancreatic cancer. The authors demonstrated that sHHI can enhance tumor deposition and efficacy of drug-containing nanoparticles consisting sterically stabilized liposomes containing doxorubicin. This treatment approach significantly increased median tumor progression time providing proof of concept that short-term sHHI treatment sequenced with nanoparticulate drug carriers constitutes a potential strategy to enhance efficacy of pancreatic cancer therapies.48
The efficacy of tumor-targeting Salmonella typhimurium AR-1 (AR-1) compared to gemcitabine on pancreatic cancer was tested using pancreatic cancer PDX models. AR-1 treatment led to significantly reduced tumor weight and showed increased efficacy compared to gemcitabine.49 The efficacy of AR-1 was then further tested using PDX models following treatment with anti-VEGF therapy (bevacizumab) on VEGF-positive pancreatic cancer tumors. Treatment with gemcitabine/bevacizumab followed by AR-1 led to a significant reduction in tumor weight compared to gemcitabine/bevacizumab alone in the PDXs and in the VEGF-positive pancreatic cancer cell line MiaPaCa2. However, neither treatment was as effective in VEGF-negative tumors/cell lines.50 Clearly, studies using PDX models warrant future clinical testing for more targeted treatments for pancreatic cancer patients.
As most preclinical PDX models target actionable mutations and are generated using tumor fragments obtained during surgical resection, it is important to evolve these models to use less invasive and more timely means and to develop genomically informed therapies in patient tumor models prior to patient treatment. For example, Allaway et al developed PDX models from fine-needle aspiration biopsies obtained from primary PDAC at the time of diagnosis and characterized both primary and metastatic sites from one patient. While still in its infancy, PDX models can now be constructed from fine-needle aspiration biopsies of pancreatic cancer, which in turn can enable genomic characterization and identification of informed therapies prior to aggressive surgical and chemotherapeutic intervention.51
Gao et al recently demonstrated both the reproducibility and the clinical translatability of high-throughput screening using patient-derived tumor xenografts to predict preclinical and clinical trial drug response. This approach identified associations between a genotype and a drug response and established mechanisms of resistance, outlining its potential in rapidly improving preclinical evaluation of treatment modalities (Figure 1).52
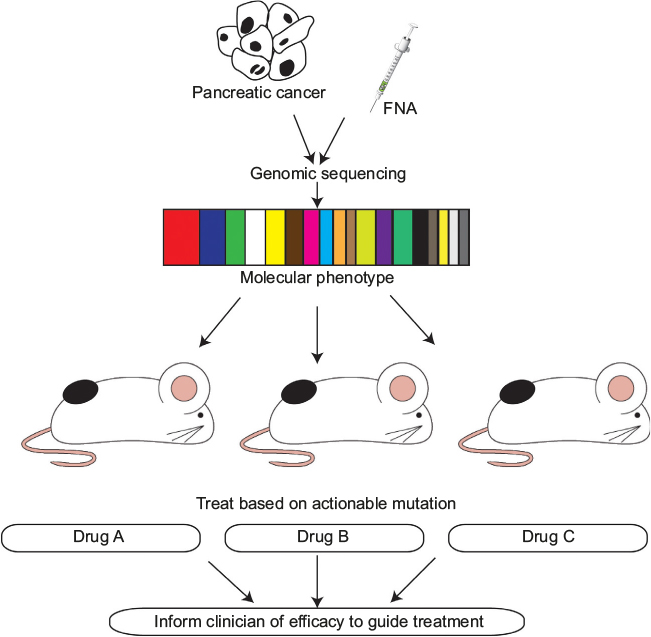 | Figure 1 Schematic outlining the use of PDXs to target actionable mutations identified in an individual’s pancreatic tumor and FNA samples to provide the clinician with a more informed decision on the potential efficacy of the selected therapeutic agent. Abbreviations: PDXs, patient-derived xenografts; FNA, fine-needle aspiration. |
PDX models to develop targeted delivery vehicles
As discussed previously, a key issue affecting the efficacy of chemotherapeutic agents on pancreatic cancer is the fibrous desmoplastic stroma. In an effort to circumvent this, Indolfi et al have exploited the PDX model to develop an implantable poly(lactic-co-glycolic)-based biodegradable device to linearly release high doses of chemotherapeutic drugs resulting in an increase in suppression of tumor growth compared to systemic delivery. By providing high concentrations of chemotherapeutic compounds locally at the tumor site, this approach could reduce the need for repeated procedures in advanced pancreatic cancer patients to debulk the tumor mass or stent the obstructed bile duct, thereby improving quality of life and outcomes for pancreatic cancer patients.53
Conclusion
Mouse models of pancreatic cancer are essential for the preclinical assessment of novel therapeutics or the repositioning of existing ones. Selection of the most appropriate model for the stage of testing as well as the experimental intent is imperative to the successful translation to human studies. Models allow a greater understanding of the biology of disease; however, all suffer limitations for various applications. Ultimately, mouse models can allow the identification of novel targets, predictive assessment of therapeutic benefit, and inform clinical trial design in terms of the expected responsive patient target subgroup. The advent of molecular technologies such as CRISPR/Cas are greatly enhancing the speed and cost-effectiveness of genetic and epigenetic manipulation of mice, and as such, more in depth mutational studies can be conducted. In addition, inducible systems offer the capacity to mimic the progressive nature in terms of acquisition of mutations and the ability to modulate both the tumor and the microenvironment through cell-specific promoters. Therefore, it is likely that future GEMMs of pancreatic cancer will focus on the complex recapitulation of the molecular subtypes identified by integrated genomic analysis of PDAC patients that can be used for preclinical assessment and finally provide some meaningful improvements in patient care. Until then, the recent advances in implementation of PDX models for preclinical assessment coupled with the study of novel immunotherapies in GEMMs offer hope for the translation of novel therapeutic strategies for pancreatic cancer.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–586. | ||
Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28(8):977–987. | ||
Wilentz RE, Albores-Saavedra J, Hruban RH. Mucinous cystic neoplasms of the pancreas. Semin Diagn Pathol. 2000;17(1):31–42. | ||
Bailey P, Chang DK, Nones K, et al; Australian Pancreatic Cancer Genome Initiative. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531(7592):47–52. | ||
Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. | ||
Waddell N, Pajic M, Patch AM, et al; Australian Pancreatic Cancer Genome Initiative. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518(7540):495–501. | ||
Biankin AV, Waddell N, Kassahn KS, et al; Australian Pancreatic Cancer Genome Initiative. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491(7424):399–405. | ||
Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis. 2014;35(7):1451–1460. | ||
Bell RH Jr, Memoli VA, Longnecker DS. Hyperplasia and tumors of the islets of Langerhans in mice bearing an elastase I-SV40 T-antigen fusion gene. Carcinogenesis. 1990;11(8):1393–1398. | ||
Ornitz DM, Hammer RE, Messing A, Palmiter RD, Brinster RL. Pancreatic neoplasia induced by SV40 T-antigen expression in acinar cells of transgenic mice. Science. 1987;238(4824):188–193. | ||
Ornitz DM, Palmiter RD, Messing A, Hammer RE, Pinkert CA, Brinster RL. Elastase I promoter directs expression of human growth hormone and SV40 T antigen genes to pancreatic acinar cells in transgenic mice. Cold Spring Harb Symp Quant Biol. 1985;50:399–409. | ||
Colvin EK, Scarlett CJ. A historical perspective of pancreatic cancer mouse models. Semin Cell Dev Biol. 2014;27:96–105. | ||
Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–450. | ||
Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–483. | ||
Plentz R, Park JS, Rhim AD, et al. Inhibition of gamma-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2009;136(5):1741.e6–1749.e6. | ||
Funahashi H, Satake M, Dawson D, et al. Delayed progression of pancreatic intraepithelial neoplasia in a conditional Kras(G12D) mouse model by a selective cyclooxygenase-2 inhibitor. Cancer Res. 2007;67(15):7068–7071. | ||
Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. | ||
Frese KK, Neesse A, Cook N, et al. nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer. Cancer Discov. 2012;2(3):260–269. | ||
Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–120. | ||
Kojima K, Vickers SM, Adsay NV, et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007;67(17):8121–8130. | ||
Izeradgene K, Combs C, Best M, et al. KrasG12D and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11(3):229–243. | ||
Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. | ||
Hruban RH, Canto MI, Goggins M, Schulick R, Klein AP. Update on familial pancreatic cancer. Adv Surg. 2010;44:293–311. | ||
Cassidy LD, Liau SS, Venkitaraman AR. Chromosome instability and carcinogenesis: insights from murine models of human pancreatic cancer associated with BRCA2 inactivation. Mol Oncol. 2014;8(2):161–168. | ||
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | ||
Royal RE, Levy C, Turner K, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–833. | ||
Feig C, Jones JO, Kraman M, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110(50):20212–20217. | ||
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–9527. | ||
Stromnes IM, Brockenbrough JS, Izeradjene K, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut. 2014;63(11):1769–1781. | ||
Bayne LJ, Beatty GL, Jhala N, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. | ||
Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. | ||
Winograd R, Byrne KT, Evans RA, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3(4):399–411. | ||
Zhu Y, Knolhoff BL, Meyer MA, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–5069. | ||
Majumder K, Arora N, Modi S, et al. A novel immunocompetent mouse model of pancreatic cancer with robust stroma: a valuable tool for preclinical evaluation of new therapies. J Gastrointest Surg. 2016;20(1):53–65. | ||
Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10(1):3–8. | ||
Rhim AD, Oberstein PE, Thomas DH, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735–747. | ||
Burrell RA, Swanton C. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol. 2014;8(6):1095–1111. | ||
Vonlaufen A, Phillips PA, Xu Z, et al. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 2008;68(19):7707–7710. | ||
Hidalgo M, Amant F, Biankin AV, et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014;4(9):998–1013. | ||
Mattie M, Christensen A, Chang MS, et al. Molecular characterization of patient-derived human pancreatic tumor xenograft models for preclinical and translational development of cancer therapeutics. Neoplasia. 2013;15(10):1138–1150. | ||
Bogden AE, Haskell PM, LePage DJ, Kelton DE, Cobb WR, Esber HJ. Growth of human tumor xenografts implanted under the renal capsule of normal immunocompetent mice. Exp Cell Biol. 1979;47(4):281–293. | ||
Cutz JC, Guan J, Bayani J, et al. Establishment in severe combined immunodeficiency mice of subrenal capsule xenografts and transplantable tumor lines from a variety of primary human lung cancers: potential models for studying tumor progression-related changes. Clin Cancer Res. 2006;12(13):4043–4054. | ||
Carter P, Presta L, Gorman CM, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci USA. 1992;89(10):4285–4289. | ||
Xue A, Julovi SM, Hugh TJ, et al. A patient-derived subrenal capsule xenograft model can predict response to adjuvant therapy for cancers in the head of the pancreas. Pancreatology. 2015;15(4):397–404. | ||
Tignanelli CJ, Herrera Loeza SG, Yeh JJ. KRAS and PIK3CA mutation frequencies in patient-derived xenograft models of pancreatic and colorectal cancer are reflective of patient tumors and stable across passages. Am Surg. 2014;80(9):873–877. | ||
Kim H, Samuel S, Lopez-Casas P, et al. SPARC-independent delivery of nab-paclitaxel without depleting tumor stroma in patient-derived pancreatic cancer xenografts. Mol Cancer Ther. 2016;15(4):680–688. | ||
Roy Chaudhuri T, Straubinger NL, Pitoniak RF, et al. Tumor-priming smoothened inhibitor enhances deposition and efficacy of cytotoxic nanoparticles in a pancreatic cancer model. Mol Cancer Ther. 2016;15(1):84–93. | ||
Hiroshima Y, Zhao M, Maawy A, et al. Efficacy of Salmonella typhimurium A1-R versus chemotherapy on a pancreatic cancer patient-derived orthotopic xenograft (PDOX). J Cell Biochem. 2014;115(7):1254–1261. | ||
Hiroshima Y, Zhang Y, Murakami T, et al. Efficacy of tumor-targeting Salmonella typhimurium A1-R in combination with anti-angiogenesis therapy on a pancreatic cancer patient-derived orthotopic xenograft (PDOX) and cell line mouse models. Oncotarget. 2014;5(23):12346–12357. | ||
Allaway RJ, Fischer DA, de Abreu FB, et al. Genomic characterization of patient-derived xenograft models established from fine needle aspirate biopsies of a primary pancreatic ductal adenocarcinoma and from patient-matched metastatic sites. Oncotarget. 2016;7(13):17087–17102. | ||
Gao H, Korn JM, Ferretti S, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21(11):1318–1325. | ||
Indolfi L, Ligorio M, Ting DT, et al. A tunable delivery platform to provide local chemotherapy for pancreatic ductal adenocarcinoma. Biomaterials. 2016;93:71–82. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.