")
Back to Archived Journals » Advances in Genomics and Genetics » Volume 5
Anaplastic thyroid cancer – an overview of genetic variations and treatment modalities
Authors Reddi H, Kumar A, Kulstad R
Received 7 June 2014
Accepted for publication 23 August 2014
Published 16 January 2015 Volume 2015:5 Pages 43—52
DOI https://doi.org/10.2147/AGG.S53448
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr John Martignetti
Honey V Reddi,1 Anirudh Kumar,2 Roger Kulstad3
1Transgenomic, Inc., New Haven, CT, USA; 2University of Toronto, Toronto, ON, Canada; 3Division of Endocrinology, Marshfield Clinic, Marshfield, WI, USA
Abstract: Anaplastic thyroid carcinoma (ATC) is a rare but highly aggressive malignancy that accounts for about 1%–2% of all thyroid cancer diagnoses but is responsible for up to 30%–40% of thyroid cancer deaths. ATCs are poorly differentiated tumors that develop on the background of preexisting, often undiagnosed, papillary thyroid carcinoma or follicular thyroid carcinoma, through progressive accumulation of changes in several oncogenic and tumor suppressor pathways, including p53, RAS, RAF, Wnt-β-catenin and the PTEN-AKT pathways. Consequently, the 1-year survival rate after diagnosis ranges from 5% to 15%. Current therapeutic approaches are aimed at common late oncogenic changes and involve inhibition of MAPK and PI3K cell proliferation pathways or restoration of p53 and PTEN tumor suppressor pathways. Since single-modality therapy has limited effect on anaplastic thyroid cancer, aggressive multimodal treatments are now the treatment of choice, in spite of which, the mean survival time from diagnosis to death continues to remain at about 6 months. The current review attempts to summarize the genetics involved in the development and progression of ATC and provides some insight into the therapeutic options being evaluated for this aggressive cancer.
Keywords: genetic alterations, treatment strategies, diagnostic testing
Introduction
Thyroid cancer incidence has been slowly increasing for the past few decades.1 Compared with the 20,000 cases observed in 2003, in 2014 it is expected that an estimated 62,980 new cases of thyroid cancer will be diagnosed in the United States, with about 1,890 deaths. Current statistics make it the ninth most common cancer in women, with 213,000 cases worldwide,2 representing 3.8% of all new cases,3 with a projection of becoming the fourth most common cancer diagnosis by 2030.4 A significant majority of thyroid malignancies are differentiated tumors which include those arising from the follicular thyrocytes such as papillary thyroid carcinoma (PTC) or follicular thyroid carcinoma (FTC), while medullary thyroid cancer arises from the parafollicular or calcitonin-producing C-cells. Of the different morphotypes that occur within the follicular cells, PTC is the most common, followed by FTC, accounting for 80% and 20%–40% of the cases, respectively.5 Anaplastic thyroid carcinoma (ATC) is the rarest but most aggressive morphotype, accounting for ~1%–2% of all thyroid cancer cases6 and ~20%–40% of the annual thyroid cancer deaths in the USA.7,8
ATC comprises undifferentiated tumors that arise as such or by dedifferentiated progression from PTC or FTC9 (Figure 1) and are observed in the sixth or seventh decade of life. Progression from a normal thyrocyte to an aggressive morphotype such as ATC is thought to occur due to the progressive accumulation of hits to multiple oncogenic pathways.10,11 ATCs are often found in association with histologically well differentiated thyroid cancer elements,12 while showing persistent genetic changes of differentiated carcinoma, coupled with additional abnormalities in several canonical pathways, including the PTEN-AKT, retinoblastoma, and p53 pathways.9,13 During the transformation from PTC or FTC to ATC, thyroid tumors lose their differentiated characteristics,14 such as the ability to produce thyroglobulin and their avidity for radioactive iodine, and develop more aggressive cancer characteristics.15
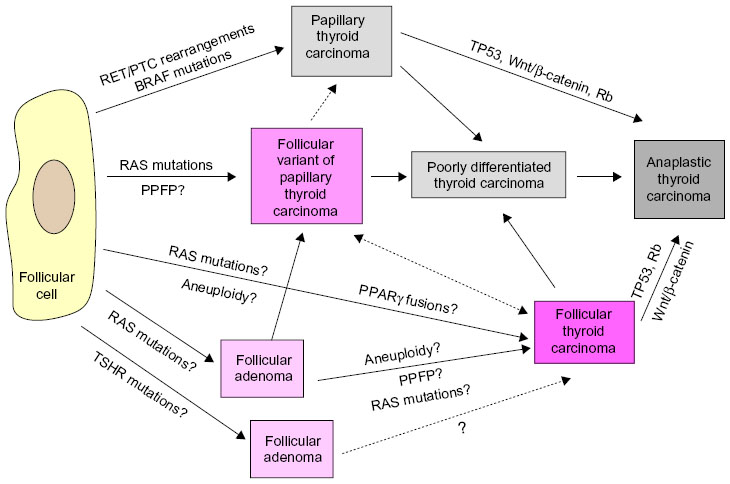 | Figure 1 Schematic of progression of a normal thyrocyte to ATC. Development of ATC is thought to occur as such or from differentiated PTC or FTC to an undifferentiated aggressive morphotype due to the progressive accumulation of hits to multiple oncogenic pathways. |
Most of the ATC tumors are inoperable at the time of diagnosis and prove to be fatal. The average survival rate is 5%–15%16,17 at 3 years.18 The mean survival time from diagnosis to death of 6 months,19 even with aggressive, multimodal therapy involving surgery, radiotherapy, and chemotherapy,7,20–22 probably due to synchronous lung metastases in 20%–50% of cases.18 Different therapeutic approaches have been used in the recent years with the hope of leading to at least reduced tumor mass and palliative control of the cancer. Small molecule inhibitors that target oncoproteins in key pathways such as mitogen-activated protein kinase (MAPK) and PI3K,23 viro-therapies including replacement of tumor suppressor genes such as TP53, and administration of oncolytic viruses are being evaluated in preclinical studies as well as clinical trials. Considering the aggressive nature of this cancer, and the minimal treatment window, there is an imperative need for the development and evaluation of novel treatment strategies.
Genetics of ATC
Anaplastic thyroid tumors exhibit considerable derangement that leads to uncontrolled cellular proliferation and the development of genomic instability.24–26 Multiple studies using arrayCGH (array comparative genomic hybridization) has shown that besides copy number changes,8,27 chromosomal gains and losses as well as gene rearrangements involving the RET proto-oncogene are also present in ATC.28 In an attempt to identify novel markers or therapeutic targets, immunohistochemistry as a tool has been used extensively to evaluate loss or overexpression of various signaling molecules.14 Elevated expression of tumor suppressors such as TP53, signaling molecules including SOX2,8,29 laminin-5γ-2 (LAMC2),30 FOXM1,31 or oncogenes such as FOXA132 are believed to be evidence for deregulation and may reflect loss of function of the relevant pathways, serving as potential therapeutic targets.
Currently no studies evaluating ATC are listed on The Cancer Genome Atlas (TCGA) website. TCGA has the evaluation of ~500 thyroid cancer genomes, including PTC and FTC, but not ATC, deposited in their public website. This should provide a more detailed, multidimensional genomic characterization of the more prevalent PTC and FTC. Limited studies evaluating ATC cell lines or patient samples have been reported. Altered expression of genes using microarrays on ATC-derived cell lines demonstrates the upregulation of DSTN, HSPA8, stathmin, LDH-A, ATP5A1, PSM B6, B23, HDP-1, and LDH-B, while TG, PBP, and FES are downregulated.33 Evaluation of thyroid-specific messenger RNA (mRNA) expression levels across all thyroid tumor types determined that NIS and PAX8 mRNA levels were significantly reduced in all types of thyroid cancer, especially in ATCs.34 Limited epigenetic studies looking at microRNA and promoter methylation have also been performed on ATC tumors with some promise, but they are yet to come to the forefront as potent diagnostic markers.28
ATCs are typified by hits to multiple signal transduction and cell cycle pathways (Table 1) such as TP53, PTEN-PI3K/AKT, RAS/RAF/MAPK, and Wnt/β-catenin (Figure 2A). Mutations in RAS (N- or H-) and BRAF which are also present in PTC suggest that they could be early oncogenic events, compounded by those in TP53, PI3K, and β-catenin resulting in progression to the aggressive dedifferentiated ATC phenotype.16,35 All these pathways intersect at some point with each other (Figure 2). Dysregulation of any one of these pathways results in cellular disorganization, increased proliferation, and survival culminating in aggressive disease.36 Recent evidence has also demonstrated the presence of mutations in novel signaling molecules (Table 2 and Figure 3B) such as CDK1 family members,37 MET,38 RASAL1,39 ALK,40 AXIN,41 IDH1,42 and TERT promoter.43
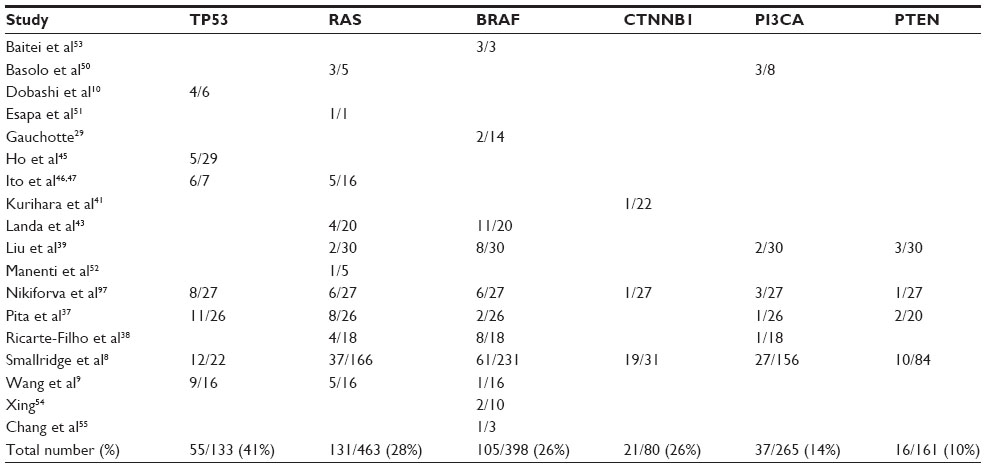 | Table 1 Mutations in canonical pathways observed in anaplastic thyroid carcinomas |
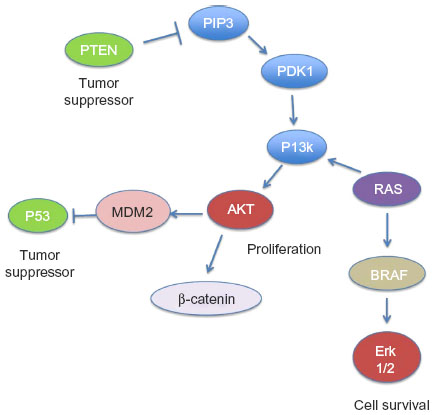 | Figure 2 Schematic of the different tumor suppressor and oncogenic pathways in anaplastic thyroid carcinoma and their points of intersection within the cell. |
 | Table 2 Novel mutations observed in anaplastic thyroid carcinomas – single reports |
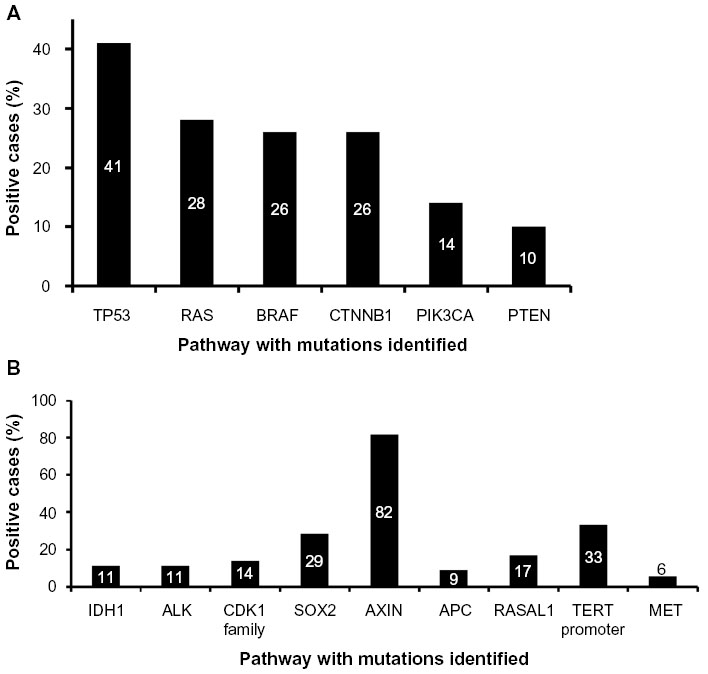 | Figure 3 Reported incidence of the different mutations that occur in anaplastic thyroid carcinoma. (A) Mutations in canonical tumor suppressor and oncogenic pathways such as p53, RAS, BRAF, Wnt/β-catenin, PTEN, and AKT. (B) Novel mutations that have been recently reported in single-cohort studies. Occurrence is reported as a percentage (%) calculated based on the number of positive cases identified, per total number of cases evaluated. |
Mutations in canonical signaling pathways
TP53 pathway
The TP53 pathway is the most common pathway found to be inactivated in most human cancers. TP53 is a transcription factor that functions as a tumor suppressor by regulation of several genes such as p21 within the cell, performing a broad range of functions, including DNA repair, metabolism, cell cycle arrest, apoptosis, and senescence.44 Of all the genetic changes that are observed to occur in ATC, TP53 mutations are the most common, seen in about 17%–80% of ATCs,8–10,37,45–48 averaging to 44% of cases (Table 1 and Figure 3A). In addition to direct mutations in TP53 that deregulate its tumor suppressor function, mutations in molecules that negatively impact TP53 function such as HMGA1 (high-mobility group A1) and p73, or proteins fostering p53 protein degradation, like MDM2, are also observed.24
RAS/RAF pathway
The RAS/RAF pathway plays a fundamental role in the regulation of cell proliferation and survival, and its aberrant activation results in tumorigenesis. RAS and RAF are part of the MAPK pathway. RAS is also an important regulator of the PI3K/Akt pathway through its interactions with the RAS-binding domains of PI3K subunits. Both RAS and RAF are present downstream of the EGFR (epithelial growth factor receptor) within the same pathway. Activated RAS triggers the protein kinase activity of RAF kinase, which phosphorylates and activates MEK (MEK1 and MEK2). MEK phosphorylates and activates a MAPK. Mutation in any one of the molecules results in their constitutive activation and a consequential increase in cell proliferation.49 Mutations in RAS8,9,38,39,43,47,48,50–52 and BRAF8,9,29,37–39,43,48,53–55 are observed in about 28% (126 of 447) and 26% (105 of 398) of cases of ATCs, respectively (Table 1 and Figure 2A), making them the second most commonly observed mutations after TP53 in these tumors.
Wnt/β-catenin pathway
Epithelial-cadherin (E-cadherin) is a transmembrane glycoprotein, the cytoplasmic portion of which is linked to the actin cytoskeleton of the cell by the catenins, resulting in cell adhesion. Decreased expression or even loss of E-cadherin/β-catenin expression, either quantitatively or qualitatively, leads to cellular disorganization and detachment resulting in the development of epithelial tumor invasion and metastasis.56 Mutations in β-catenin are believed to occur late in the progression cycle and are considered to be involved in the transformation from differentiated thyroid cancer to ATC.35 About 26% (21 of 80) of ATCs are shown to have mutations in β-catenin8,41,48 (Table 1 and Figure 2A).
PI3K/Akt-PTEN pathway
As with the RAS/RAF pathway, the PI3K/Akt pathway also plays a pivotal role in the regulation of cell growth, division, and survival, and therefore, when disarranged, results in tumorigenesis (Figure 2). Signaling of this pathway involves PI3K-catalyzed generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3), which recruits phosphoinositide-dependent kinase 1 (PDK1). PDK1 subsequently activates the protein kinase Akt through phosphorylation at the cell membrane, which in turn phosphorylates and regulates downstream effector proteins and promotes cell proliferation and survival.57 Phosphatase and tensin homolog deleted from chromosome 10 (PTEN) negatively regulates this pathway by degrading PIP3 and terminating the signaling.58 In contrast to the prevalence of TP53 and RAS/RAF mutations in ATC, only a small component of carcinomas, 14% (37 of 265) and 10% (16 of 161), harbor mutations in PIK3CA and PTEN respectively.8,37–39,48,50 (Table 1 and Figure 2A).
Clinical testing and molecular diagnostics for thyroid cancer
Evaluation of thyroid nodules involves a multistep process including clinical assessment by a physician, measurement of thyrotropin (TSH), ultrasound evaluation, and biopsy of nodules selected according to size and ultrasound characteristics based on recommendations of the American Thyroid Association and The Association of Clinical Endocrinologists.59 For nodules that require biopsy, cytological analysis of fine needle aspirate biopsy (FNAB) is standard of care for early detection of malignancy. Cytopathologists can reliably diagnose benign and overtly malignant nodules using standard microscopy techniques. Unfortunately, cytopathology techniques are inherently unable to classify so-called indeterminate nodules as benign or malignant because this requires visualization of the nodule margins.
The prevalence of indeterminate nodules varies by report but is generally accepted to be 15%–35% of all nodules.60 This category includes the subtypes of Atypia of Undetermined Significance or Follicular Lesion of Undetermined Significance (AUS/FLUS), (Suspicious for) Follicular or Hürthle Cell Neoplasm (FN/HCN), and Suspicious for Malignancy.61–63 The pre-test probability of malignancy for the Suspicious for Malignancy category is close to 85%,64 and thus, surgery is usually recommended for these patients. However, only 15%–35% of other indeterminate nodules prove to be malignant on histological evaluation,62 driving the need for molecular testing to prevent unnecessary surgery to the patient.
Mutation panels using different methodologies are commercially available for pre-operative evaluation of FNAB by various companies such as Asuragen (miRInform™), Qiagen (qBiomarker; 13 genes), Quest (4 markers), University of Pittsburg Medical Center (Thyroseq; 15 genes), and Veracyte Inc. (Affirma™ Gene Expression Classifier, which tests for 142 genes). Most panels evaluate the presence of point mutations in TP53, AKT, PTEN, RB, CTNNB1, RAS, and BRAF, and gene rearrangements such as RET/PTC and PAX8/PPARγ. However, the panels differ in the way they approach malignancy risk. Most are designed to detect certain high risk mutations that may be associated with malignant disease. In these cases, a positive test would drive the decision for surgery or perhaps justify a more extensive resection (ie, total thyroidectomy, instead of a unilateral lobectomy). A negative test, however, would not rule out malignancy, and hence diagnostic surgery would still be required.65
The Veracyte Inc. Affirma™ Gene Expression Classifier approaches these nodules differently. This panel sacrifices specificity and positive predictive value for the sake of sensitivity. The panel tests for the expression of 142 markers. A nodule classified as “low risk” can be followed clinically and with serial ultrasonography in lieu of surgery. Nodules classified as “suspicious” are still resected albeit with the a priori knowledge that there is still a high probability of benign histology.
Since significant progress has not been made in the identification of novel diagnostic markers specific to ATC due to the rarity of this cancer, the limited studies28 that have identified some potential markers will hopefully make the cut to diagnostic markers in the near future. Meanwhile, current genetic testing of thyroid nodules is much more applicable to the detection of differentiated cancers, given that ATC does seem to develop on the background of PTC and FTC, genetic testing is indeed beneficial in early detection of these morphotypes of thyroid cancer. The aim being, the ability to detect these morphotypes early enough to allow for surgical and therapeutic intervention, thereby potentially preventing their progression to aggressive, fatal ATC.
Preclinical and clinical studies evaluating potential therapies
Most studies evaluating therapies for ATC have concentrated on the use of small-molecule inhibitors, viro-therapeutics including oncolytic viruses, and gene replacement vectors, which can inhibit the growth of ATC cell lines in culture and in mouse tumor explant models.66–69 In the recent past, a number of transgenic mice have been developed that mimic ATC in vivo70–73 and serve as much more clinically relevant models for preclinical testing. These models have primarily been used to elucidate the hypothesis that ATCs arise as a progression from PTC or FTC due to hits to multiple signaling pathways and therefore provide the closest simulation environment reflective of direct in human testing.
A significant number of preclinical studies and clinical trials have been conducted evaluating the therapeutic efficacy of small-molecule inhibitors of transcription factors and signaling pathways both in vitro and in vivo. Studies using the tyrosine kinase inhibitors sorafenib, combretastatin, and axitinib in ATC have shown partial encouraging results but are hindered by small numbers.23 These studies have been extensively reviewed12,23,74 and will therefore not be discussed in detail in the current manuscript.
Radioactive iodine is useful for the treatment of well differentiated thyroid cancers, though it has not been useful in the treatment of ATC. There are no randomized trial data to conclusively demonstrate that survival is improved in ATC patients in response to treatment with systemic chemotherapy. There have been several trials evaluating strategies to redifferentiate radioiodine-resistant well differentiated thyroid cancers in an attempt to render these tumors responsive to this treatment modality.75,76 A recent study by Ho et al77 successfully used selumetinib, a selective allosteric MEK1 and MEK2 inhibitor as a means to reestablish radioiodine avidity in non-iodine avid advanced papillary thyroid cancer. The activation of the MAPK or MEK is thought to play a critical role in the pathogenesis of radioiodine resistance. Activation of this pathway inhibits the expression of thyroid hormone biosynthesis genes including the sodium-iodine symporter and thyroid peroxidase which facilitate iodine uptake and organification, respectively.78–80 Using I-124 PET-CT scintigraphy for precise quantification of iodine uptake, the investigators were able to show that selumetinib increased lesional uptake of radioactive iodine in 12 of 20 patients (four of nine patients with BRAF mutations, and five of five patients with NRAS mutations). Of the 12 patients who showed increased lesional uptake, eight reached the dosimetry threshold for radioiodine therapy. Five of these eight patients had confirmed partial response to therapy, and three had clinically stable disease at the end of the study period. While this study applies to patients with papillary, not anaplastic thyroid cancer, it serves as an important proof of concept that re-differentiation strategies may hold promise as treatments for thyroid cancers including ATC.
Oncolytic viruses on the other hand have been extensively used as therapy for several cancers;81 however, limited efforts have thus far gone into understanding their potential for the therapy of ATC. Adeno82,83 measles,69 herpes simplex,84,85 and vaccinia viruses86,87 have been tested in vitro as well as in vivo in mouse explant models (Table 3). Conditionally replicative viruses include those that target cells with specific tumor suppressor pathways that have been88–90 or are directed to negatively impact tumor growth such as targeting of angiogenesis.91 Non-replicative adenoviruses, used as vectors to replace tumor suppressor genes such as TP53,92 TTF1,93 or to express genes with therapeutic potential such as the sodium iodide symporter (NIS) to enable dual targeting,69,86 have also been evaluated (Table 3).
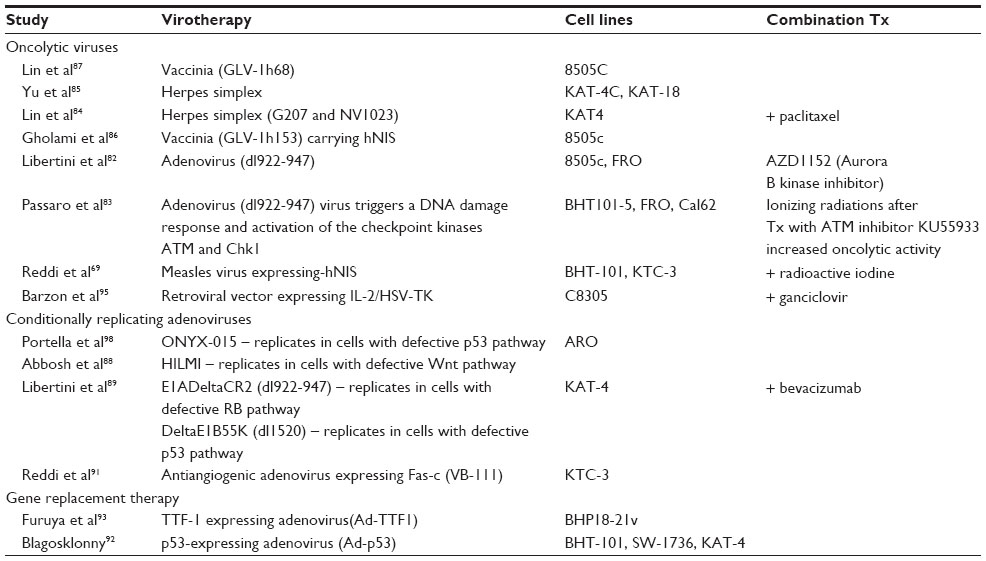 | Table 3 Preclinical studies evaluating the efficacy of virotherapies in anaplastic thyroid carcinoma cell lines using mouse explant models |
As single-modality therapy has been shown to have a limited effect on ATC, multimodality therapy has become the treatment of choice in recent years. Latest studies have therefore looked at dual therapeutics with combinations of viruses, along with either small-molecule inhibitors or additional therapies such as radiation or radioiodine therapy. Administration of an ATM (ataxia telangiectasia mutated) inhibitor KU55933 increases the oncolytic activity of the adenovirus dl922-947 in combination with ionizing radiations.83 Oncolytic viruses (measles, vaccinia, and lentiviruses) expressing the NIS gene have been used to induce cell death as well as facilitate noninvasive imaging and radioiodine therapy.69,86,94 Combinatorial therapeutic options using small molecule inhibitors such as paclitaxel,84 bevacizumab,89 gancliclovir,95 and aurora kinase inhibitor AZD1152,82 along with independent virus treatments, have also been evaluated preclinically.
In spite of the demonstrated efficacy of virotherapies in preclinical studies, sadly there has been an incredible vacuum in terms of translation of these potential therapies for clinical testing in ATC patients. Possible reasons could be the limited efficacy of the viruses after systemic administration or the potential biohazard implications. Small-molecule inhibitors continue to gain momentum in use, with the hope of leading to at least reduced tumor mass and palliative control of the cancer.
Current treatment regimens and management of ATC patients
Current therapeutic strategies for ATC focus on the use of combination therapy with surgery, chemotherapy using taxanes, anthracyclines, and platins, and external beam radiation therapy. Given the almost fatal progression of the cancer, the American Thyroid Association has developed the first comprehensive guidelines for the management of ATC patients.96 Rapid evaluation and establishment of treatment goals are extremely important for optimum patient management and requires a multidisciplinary team approach. The guidelines include 65 recommendations, which were derived based on extensive evaluation of available literature on the clinicopathological features of ATC, survival times, and therapeutic response rates. The guidelines suggest that patients with stage IVA/IVB resectable disease have the best prognosis, particularly if a multimodal approach of surgery, radiation, and systemic therapy is used. Patients with stage IVC disease should be considered for a clinical trial or hospice/palliative care, depending upon their preference. New drugs that target important molecular characteristics of ATC, such as agents that target the cell cycle and angiogenesis, should be rapidly adopted into clinical practice post preclinical evaluation. The treatment of ATC with targeted therapeutics alone, in combination, or as an adjuvant to traditional cytotoxic chemotherapy and radiation therapy, warrants further clinical study and may offer new hope for individuals diagnosed with this fatal thyroid malignancy.
Conclusion
Progress on the diagnosis and treatment of ATC has been extremely slow, though ATC is known to be one of the most aggressive tumor types, primarily due to the limited information currently available on this tumor type. Factors that have restricted the expansion of knowledge include the rarity of disease, which limits the availability of material to work with, efforts being focused on the bigger killer cancers, and the absence of advocacy. The need for genomic studies of ATC for diagnosis as well as treatment modalities to be developed is clearly apparent. We hope that the recent advances in technology, and a global effort to put in check all cancers, drives the advancement of understanding of tumor biology in ATC, resulting in effective diagnostic markers and the identification of novel targeted treatment modalities.
Disclosure
The authors report no conflicts of interest in this work.
References
Kilfoy BA, Zheng T, Holford TR, et al. International patterns and trends in thyroid cancer incidence, 1973–2002. Cancer Causes Control. 2009;20:525–531. | |
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. | |
Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2011. Bethesda, MD: National Cancer Institute; 2014. | |
Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913–2921. | |
Mathur A, Olson MT, Zeiger MA. Follicular lesions of the thyroid. Surg Clin North Am. 2014;94:499–513. | |
Gilliland FD, Hunt WC, Morris DM, et al. Prognostic factors for thyroid carcinoma. A population-based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program1973–1991. Cancer. 1997;79:564–573. | |
McIver B, Hay ID, Giuffrida DF, et al. Anaplastic thyroid carcinoma: a 50-year experience at a single institution. Surgery. 2001;130:1028–1034. | |
Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer. 2009;16:17–44. | |
Wang HM, Huang YW, Huang JS, et al. Anaplastic carcinoma of the thyroid arising more often from follicular carcinoma than papillary carcinoma. Ann Surg Oncol. 2007;14:3011–3018. | |
Dobashi Y, Sakamoto A, Sugimura H, et al. Overexpression of p53 as a possible prognostic factor in human thyroid carcinoma. Am J Surg Pathol. 1993;17:375–381. | |
Wiseman SM, Griffith OL, Deen S, et al. Identification of molecular markers altered during transformation of differentiated into anaplastic thyroid carcinoma. Arch Surg. 2007;142:717–727; discussion 727–729. | |
Smallridge RC, Copland JA. Anaplastic thyroid carcinoma: pathogenesis and emerging therapies. Clin Oncol. 2010;22:486–497. | |
Catalano MG, Fortunati N, Boccuzzi G. Epigenetics modifications and therapeutic prospects in human thyroid cancer. Front Endocrinol (Lausanne). 2012;3:40. | |
Wiseman SM, Masoudi H, Niblock P, et al. Anaplastic thyroid carcinoma: expression profile of targets for therapy offers new insights for disease treatment. Ann Surg Oncol. 2007;14:719–729. | |
Sherman SI. Targeted therapies for thyroid tumors. Mod Pathol. 2011;24 Suppl 2:S44–S52. | |
Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6:292–306. | |
Sherman SI. Thyroid carcinoma. Lancet. 2003;361:501–511. | |
Ursino S, Fiorica F, Stefanelli A, et al. Anaplastic thyroid cancer: a case report of a long term survival patient and review of literature data. Eur Rev Med Pharmacol Sci. 2014;18:1368–1372. | |
Nix PA, Nicolaides A, Coatesworth AP. Thyroid cancer review 3: management of medullary and undifferentiated thyroid cancer. Int J Clin Pract. 2006;60:80–84. | |
Haigh PI. Anaplastic thyroid carcinoma. Curr Treat Options Oncol. 2000;1:353–357. | |
Hundahl SA, Fleming ID, Fremgen AM, et al. A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the US, 1985–1995 [see comments]. Cancer. 1998;83:2638–2648. | |
Veness MJ, Porter GS, Morgan GJ. Anaplastic thyroid carcinoma: dismal outcome despite current treatment approach. ANZ J Surg. 2004;74:559–562. | |
Antonelli A, Fallahi P, Ulisse S, et al. New targeted therapies for anaplastic thyroid cancer. Anticancer Agents Med Chem. 2012;12:87–93. | |
Salvatore G, Nappi TC, Salerno P, et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res. 2007;67:10148–10158. | |
Wreesmann VB, Ghossein RA, Patel SG, et al. Genome-wide appraisal of thyroid cancer progression. Am J Pathol. 2002;161:1549–1556. | |
Zitzelsberger H, Thomas G, Unger K. Chromosomal aberrations in thyroid follicular- cell neoplasia: in the search of novel oncogenes and tumour suppressor genes. Mol Cell Endocrinol. 2010;321:57–66. | |
Liu Z, Liu D, Bojdani E, et al. IQGAP1 plays an important role in the invasiveness of thyroid cancer. Clin Cancer Res. 2010;16:6009–6018. | |
Lee J, Hwang JA, Lee EK. Recent progress of genome study for anaplastic thyroid cancer. Genomics Inform. 2013;11:68–75. | |
Gauchotte G, Philippe C, Lacomme S, et al. BRAF, p53 and SOX2 in anaplastic thyroid carcinoma: evidence for multistep carcinogenesis. Pathology. 2011;43:447–452. | |
Garg M, Kanojia D, Okamoto R, et al. Laminin-5γ-2 (LAMC2) is highly expressed in anaplastic thyroid carcinoma and is associated with tumor progression, migration, and invasion by modulating signaling of EGFR. J Clin Endocrinol Metab. 2014;99:E62–E72. | |
Bellelli R, Castellone MD, Garcia-Rostan G, et al. FOXM1 is a molecular determinant of the mitogenic and invasive phenotype of anaplastic thyroid carcinoma. Endocr Relat Cancer. 2012;19:695–710. | |
Nucera C, Eeckhoute J, Finn S, et al. FOXA1 is a potential oncogene in anaplastic thyroid carcinoma. Clin Cancer Res. 2009;15:3680–3689. | |
Onda M, Emi M, Yoshida A, et al. Comprehensive gene expression profiling of anaplastic thyroid cancers with cDNA microarray of 25 344 genes. Endocr Relat Cancer. 2004;11(4):843–854. | |
Passon N, Puppin C, Lavarone E, et al. Cyclic AMP-response element modulator inhibits the promoter activity of the sodium iodide symporter gene in thyroid cancer cells. Thyroid. 2012;22(5):487–493. | |
Wiseman SM, Masoudi H, Niblock P, et al. Derangement of the E-cadherin/catenin complex is involved in transformation of differentiated to anaplastic thyroid carcinoma. Am J Surg. 2006;191:581–587. | |
Steelman LS, Chappell WH, Abrams SL, et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy- implications for cancer and aging. Aging. 2011;3:192–222. | |
Pita JM, Figueiredo IF, Moura MM, et al. Cell cycle deregulation and TP53 and RAS mutations are major events in poorly differentiated and undifferentiated thyroid carcinomas. J Clin Endocrinol Metab. 2014;99:E497–E507. | |
Ricarte-Filho JC, Ryder M, Chitale DA, et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009;69:4885–4893. | |
Liu D, Yang C, Bojdani E, et al. Identification of RASAL1 as a major tumor suppressor gene in thyroid cancer. J Natl Cancer Inst. 2013;105:1617–1627. | |
Murugan AK, Xing M. Anaplastic thyroid cancers harbor novel oncogenic mutations of the ALK gene. Cancer Res. 2011;71:4403–4411. | |
Kurihara T, Ikeda S, Ishizaki Y, et al. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid. 2004;14:1020–1029. | |
Murugan AK, Bojdani E, Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010;393:555–559. | |
Landa I, Ganly I, Chan TA, et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab. 2013;98:E1562–E1566. | |
Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. | |
Ho YS, Tseng SC, Chin TY, et al. p53 gene mutation in thyroid carcinoma. Cancer Lett. 1996;103:57–63. | |
Ito T, Seyama T, Mizuno T, et al. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res. 1992;52:1369–1371. | |
Ito T, Seyama T, Mizuno T, et al. Genetic alterations in thyroid tumor progression: association with p53 gene mutations. Jpn J Cancer Res. 1993;84:526–531. | |
Nikiforova MN, Wald AI, Roy S, et al. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98:E1852–E1860. | |
Brzezianska E, Pastuszak-Lewandoska D. A minireview: the role of MAPK/ERK and PI3K/Akt pathways in thyroid follicular cell-derived neoplasm. Front Biosci (Landmark Ed). 2011;16:422–439. | |
Basolo F, Pisaturo F, Pollina LE, et al. N-ras mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid. 2000;10:19–23. | |
Esapa CT, Johnson SJ, Kendall-Taylor P, et al. Prevalence of Ras mutations in thyroid neoplasia. Clin Endocrinol. 1999;50:529–535. | |
Manenti G, Pilotti S, Re FC, et al. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur J Cancer. 1994;30A:987–993. | |
Baitei EY, Zou M, Al-Mohanna F, et al. Aberrant BRAF splicing as an alternative mechanism for oncogenic B-Raf activation in thyroid carcinoma. J Pathol. 2009;217:707–715. | |
Xing M. BRAF V600E mutation and papillary thyroid cancer. JAMA. 2013;310:535. | |
Chang YS, Lin IL, Yeh KT, et al. Rapid detection of K-, N-, H-RAS, and BRAF hotspot mutations in thyroid cancer using the multiplex primer extension. Clin Biochem. 2013;46:1572–1577. | |
Rosenbluh J, Wang X, Hahn WC. Genomic insights into WNT/β-catenin signaling. Trends Pharmacol Sci. 2014;35:103–109. | |
Hassan B, Akcakanat A, Holder AM, et al. Targeting the PI3-kinase/Akt/mTOR signaling pathway. Surg Oncol Clin N Am. 2013;22:641–664. | |
Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39:183–190. | |
McIver B, Castro MR, Morris JC, et al. An Independent Study of a Gene Expression Classifier (Afirma™) in the evaluation of cytologically indeterminate thyroid nodules. J Clin Endocrinol Metab. Epub April 29, 2014. | |
Alexander EK, Kennedy GC, Baloch ZW, et al. Preoperative diagnosis of benign thyroid nodules with indeterminate cytology. N Engl J Med. 2012;367:705–715. | |
Cibas ES, Ali SZ. The Bethesda System for reporting thyroid cytopathology. Thyroid. 2009;19:1159–1165. | |
Cibas ES, Ali SZ; NCI Thyroid FNA State of the Science Conference. The Bethesda System for reporting thyroid cytopathology. Am J Clin Pathol. 2009;132:658–665. | |
Layfield LJ, Cibas ES, Gharib H, Mandel SJ. Thyroid aspiration cytology: current status. CA Cancer J Clin. 2009;59:99–110. | |
Baloch ZW, Mandel SJ, LiVolsi VA. Are we ready to modify the Bethesda thyroid fine-needle aspiration classification scheme? Cancer Cytopathol. 2013;121:171–174. | |
Mon SY, Hodak SP. Molecular diagnostics for thyroid nodules: the current state of affairs. Endocrinol Metab Clin N Am. 2014;43:345–365. | |
Nehs MA, Nucera C, Nagarkatti SS, et al. Late intervention with anti-BRAF(V600E) therapy induces tumor regression in an orthotopic mouse model of human anaplastic thyroid cancer. Endocrinology. 2012;153:985–994. | |
Nucera C, Nehs MA, Mekel M, et al. A novel orthotopic mouse model of human anaplastic thyroid carcinoma. Thyroid. 2009;19:1077–1084. | |
Reddi HV, Driscoll CB, Madde P, et al. Redifferentiation and induction of tumor suppressors miR-122 and miR-375 by the PAX8/PPARgamma fusion protein inhibits anaplastic thyroid cancer: a novel therapeutic strategy. Cancer Gene Ther. 2013;20:267–275. | |
Reddi HV, Madde P, McDonough SJ, et al. Preclinical efficacy of the oncolytic measles virus expressing the sodium iodide symporter in iodine non-avid anaplastic thyroid cancer: a novel therapeutic agent allowing noninvasive imaging and radioiodine therapy. Cancer Gene Ther. 2012;19:659–665. | |
Antico Arciuch VG, Russo MA, Dima M, et al. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget. 2011;2:1109–1126. | |
Charles RP, Silva J, Iezza G, Phillips WA, McMahon M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol Cancer Res. 2014;12(7):979–986. | |
McFadden DG, Vernon A, Santiago PM, et al. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A. 2014;111:E1600–E1609. | |
Vanden Borre P, McFadden DG, Gunda V, et al. The next generation of orthotopic thyroid cancer models: immunocompetent orthotopic mouse models of BRAF V600E-positive papillary and anaplastic thyroid carcinoma. Thyroid. 2014;24:705–714. | |
Fallahi P, Ferrari SM, Vita R, et al. Thyroid dysfunctions induced by tyrosine kinase inhibitors. Expert Opin Drug Saf. 2014;13:723–733. | |
Coelho SM, Carvalho DP, Vaisman M. New perspectives on the treatment of differentiated thyroid cancer. Arq Bras Endocrinol Metabol. 2007;51(4):612–624. | |
Kukulska A, Krajewska J, Gawkowska-Suwiska M, et al. Radioiodine thyroid remnant ablation in patients with differentiated thyroid carcinoma (DTC): prospective comparison of long-term outcomes of treatment with 30, 60 and 100 mCi. Thyroid Res. 2010;3(1):9. | |
Ho AL, Grewal RK, Leboeuf R, et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med. 2013;368(7):623–632. | |
Durante C, Puxeddu E, Ferretti E. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab. 2007;92(7):2840–2843. | |
Knauf JA, Ouyang B, Croyle M, Kimura E, Fagin JA. Acute expression of RET/PTC induces isozyme-specific activation and subsequent downregulation of PKCepsilon in PCCL3 thyroid cells. Oncogene. 2003;22(44):6830–6838. | |
Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res. 2007;13(4):1341–1349. | |
Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. | |
Libertini S, Abagnale A, Passaro C, et al. AZD1152 negatively affects the growth of anaplastic thyroid carcinoma cells and enhances the effects of oncolytic virus dl922-947. Endocr Relat Cancer. 2011;18:129–141. | |
Passaro C, Abagnale A, Libertini S, et al. Ionizing radiation enhances dl922-947-mediated cell death of anaplastic thyroid carcinoma cells. Endocr Relat Cancer. 2013;20:633–647. | |
Lin SF, Gao SP, Price DL, et al. Synergy of a herpes oncolytic virus and paclitaxel for anaplastic thyroid cancer. Clin Cancer Res. 2008;14:1519–1528. | |
Yu Z, Eisenberg DP, Singh B, Shah JP, Fong Y, Wong RJ. Treatment of aggressive thyroid cancer with an oncolytic herpes virus. Int J Cancer. 2004;112:525–532. | |
Gholami S, Haddad D, Chen CH, et al. Novel therapy for anaplastic thyroid carcinoma cells using an oncolytic vaccinia virus carrying the human sodium iodide symporter. Surgery. 2011;150:1040–1047. | |
Lin SF, Price DL, Chen CH, et al. Oncolytic vaccinia virotherapy of anaplastic thyroid cancer in vivo. J Clin Endocrinol Metab. 2008;93:4403–4407. | |
Abbosh PH, Li X, Li L, et al. A conditionally replicative, Wnt/β-catenin pathway-based adenovirus therapy for anaplastic thyroid cancer. Cancer Gene Ther. 2007;14:399–408. | |
Libertini S, Iacuzzo I, Perruolo G, et al. Bevacizumab increases viral distribution in human anaplastic thyroid carcinoma xenografts and enhances the effects of E1A- defective adenovirus dl922-947. Clin Cancer Res. 2008;14:6505–6514. | |
Portella G, Scala S, Vitagliano D, et al. ONYX-015, an E1B gene-defective adenovirus, induces cell death in human anaplastic thyroid carcinoma cell lines. J Clin Endocrinol Metab. 2002;87:2525–2531. | |
Reddi HV, Madde P, Cohen YC, et al. Antitumor activity of VB-111, a novel antiangiogenic virotherapeutic, in thyroid cancer xenograft mouse models. Genes Cancer. 2011;2:993–995. | |
Blagosklonny MV, Giannakakou P, Wojtowicz M, et al. Effects of p53-expressing adenovirus on the chemosensitivity and differentiation of anaplastic thyroid cancer cells. J Clin Endocrinol Metab. 1998;83:2516–2522. | |
Furuya F, Shimura H, Miyazaki A, et al. Adenovirus-mediated transfer of thyroid transcription factor-1 induces radioiodide organification and retention in thyroid cancer cells. Endocrinology. 2004;145:5397–5405. | |
Kim JE, Ahn BC, Hwang MH, et al. Combined RNA interference of hexokinase II and (131)I-sodium iodide symporter gene therapy for anaplastic thyroid carcinoma. J Nucl Med. 2011;52:1756–1763. | |
Barzon L, Bonaguro R, Castagliuolo I, et al. Gene therapy of thyroid cancer via retrovirally-driven combined expression of human interleukin-2 and herpes simplex virus thymidine kinase. Eur J Endocrinol. 2003;148:73–80. | |
Smallridge RC, Ain KB, Asa SL, et al; American Thyroid Association Anaplastic Thyroid Cancer Guidelines Taskforce. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104–1139. | |
Nikiforova MN, Wald AI, Roy S, Durso MB, Nikiforov YE. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J Clin Endocrinol Metab. 2013;98(11):1852–1860. | |
Portella G, Pacelli R, Libertini S, et al. ONYX-015 enhances radiation-induced death of human anaplastic thyroid carcinoma cells. J Clin Endocrinol Metab. 2003;88(10):5027–5032. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.