")
Back to Journals » International Journal of General Medicine » Volume 13
Anaplastic Pleomorphic Xanthoastrocytoma: A Case Report and Literature Review
Received 23 October 2020
Accepted for publication 7 December 2020
Published 16 December 2020 Volume 2020:13 Pages 1581—1587
DOI https://doi.org/10.2147/IJGM.S285989
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jing Liu, Yanhua Sun, Xia Liu
Department of Pathology, Shenzhen University 1st Affiliated Hospital, Shenzhen Second People’s Hospital, Shenzhen University School of Medicine, Shenzhen 518035, People’s Republic of China
Correspondence: Xia Liu
Department of Pathology, Shenzhen University 1st Affiliated Hospital, Shenzhen Second People’s Hospital, Shenzhen University School of Medicine, Shenzhen 518035, People’s Republic of China
Tel/Fax +86 755 88698000
Email [email protected]
Background: With an incidence of less than 1% among astrocytomas, pleomorphic xanthoastrocytoma (PXA) is rare. When its mitotic activity exceeds 5 mitoses/10 high-power fields, PXA is defined as anaplastic pleomorphic xanthoastrocytoma (APXA). This report documents the clinical manifestations and histopathological characteristics of APXA to help prevent future misdiagnoses.
Case Presentation: A 28-year-old male patient had a sudden limb twitch and visited a local hospital. A head magnetic resonance imaging scan showed large patches of abnormal signal intensity that were approximately 6.0× 3.3 cm in size in the right frontal and parietal lobes, with iso- to slightly hypointense signals on T1-weighted images (T1WI) and mixed hyperintense signals on T2-weighted images (T2WI). Optical microscopic imaging found pleomorphic tumor cells with sheet-like growth, as well as foamy tumor cells, multinucleated giant cells, pleomorphic cells with atypical nuclei, and acidophilic bodies. Some areas were densely packed with obvious atypia and visible mitoses. The patient tested positive for glial fibrillary acidic protein (GFAP), vimentin (Vim), neuronal nuclear antigen (NeuN), P53, oligodendrocyte transcription factor-2 (OLIG-2), and ATRX, while he tested negative for synaptophysin (Syn), CD34, S-100, BRAF V600E, and IDH1 R132H. The Ki-67 labeling index was 15%. Genetic sequencing showed that IDH1 and IDH2 genes were wild-type, but that his BRAF gene harbored the V600E mutation.
Conclusion: APXA is a WHO grade III astrocytoma that can be distinguished from WHO grade II PXA according to the level of mitosis. Imaging may help to inform the difficult differentiation between APXA and epithelioid glioblastoma. Nonetheless, a clear diagnosis warrants carrying out a comprehensive analysis, including histomorphological, immunophenotypic, and molecular assessments.
Keywords: pleomorphic xanthoastrocytoma, anaplastic, immunohistochemistry, pathology
Introduction
Pleomorphic xanthoastrocytoma (PXA) is a rare, clearly delineated type of astrocytoma on the surface of the brain. The proportion of PXA is less than 1% among all astrocytomas, and it is mostly seen in children and adolescents, with a median age of onset of 22 years old.1 PXA was first described in 1979 as a tumor with a relatively good prognosis in young patients.2 While the tumor can develop in different regions across the brain, it usually originates in the temporal lobe and induces epilepsy. Histopathological analyses of the tumors usually show pleomorphism. There may be a mixture of spindle cells, monocyte-like cells, and multinucleated giant cells. The tumors often exhibit prominent atypia, in which some cells have cytoplasmic vacuolization, lipophilic changes, and visible acidophilic bodies. Some cases may also have areas of epithelioid cells. Abundant lymphoid sleeves can be seen around the tumor blood vessels, and lymphocytes can infiltrate the tumors. In 2016, the World Health Organization (WHO) defined PXA with a mitotic activity of a rate exceeding 5 mitoses/10 high-power field (HPF) as “anaplastic pleomorphic xanthoastrocytoma (APXA)” and classified it as WHO grade III. In this study, we reported a case of primary APXA and reviewed the literature relevant to APXA published in China and abroad. We summarized the clinical and pathological characteristics of APXA and its prognosis to gain a deeper understanding of the disease.
Case Presentation
Four days prior to visiting a hospital, a 28-year-old male reported that he was unconscious and had a brief limb twitch for about five minutes without any obvious cause. Examination and head magnetic resonance imaging (MRI) performed at a local hospital revealed a space-occupying lesion in the right frontal lobe that could be a glioma. The specific diagnosis and treatment processes at the local hospital were unknown. The patient had no dizziness, vertigo, nausea, vomiting, impaired consciousness, dysphagia, or choking when drinking water. He came to our hospital for further diagnosis and treatment with a preliminary diagnosis of “space-occupying lesion in the right frontal lobe: possible glioma.” Since the onset of the disease, the patient’s sleep, mental status, diet, and bodily functions were normal, and his weight had not undergone any recent significant changes. The patient agreed for publication of the case details was informed consent for publication of the case details and provided “written INFORMED consent”.
Physical Examination After Admission
The patient had a blood pressure of 130/84 mmHg (1 mmHg = 0.133 kPa), was conscious and fluent in his speech, and could understand and answer our questions. Of equal size, his pupils were round (diameter, approximately 3 mm) and were sensitive to light reflection. He had normal eye movements, with no nystagmus. His nasolabial folds were symmetrical, his tongue was in the middle when extended, his limbs had a grade-5 muscle strength, his muscle tension and tendon reflexes were normal, and his pathological signs were negative. He had roughly symmetrical pain sensation and normal motor coordination. There was no resistance in the neck, and no signs of Klinefelter syndrome. The sound localization test was normal. There was no obvious abnormality in the heart, lungs, or abdomen.
Auxiliary Examination
A head MRI showed large patches of about 6.0 cm×3.3 cm in size and abnormal signal intensity in the right frontal and parietal lobes, with iso- to slightly hypointense signals on T1-weighted images (T1WI), mixed hyperintense signals on T2-weighted images (T2WI), hyperintense signals on FLAIR images, and slightly hyperintense signals on DTI images. Contrast-enhanced imaging of the lesions showed scattered strips of mild enhancements and strong round enhancements (diameter, 0.5 cm) in the limbic gyrus. The brain tissue around the lesions was swollen. The meninges in the left frontotemporal lobe were greatly thickened and showed strong enhancements. A patch of abnormal signal intensity was seen in the left corona radiata, with hypointense signals on T1WI images, and hyperintense signals on T2WI and FLAIR images. There was no abnormal signal or space-occupying lesion in the rest of the brain. No midline shift was observed. The ventricle system had normal morphology and size. There was no widening of the cisterns, fissures, or sulci. No abnormal signal intensity was found in the pericranial soft tissues (Figure 1). The final clinical diagnosis was: “space-occupying lesion in the right frontal lobe, possible glioma.”
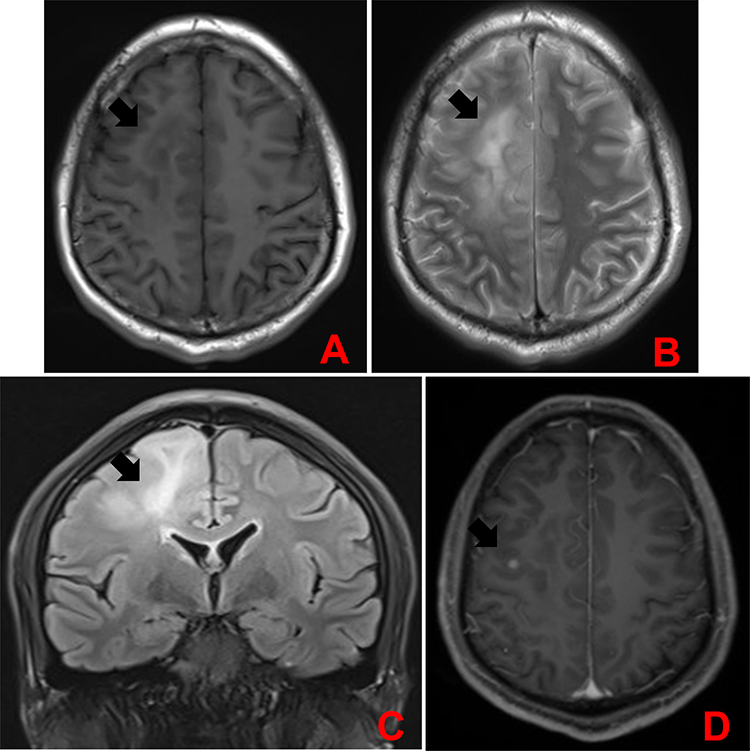 |
Figure 1 Head MRI. (A) Cross-sectional T1WI showed large areas of iso- to slightly hypointense signals in the right frontal and parietal lobes (indicated by the arrow). (B) Cross-sectional T2WI showed mixed hyperintense signals in the right frontal and parietal lobes (indicated by the arrow). (C) Coronal FLAIR images showed hyperintense signals on in the right frontal and parietal lobes (indicated by the arrow). (D) Cross-sectional T1W enhanced image showed scattered strips of mild enhancements and strong round enhancements (diameter, 0.5 cm) in the limbic gyrus (indicated by the arrow). |
Diagnosis and Treatment
The patient underwent surgery for the resection of the lesion under general anesthesia and local infiltration of the scalp nerves at the operative site. During the surgery, it was found that the posterior part of the tumor had expanded to the precentral gyrus (the area that controls the movement of left upper extremity), and the surface capillaries of the right superior frontal gyrus and precentral gyrus had proliferated considerably. The gyrus showed hypertrophy, but the sulci were still present. The tumor was tough in texture and had a moderate blood supply. Considering that the posterior part of the tumor resided in the vicinity of the precentral gyrus that controls the left upper limb, we decided to preserve the precentral gyrus. Along the anterior side of the precentral sulcal vein, the tumor was removed anteriorly along its apparent border, 5.5 cm lateral to the midline. The resection reached the corpus callosum. The excised specimen was sent for the following histopathological tests: (1) Observation of the gross specimen: The resected tissue was grayish-yellow (size, 5.4 cm×4 cm×2 cm). The cross-section was grayish-white with a moderately tough texture. (2) Hematoxylin and eosin (HE) staining: The tumor cells were pleomorphic with sheet-like growth, and included foamy tumor cells, multinucleated giant cells, pleomorphic cells with atypical nuclei, and acidophilic bodies. Some areas were densely packed with obvious atypia and visible mitoses. There was no necrosis or vascular endothelial hyperplasia in the tumor tissue (Figure 2). (3) Immunohistochemical staining: The SP two-step method was used, with detection kits and antibodies purchased from Beijing Zhongshan Jinqiao Biotechnology Co., Ltd. The expression patterns of ATRX protein (Figure 3A), glial fibrillary acidic protein (GFAP, Figure 3B), oligodendrocyte transcription factor-2 (OLIG-2, Figure 3C), vimentin (Vim, Figure 3D), neuronal nuclear antigen (NeuN, Figure 3E), and P53 protein were positive. The expression patterns of synaptophysin (Syn), CD34 protein, BRAF V600E (Figure 3G), IDH1 R132H, and S-100 protein were negative. The Ki-67 antigen labeling index was about 15% (Figure 3F). (4) Genetic testing: The BRAF locus was amplified by polymerase chain reaction (PCR). The results showed that the tumor had a BRAF V600E mutation (Figure 3H). The final pathological diagnosis identified (space-occupying lesion in the right frontal lobe) APXA, WHO grade III. After the surgery, temozolomide was given as a regular chemotherapy drug, along with anti-epileptic drugs. The patient has been followed-up since the surgery, and at the time of writing, there was no tumor recurrence, postoperative complications, or seizures.
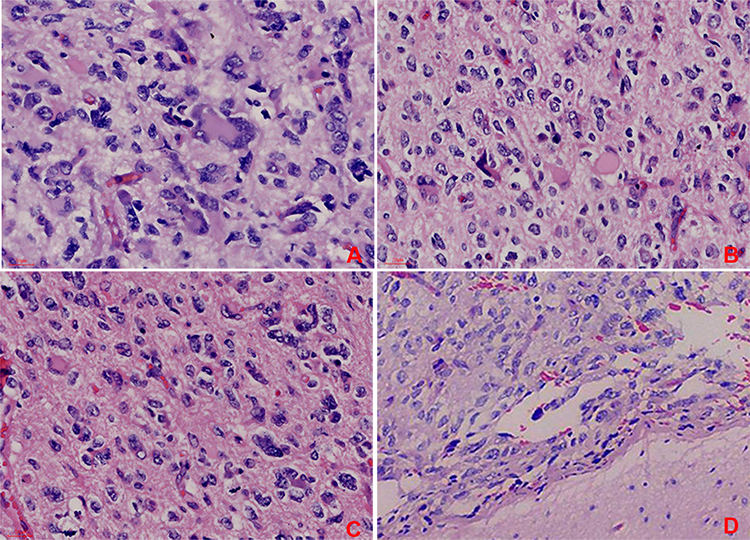 |
Figure 2 HE staining under light microscope at 400× magnification. (A) Tumor cells are pleomorphic and consist of monocytes, multinucleated giant cells, and cells with atypical nuclei, with prominent atypia. (B) Some areas of tumor cells are vacuous with acidophilic bodies. (C) Cells with atypical nuclei and proliferation of reticular fibers around vacuolated cells. (D) The tumor is located in the superficial cortex. |
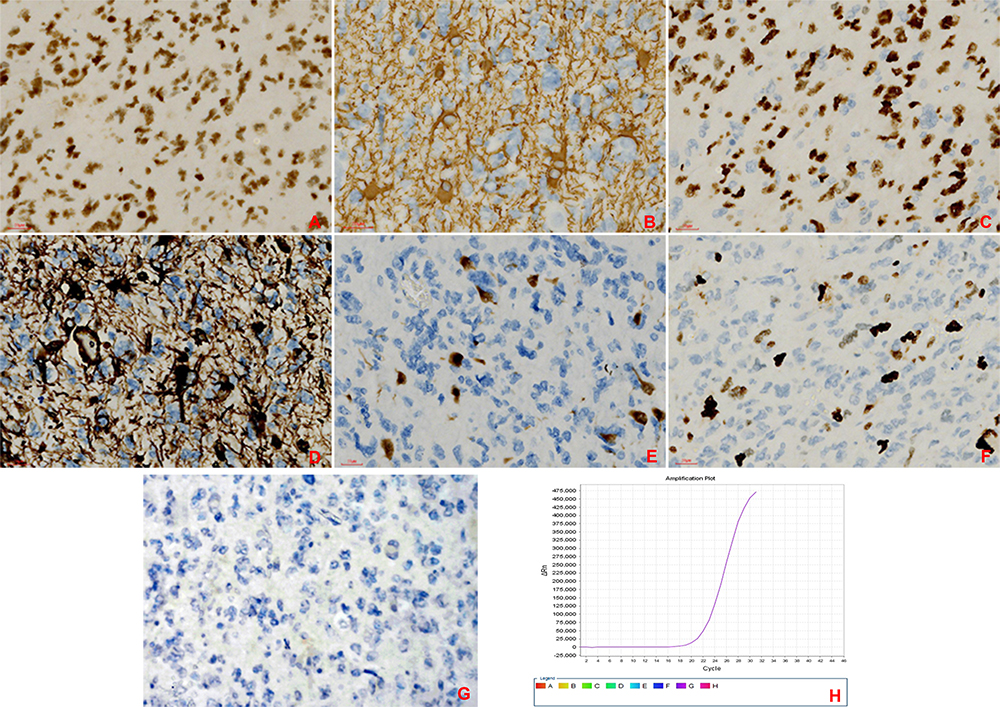 |
Figure 3 Immunohistochemical staining (SP two-step method) under light microscope at 400× magnification. (A) There was no loss of ATRX in tumor cells. (B) Some tumor cells expressed GFAP. (C) Some tumor cells expressed OLIG-2. (D) Some tumor cells expressed Vim. (E) A few tumor cells expressed NeuN. (F) Ki-67 antigen marking of the tumor cells showed areas with higher cell proliferation indices. (G) BRAF V600E antigen marking of the tumor cells showed no expression in tumor cells. (H) The BRAF locus was amplified by polymerase chain reaction (PCR) and showed that the tumor had a BRAF V600E mutation. |
Discussion and Conclusions
PXA accounts for about 1% of astrocytomas. It mainly arises in the outer parts of the cerebral cortex, and most often in the temporal lobe. The lesions are often localized, manifesting as solid or cystic masses, and can affect the pia mater. The clinical symptoms mainly consist of epilepsy. There have also been reports of cases of PXA in other regions, including the cerebellum,3–5 spinal cord,6 tectorial area,7 meninges,8 and retina.9 Electron microscopy studies have found bidirectional differentiation of glial neurons, as well as expression of CD34 and other neuronal cell markers, in PXA cases. Thus, they proposed that PXA might originate from neuroepithelial cells, which needs to be confirmed by further research.10,11 The typical histopathological features of PXA include a prominent pleomorphism, with multinucleated giant cells, giant cells with atypical nuclei, and foamy tumor cells. In some cases, there can also be local epithelioid tumor cells, which need to be distinguished from epithelioid glioblastoma. Lymphocytes can scatter among the tumor cells and form an interstitial perivascular lymphoid sleeve, where acidophilic bodies can be found. In 2016, the WHO defined PXA with a mitotic activity ≥5 mitoses/10 HPF as “anaplastic pleomorphic xanthoastrocytoma (APXA),” classifying it as WHO grade III. The location, morphology, and immunological markers of APXA are similar to those of PXA. Thus, the cut-off value for mitotic activity of ≥5 mitoses/10 HPF is key to their differentiation.
APXA is a rare type of tumor. In 2016, Choudry et al collected and compiled the clinical and pathological features of adult-onset primary APXA cases reported from 1979 to 2016. They found that the average time from the initial diagnosis of APXA to recurrence was 14 months. Pleomorphism of tumor cells was found in all cases. Atypical nuclei, multinucleated giant cells, lipid-containing vacuoles in the cytoplasm, and mitoses were found in most cases. In addition, there were abundant reticular fibers and lymphocyte infiltrations around the blood vessels.12 About 50%–78% of PXA cases have BRAF gene mutations, most of which are BRAF V600E mutations. Compared with PXA, APXA has a lower BRAF mutation rate. Studies have shown that 9 out of 19 APXA patients have a BRAF M600E mutation (mutation rate, 47.4%), while 30 of 50 PXA patients have a BRAF M600E mutation (mutation rate, 75%). However, it remains unclear as to whether a BRAF mutation is related to prognosis.13,14 The mutation rate of BRAF V600E is not significantly different between PXA and APXA cases.14 Pradhan et al reviewed the clinical radiology, histomorphology, and immunohistochemistry characteristics of five cases of APXA. During the follow-up, they found that one case converted to glioblastoma and metastasized, spreading to the spinal cord.15 In China, Sun et al reported a case of PXA with anaplastic characteristics. The patient was 58-years-old and died 11 days after the surgery due to respiratory and circulatory failure.16 Shao et al reported an 11-year-old child with APXA who did not receive adjuvant radiation therapy or chemotherapy after surgery but achieved good recovery and was in a stable condition at the time of discharge. However, the patient was lost to follow-up after 9 months.17 Based on these reports, we speculated that the prognosis of APXA may be good in young patients, but poor in elderly patients.
Due to its atypical morphology, prominent atypia, high mitotic activity, areas of high cell density, necrosis, and vascular endothelial hyperplasia, APXA can be frequently misdiagnosed as glioblastoma (WHO grade IV), especially giant cell glioblastoma and epithelioid glioblastoma. Here, we summarized the main diagnostic criteria of APXA, alongside its key differences from giant cell glioblastoma and epithelioid glioblastoma. (1) Giant cell glioblastoma (GCG): GCG is a relatively rare subtype of IDH-wildtype glioblastoma, which usually has a TP53 mutation and does not express CD34 or neuronal markers. Its prognosis is better than that of a normal glioblastoma.18,19 The histopathological and morphological characteristics of GCG partially overlap with those of PXA. Both have a large number of pleomorphic multinucleated giant cells, small spindle cells, reticular fibers, and interstitial lymphocyte infiltration. However, in GCG, tumor cells grow around blood vessels to form a “rosette” structure, and microvascular proliferation is uncommon. GCG and PXA are very similar in morphology. If the tumor is on the surface of the brain, and the patient is relatively young and has a BRAF V600E mutation, the diagnosis is more likely to be APXA if the mitotic activity exceeds 5 mitoses/10 HPF. In addition, APXA tumor cells can also be distinguished by the expression patterns of CD34, NeuN, Syn, and other neuronal markers. (2) Epithelioid glioblastoma (eGBM): The location, age of onset, molecular changes, as well as part of the morphological characteristics of eGBM overlap with those of APXA, and it is difficult to distinguish between the two. About 50% of eGBM cases have a BRAF V600E mutation, and the mutation rate is similar to that of APXA.20 The morphological characteristics of eGBM involve large areas of epithelioid or striated muscle-like cells, poor cell adhesion, red staining of the cytoplasm, and nuclear deviation. Some areas can be similar to the morphological aspects of PXA, but these generally do not exceed 50%. Necrosis is common in eGBM, and mainly manifests as patches of necrosis, rather than “pseudopalisading” necrosis. APXA can also have some epithelioid components, making it difficult to distinguish it from eGBM, but these components generally do not exceed 50%. APXA and eGBM overlap greatly in their morphology, molecular changes, and immunological characteristics. There are also reports that PXA can transform to eGBM,21 indicating that the two are linked, an interesting association which warrants further research. In addition to the amount of epithelioid cell components, another main difference between the two is that APXA usually has many low-grade PXA components and acidophilic bodies, which are uncommon in eGBM. In this context, necrosis and the Ki-67 index can also provide information but are not absolute.
As a result of its distinctive molecular genetic changes, APXA is associated with a BRAF V600E mutation. Thus, it needs to be differentiated from other types of gliomas in whose pathogenesis BRAF V600E mutations have been implicated, such as pilocytic astrocytoma, ganglion cell glioma, anaplastic ganglion cell glioma, and dysembryoplastic neuroepithelial tumors. Although these gliomas all have BRAF V600E mutations, they have their own characteristic histological and morphological changes, and none of them exhibit pleomorphisms or lipid-rich multinucleated giant cells, which facilitates their distinction from APXA.
PXA usually has a good prognosis. Studies have shown the five-year relapse-free survival rate to be 70.9% and the five-year overall survival rate to be 90.4%.13 However, the prognosis of APXA is poor, while the five-year survival rate is significantly lower than that of PXA. The implication of a BRAF V600E mutation in PXA and APXA remains unclear, and further research is needed. There is not much difference between APXA and PXA in terms of histological or morphological characteristics. They cannot be distinguished by pleomorphism or cell density, and they both have small amounts of necrosis and vascular endothelial hyperplasia. The only difference between the two is nuclear division. In conclusion, it is important to know the histopathological characteristics of PXA, so as not to misdiagnose it as glioblastoma based on the prominent necrosis and cell atypia. Immunohistochemistry and molecular testing can be helpful for this differentiation. There are reports of recurrence cases where PXA develops into eGBM,21 and it is common to see areas of PXA in eGBM. Furthermore, both can have BRAF V600E mutations, suggesting that they may be linked, an association which warrants investigation in further studies.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Consent to Participate
This study was performed in accordance with the principles of the Helsinki Declaration and approved by the Ethics Committee of the Shenzhen Second People’s Hospital.
Consent for Publication
The patient signed his consent for the publication of this report.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Shenzhen Science and Technology Innovation Commission (grant number JCYJ20170306090714854). The funder had no role in the manuscript.
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Giannini C, Scheithauer BW, Burger PC, et al. Pleomorphic xanthoastrocytoma: what do we really know about it? Cancer. 1999;85(9):2033–2045. doi:10.1002/(SICI)1097-0142(19990501)85:9<2033::AID-CNCR22>3.0.CO;2-Z
2. Kepes JJ, Rubinstein LJ, Eng LF. Pleomorphic xanthoastrocytoma: a distinctive meningo-cerebral glioma of young subjects with relatively favourable prognosis. A study of 12 cases. Cancer. 1979;44:1839–1852. doi:10.1002/1097-0142(197911)44:5<1839::AID-CNCR2820440543>3.0.CO;2-0
3. Lindboe CF, Cappelen J, Kepes JJ. Pleomorphic xanthoastrocytoma as a component of a cerebellar ganglioglioma: case Report. Neurosurgery. 1992;31(2):353–355. doi:10.1227/00006123-199208000-00023
4. Wasdahl DA, Scheithauer BW, Andrews BT, Jeffrey RA. Cerebellar pleomorphic xanthoastrocytoma: case report. Neurosurgery. 1994;35(5):947–951. doi:10.1227/00006123-199411000-00022
5. Gupta S, Mehrotra A, Pal L, Bhaisora KS, Jaiswal AK, Kumar R. An infratentorial pure pleomorphic xanthoastrocytoma arising from middle cerebellar peduncle: a rare location of an uncommon tumor. World Neurosurg. 2018;111:335–340. doi:10.1016/j.wneu.2017.12.125
6. Herpers MJ, Freling G, Beuls EA. Pleomorphic xanthoastrocytoma in the spinal cord. Case report. J Neurosurg. 1994;80:564–569. doi:10.3171/jns.1994.80.3.0564
7. Vizcaíno MA, Caccamo DV, Fox E, Rodriguez FJ. Pleomorphic xanthoastrocytoma: report of two cases with unconventional clinical presentations. Clin Neuropathol. 2014;33(11):380–387. doi:10.5414/NP300766
8. Dadhich H, Sharma R, Borkar SA, Dash A, Mahajan S, Sharma MC. Solitary extra-axial intracranial primary meningeal pleomorphic xanthoastrocytoma: an extremely rare case. World Neurosurg. 2019;130:386–390. doi:10.1016/j.wneu.2019.06.218
9. Zarate JO, Sampaolesi R. Pleomorphic xanthoastrocytoma of the retina. Am J Surg Pathol. 1999;23(1):79–81. doi:10.1097/00000478-199901000-00008
10. Hirose T, Giannini C, Scheithauer BW. Ultra-structural features of pleomorphic xanthoastrocytoma: a comparative study with glioblastoma multiforme. Ultrastruct Pathol. 2001;25(6):469–478. doi:10.1080/019131201753343502
11. Koelsche C, Sahm F, Wöhrer A, et al. BRAF-mutated pleomorphic xanthoastrocytoma is associated with temporal location, reticulin fiber deposition and CD34 expression. Brain Pathol. 2014;24:221–229. doi:10.1111/bpa.12111
12. Choudry UK, Khan SA, Qureshi A, Bari E. Primary anaplastic pleomorphic xanthoastrocytoma in adults. Case report and review of literature. Int J Surg Case Rep. 2016;27:183–188. doi:10.1016/j.ijscr.2016.08.022
13. Ida CM, Rodriguez FJ, Burger PC, et al. Pleomorphic xanthoastrocytoma: natural history and long-term follow-up. Brain Pathol. 2015;25(5):575–586. doi:10.1111/bpa.12217
14. Schmidt Y, Kleinschmidt-DeMasters BK, Aisner DL, Lillehei KO, Damek D. Anaplastic PXA in adults: case series with clinicopathologic and molecular features. Neurooncol. 2013;111:59–69. doi:10.1007/s11060-012-0991-4
15. Pradhan P, Dey B, Srinivas BH, Jacob SE, Rathakrishnan RKV. Clinico-histomorphological and immunohistochemical profile of anaplastic pleomorphic xanthoastrocytoma: report of five cases and review of literature. Int J Hematol Oncol Stem Cell Res. 2018;12:265–272.
16. Sun CY, Yu SZ. Pleomorphic xanthoastrocytoma with anaplastic features: one case report and review of literature. Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi. 2014;14:1091–1095.
17. Shao LW, Wang FL. Anaplastic pleomorphic xanthoastrocytoma. Zhongguo Xian Dai Shen Jing Ji Bing Za Zhi. 2017;17:616–625.
18. Peraud A, Watanabe K, Schwechheimer K, Yonekawa Y, Kleihues P, Ohgaki H. Genetic profile of the giant cell glioblastoma. Lab Invest. 1999;79:123–129.
19. Ortega A, Nuño M, Walia S, Mukherjee D, Black KL, Patil CG. Treatment and survival of patients harboring histological variants of glioblastoma. J Clin Neurosci. 2014;21(10):1709–1713. doi:10.1016/j.jocn.2014.05.003
20. Kleinschmidt-DeMasters BK, Aisner DL, Birks DK, Foreman NK. Epithelioid GBMs show a high percentage of BRAF V600E mutation. Am J Surg Pathol. 2013;37(5):685–698. doi:10.1097/PAS.0b013e31827f9c5e
21. Tanaka S, Nakada M, Nobusawa S, et al. Epithelioid glioblastoma arising from pleomorphic xanthoastrocytoma with the BRAF V600E mutation. Brain Tumor Pathol. 2014;31(3):172–176. doi:10.1007/s10014-014-0192-2
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.