")
Back to Journals » OncoTargets and Therapy » Volume 10
Anaplastic lymphoma kinase inhibition in metastatic non-small cell lung cancer: clinical impact of alectinib
Authors Muller IB, de Langen AJ, Giovannetti E, Peters GJ
Received 26 May 2017
Accepted for publication 11 August 2017
Published 13 September 2017 Volume 2017:10 Pages 4535—4541
DOI https://doi.org/10.2147/OTT.S109493
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tohru Yamada
Ittai B Muller,1 Adrianus J de Langen,2,3 Elisa Giovannetti,4,5 Godefridus J Peters4
1Department of Clinical Chemistry, 2Department of Pulmonology, VU University Medical Center, 3Department of Thoracic Oncology, Netherlands Cancer Institute, 4Department of Medical Oncology, VU University Medical Center, Amsterdam, the Netherlands; 5Cancer Pharmacology Lab, AIRC Start-Up Unit, University of Pisa, Pisa, Italy
Abstract: A subset of non-small cell lung cancer (NSCLC) tumors (5%) harbors an anaplastic lymphoma kinase (ALK) translocation that drives tumorigenesis. The clinically approved first-line treatment crizotinib specifically inhibits ALK and improves progression-free survival (PFS) in treated and untreated patients by 4 months compared to standard chemotherapy. While some patients relapse after crizotinib treatment due to resistance mutations in ALK, second-generation ALK inhibitors effectively induce tumor response and prolong PFS. Alectinib, a second-generation ALK inhibitor, has recently been approved for ALK-rearranged NSCLC after patients progressed on crizotinib. Alectinib is able to inhibit several crizotinib- and ceritinib-resistant ALK mutations in vitro. Furthermore, alectinib is a more potent tyrosine kinase inhibitor (TKI), with favorable safety profile, and has increased penetration into the central nervous system, inhibiting crizotinib-resistant brain metastases. The discovery of effective personalized therapies to combat ALK-rearranged NSCLC such as alectinib is an example of the importance of genomic profiling of NSCLC and provides an excellent template for future discoveries in managing these tumors.
Keywords: crizotinib, acquired resistance, alectinib, anaplastic lymphoma kinase, tyrosine kinase inhibitors, non-small cell lung cancer
Introduction
Lung cancer is the leading cause of cancer-related death worldwide, with ~1.6 million deaths reported globally in 2012.1 More than 85% of these cases are diagnosed as non-small cell lung cancer (NSCLC) in which a subgroup of tumors, ~5%, exhibits a fusion-type protein kinase called echinoderm microtubule-associated protein-like 4 (EML4)–anaplastic lymphoma kinase (ALK). In these EML–ALK-positive NSCLC patients, the EML4–ALK fusion protein is the dominant oncogenic driver. This constitutively active fusion protein originates from an inversion within the short arm of human chromosome 2.2,3
ALK belongs to the insulin receptor superfamily, and under normal conditions, ligand binding to ALK results in dimerization and subsequent transphosphorylation and downstream kinase signaling. Its structurally related ligands, pleiotrophin and midkine, play a role during the development of the nervous system and together are known as the neurite growth-promoting family.4 The fusion oncogene EML4–ALK causes ligand-independent dimerization of ALK, resulting in constitutive downstream signaling of canonical ALK pathways. Downstream of ALK, the EML4–ALK fusion oncogene activates several canonical oncogenic pathways, including the PI3K pathway, the JAK/STAT pathway, and the RAS/MEK/ERK pathway (Figure 1).5
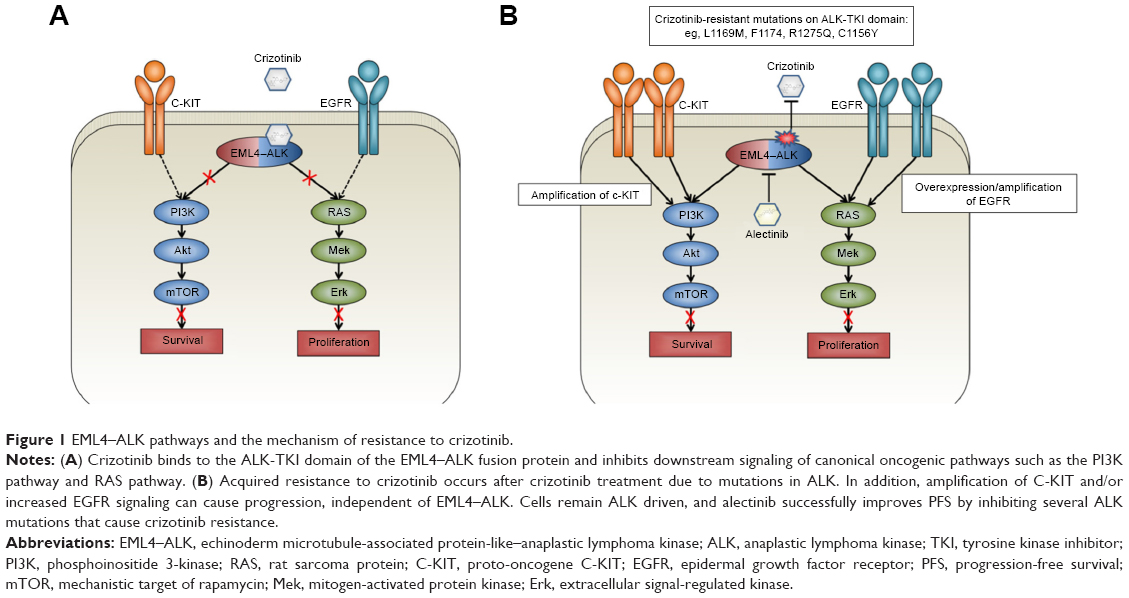 | Figure 1 EML4–ALK pathways and the mechanism of resistance to crizotinib. |
ALK inhibition in NSCLC and crizotinib resistance
Treatment of ALK-dependent NSCLC typically starts with crizotinib and more recently also with ceritinib. Crizotinib is a potent oral small-molecule tyrosine kinase inhibitor of ALK inhibitor, as well as c-MET and ROS1 kinases, that was first identified in 2010 by Kwak et al.6 Oral administration of crizotinib resulted in an objective response rate (ORR) of 57% in previously treated NSCLC patients with ALK rearrangements.6 In treatment-naive ALK-positive NSCLC, crizotinib outperformed standard first-line chemotherapy, indicating a clear and effective strategy window for targeting ALK in this patient population. However, some patients failed to respond and in others, resistance occurred after an average of 11.3 months.7
Acquired resistance to crizotinib can be caused by additional target mutations of ALK, ALK gene amplification, or activation of alternative signaling pathways.2,3 Mutations in ALK that confer resistance have been identified in up to 22% of crizotinib-resistant patients, of which the most important ones seem to inhibit binding of crizotinib to the ATP-binding pocket of ALK.8 The lack of long-term benefit from crizotinib due to the acquisition of resistance has prompted the development of increasingly potent, selective, and brain-penetrant ALK inhibitors, with differential spectrum of activity against the most common resistance mutations. Table 1 contains an overview of the different resistance mutations reported in recent literature and in which an ALK inhibitor is effective against these mutations.
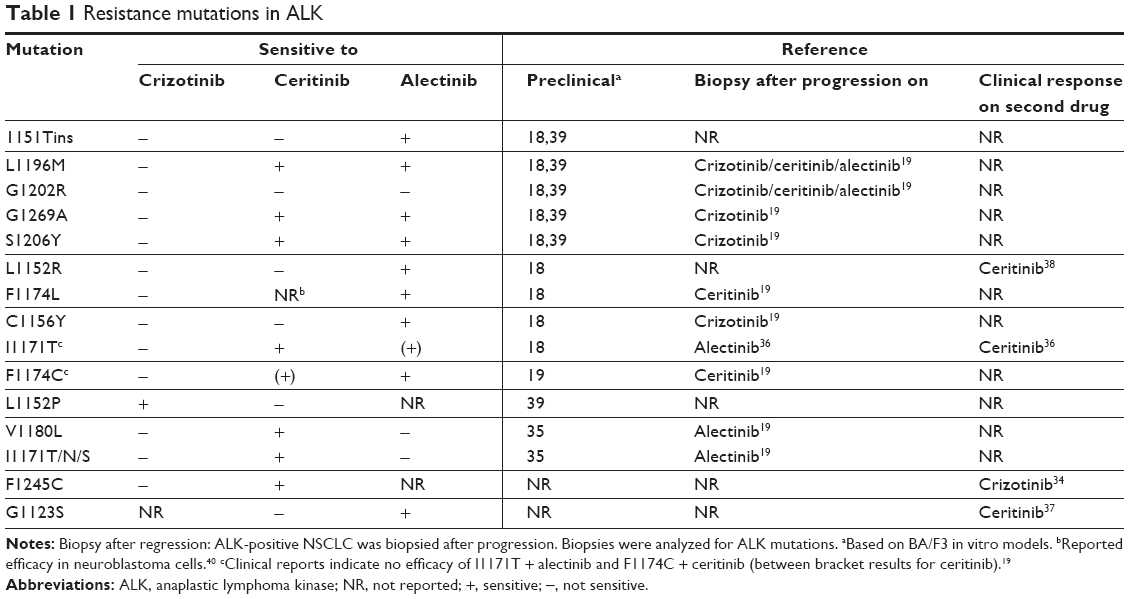 | Table 1 Resistance mutations in ALK |
Bypassing ALK signaling through increasing EGFR signaling in vitro can also cause crizotinib resistance. Approximately 50% of patients exhibit overexpression of EGFR when crizotinib resistance occurs.3 A combination therapy of crizotinib and gefitinib can resensitize these cells and inhibit growth,9 while a combination of erlotinib and crizotinib showed schedule-dependent synergism in several NSCLC cell lines, indicating a potential for combination therapy of ALK inhibitors and EGFR inhibitors.10
The most common site of relapse after crizotinib treatment is the central nervous system (CNS).11 The predisposition toward CNS progression during crizotinib treatment is attributable to poor accumulation of the drug into the CNS since this drug can be readily effluxed from the CNS through P-glycoprotein (P-gp)-mediated transport. The limited penetration of the blood–brain barrier of crizotinib is supported by the low intracranial response rate (7% ORR) in ALK-rearranged NSCLC patients.12
Preclinical efficacy of alectinib
Since resistance to crizotinib arises in all patients, several second-generation ALK inhibitors were developed to combat this resistance. After crizotinib, ceritinib gained approval as the second-line treatment, thus expanding the array of effective therapeutic options for ALK-positive NSCLC. The approval of ceritinib was based on the positive data in the ASCEND-2 and ASCEND-3 studies, in which an increased ORR was found in both crizotinib-naive and −resistant patients.13,14 In the ASCEND-4 study, progression-free survival (PFS) was 16.6 months compared to 8.1 months in the platinum-therapy arm.15 Alectinib (RO5424802/CH5424802) was designed specifically to be a more potent and selective anti-ALK therapy to bypass crizotinib resistance. Alectinib exhibits a threefold increase in in vitro ALK inhibition (53 nM alectinib versus 150.8 nM crizotinib).16 In 2016, alectinib gained the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval as a second-line therapy in ALK-rearranged NSCLC patients treated with crizotinib.17 Alectinib is effective in treating several crizotinib-resistant mutations in ALK, including L1196M, F1174L, R1275Q, and C1156Y, in vitro. L1196M is considered the gatekeeper mutation in crizotinib-resistant mutants, and alectinib can selectively inhibit growth of L1196M-driven tumors.16,18 In addition to bypassing crizotinib-resistant mutants in vitro, mouse models of human xenografts with crizotinib-resistant ALK mutations were sensitive to alectinib, which inhibited tumor growth.16 Alectinib also showed in vitro efficacy against ceritinib-resistant ALK-mutant L1198F and moderate potency against the compound mutation D1203N+F1174C.19
Alectinib has increased potency with regard to penetration of the CNS, since it effectively inhibits the growth of ALK-positive CNS lesions in an intracranial tumor implantation model. In addition, alectinib is not transported by P-gp-mediated transport and thus shows a high CNS-to-plasma ratio in this model.20 The CNS is considered as a sanctuary site after crizotinib treatment in NSCLC patients, while brain metastases were reported in ~35% of patients at the time of study entry into the PROFILE 1007 trial. Therefore, CNS penetration is crucial in combating resistance after first-line crizotinib treatment.11,21
Clinical efficacy of alectinib
In a single-arm, multicentre, open-label, Japanese Phase I–II study (AF-0001JP), patients were entered when positive for the ALK fusion gene, 24 for the Phase I and 46 for the Phase II part. In the last group, the ORR was 93.4% (complete response in two patients and partial response in 41 patients).22 After entering the last patient, treatment was continued, and after 3 years, 25/46 patients were still receiving alectinib, reaching a 3-year progression-free survival (PFS) of 62% (95% confidence interval [CI], 45%–75%) and a 3-year overall survival (OS) of 78%. Importantly of the 14 patients with brain metastases at baseline, six remained without progression.23 This study shows a long-term benefit of alectinib in non-selected ALK inhibitor-naive patients.
In a single-line multicentre Phase II study (NP28761), alectinib showed an ORR of 48% in ALK-positive, crizotinib-resistant patients. The median PFS and duration of response (DOR) in these patients were 8.1 months (95% CI, 6.2–12.6 months) and 13.5 months (95% CI, 6.7 months to not reached), respectively.24 A Phase II study (NP28673) similarly showed efficacy of alectinib in a cohort of crizotinib-resistant, ALK-positive NSCLC patients with an ORR of 50%. The median PFS in this study was 8.9 months (95% CI, 5.6–11.3 months), similar to NP28761.25
Alectinib was also effective against human CNS metastasis. In the NP28761 study, alectinib showed an ORR of 75% (12/16) in crizotinib-resistant NSCLC patients with measurable CNS lesions.24 The NP28673 study reported a CNS ORR of 57% in similar crizotinib-resistant patients.25
These studies suggest that alectinib is an effective treatment strategy in crizotinib-resistant NSCLC, in addition to ceritinib. The efficacy of alectinib corroborates the idea that crizotinib-resistant tumors stay dependent on ALK signaling, and this is especially interesting since both ceritinib and alectinib are able to inhibit ALK-positive NSCLC mutants harboring different resistance mutations.26
Furthermore, an open-label, randomized Phase III study called the J-ALEX trial compared the efficacy and safety of alectinib versus crizotinib in Japanese ALK-positive advanced or recurrent NSCLC patients with no prior ALK inhibition therapy. The report demonstrates that patients receiving alectinib had not yet reached the median PFS, while patients receiving crizotinib showed a median PFS of 10.2 months. Grade 3–4 toxicities were also less frequent in the alectinib arm (27%) compared to the crizotinib arm (52%).27
In addition to the J-ALEX trial, a Phase III randomized clinical trial that compared alectinib with crizotinib in treatment-naive ALK-positive patients was recently concluded. The authors reported similar results regarding the superiority of alectinib versus crizotinib: lower chance of progression (41% vs 68%), higher 12-month event-free survival rate (68.4% vs 48.7%), lower rate of CNS progression (12% vs 45%), higher response rate (82.9% vs 75.5%), and less adverse events (41% vs 50%).28 These results suggest a role for alectinib as a first-line treatment instead of (or in addition to) crizotinib. FDA approval for treatment in therapy-naive ALK-rearranged NSCLC patients is still pending.
Other ALK inhibitors are also in development such as lorlatinib and brigatinib, the latter received regulatory approval recently.29 A targeted therapeutic approach, aimed at elucidating the specific ALK mutations that cause resistance to crizotinib, could be of vital importance in the future of personalized NSCLC therapy.
Safety profile of alectinib
The most common adverse events in the NP28761 and NP28673 trials were constipation (34%), fatigue (41%), peripheral edema (30%), and myalgia (29%).17 Compared to ceritinib, alectinib seems well tolerated. A much larger percentage of ceritinib-treated patients exhibited adverse events such as diarrhea (86%), nausea (80%), vomiting (60%), and abdominal pain (54%).30 Alectinib-related deaths were reported in one patient in each study. In NP28761, a patient died of a treatment-related hemorrhage, and in NP28673, one patient died of intestinal perforation related to alectinib treatment. Although most patients tolerated the maximum dose of 600 mg alectinib (mean dose intensity of 97%), dose reductions in NP28761 and NP28673 were necessary in 16% and 21% of patients, respectively.24,25 In the AF-001JP in which ALK-positive crizotinib-naive patients received alectinib as first line, the drug was well tolerated with no grade 4 or 5 toxicity; serious adverse events were reported in 24.1% of the patients in the safety population and 21.7% of the patients in the Phase II. Onset was usually in the first 6 months and did not worsen in the following 3-year period. Only all-grade diarrhea onset occurred throughout treatment.23 This study demonstrated that long-term alectinib was well tolerated.
Pharmacokinetics of alectinib
Alectinib is metabolized by cytochrome P450 3A4 (CYP3A4), and its major active metabolite is M4. Both alectinib and M4 circulate in the plasma of alectinib-treated patients spiked with radioactive alectinib, constituting 76% of the total radioactivity in plasma from alectinib in the pharmacokinetic study evaluated by the FDA for registration (Table 2).31 In patients with ALK-rearranged NSCLC treated twice daily with 600 mg alectinib, the geometric mean of maximum steady-state concentration (Cmax,ss) was 665 ng/mL for alectinib and 264 ng/mL for M4 with a coefficient of variation of 44% and 45%, respectively. These data suggest a high exposure of the drug compared to crizotinib (steady-state concentration of 100–135 ng/mL). The time to maximum concentration was 3–5 hours for alectinib and 5–10 hours for M4, while the time to reach steady state was 7 days for both. Similar to crizotinib and ceritinib, the majority of alectinib and M4 were bound to plasma protein (99% for both), and alectinib had a similar volume of distribution as ceritinib (4016 L for alectinib and 4230 L for ceritinib). Overall, the pharmacokinetic profile of alectinib is comparable to its predecessors crizotinib32 and ceritinib.26
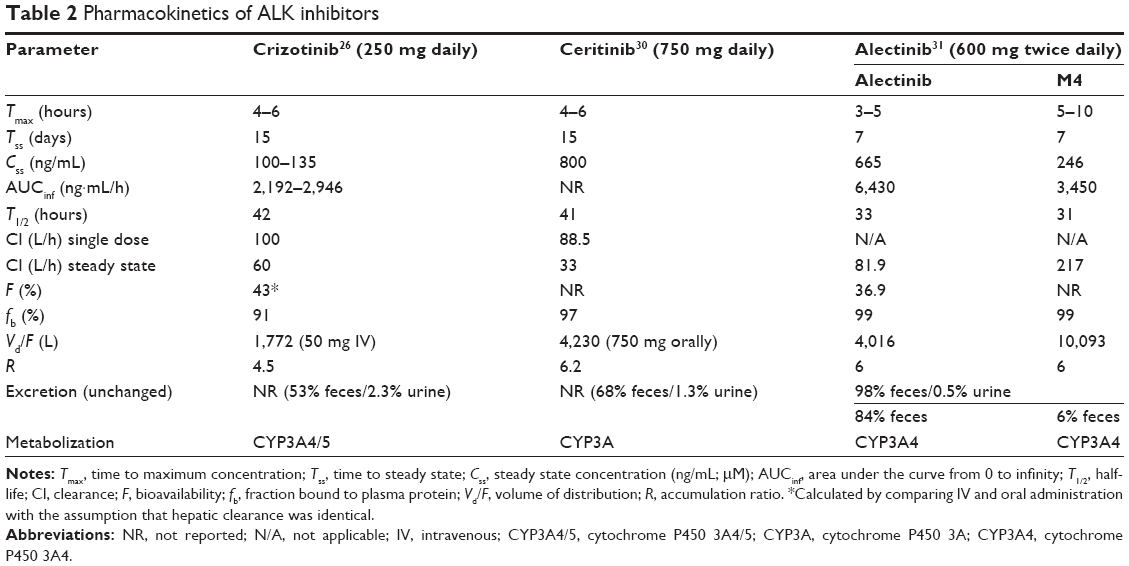 | Table 2 Pharmacokinetics of ALK inhibitors |
Patient-focused perspectives
Alectinib showed a positive safety profile compared to crizotinib in the J-ALEX trial.27 The most common adverse event in the alectinib arm was constipation (36%) while patients receiving crizotinib displayed nausea (74%), diarrhea (73%), vomiting (59%), visual impairments (55%), dysgeusia (52%), and constipation (46%). Results on median PFS in the ALEX trial indicate that alectinib may become a first-line treatment for ALK-positive NSCLC patients.28 The question then arises is; which ALK inhibitor should be given? It is important to note that different ALK inhibitors differ in their respective ALK resistance mutations after treatment. The importance of repeat biopsies upon progression may be different depending on which drug is given; the first-generation drug crizotinib or one of the second-generation drugs (ceritinib/alectinib). Gainor et al demonstrated that the refractory G1202R mutation is more common after progression on second-generation ALK inhibitors. Therefore, sequential treatment of tumors with ALK inhibitors may elicit unique responses.19 In order to accurately treat the patient, genotyping of recurrent tumors is crucial. However, repeated biopsies are not always possible.
Developing noninvasive techniques such as genotyping of circulatory DNA (ctDNA) seems to be the way forward. The ability to screen the ctDNA for relevant ALK resistance mutations during progression would allow clinicians to adjust therapy strategy with minimal invasion, improving quality of care and patient outcome.
Conclusion
It seems evident from the recent success of ceritinib and the fast-track FDA approval of alectinib that genomic profiling of NSCLC tumors is necessary to personalize the treatment of ALK-positive lung cancer patients. Especially after progression on second-generation ALK inhibitors, different mutations may occur. While initial treatment of crizotinib can grant ALK-positive patients an extra year of PFS, treatment of resistant patients with a second-generation ALK inhibitor such as alectinib afterward can prolong this survival for an extra 8.1 months. In addition, the ideal start and sequence of ALK inhibitors still need to be examined. Each ALK inhibitor (including the recently approved brigatinib) exhibits its own molecular response, and continuous surveillance on resistance mutations is crucial for an effective treatment strategy. Depending on the type of crizotinib-resistant mutations, patients can now be offered the choice between two potent and effective ALK inhibitors, and other even more potent inhibitors are under clinical investigation. If similar drugs such as lorlatinib gain FDA approval, the arsenal to treat patients increases, improving long-term treatment strategies.
Of note, in a recent report, Shaw et al showed a remarkable resensitization of a patient being retreated with crizotinib. The patient exhibited ALK rearrangement and was treated initially with crizotinib. The patient became progressive and was treated with chemotherapy and second-generation ALK inhibitor ceritinib. However, the patient appeared to be resistant to ceritinib and was given lorlatinib, a third-generation ALK inhibitor. After an initial response to lorlatinib, the patient became lorlatinib resistant and developed an L1198F mutation in ALK. Surprisingly, this mutation conferred an increase in sensitivity to crizotinib, and the patient was subsequently treated again with crizotinib and went back into remission.33
Genomic profiling of NSCLC patients could therefore result in a treatment strategy where not only a personalized sequence and combination of drugs are administered but it might also form the basis for transforming ALK-positive NSCLC into a chronic disease instead of a fatal one.
Disclosure
The authors report no conflicts of interest in this work.
References
Ferlay J, Soerjomataram I, Ervik M, et al [homepage on the Internet]. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. Lyon, France: International Agency for Research on Cancer; 2013. Available from: http://globocan.iarc.fr. Accessed February 4, 2017. | ||
Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–1739. | ||
Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19(15):4273–4281. | ||
Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263(5151):1281–1284. | ||
Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17(8):2081–2086. | ||
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | ||
Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–1482. | ||
Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4(120):120ra117. | ||
Van Der Steen N, Deschoolmeester V, Wouters A, Lardon F, Rolfo C, Pauwels P. Abstract 2593: combination therapy with EGFR-TKI and cMET-TKIs in non-small cell lung cancer: the golden duo. Cancer Res. 2015;75:2593. | ||
Gainor JF, Ou SH, Logan J, Borges LF, Shaw AT. The central nervous system as a sanctuary site in ALK-positive non-small-cell lung cancer. J Thorac Oncol. 2013;8(12):1570–1573. | ||
Costa DB, Shaw AT, Ou SH, et al. Clinical experience with crizotinib in patients with advanced ALK-rearranged non-small-cell lung cancer and brain metastases. J Clin Oncol. 2015;33(17):1881–1888. | ||
Shaw AT, Spigel DR, Tan DS, et al. MINI01.01: whole body and intracranial efficacy of ceritinib in ALK-inhibitor naive patients with ALK+ NSCLC and brain metastases: results of ASCEND 1 and 3: topic: medical oncology. J Thorac Oncol. 2016;11(11S):S256. | ||
Crino L, Ahn MJ, De Marinis F, et al. Multicenter phase II study of whole-body and intracranial activity with ceritinib in patients with ALK-rearranged non-small-cell lung cancer previously treated with chemotherapy and crizotinib: results from ASCEND-2. J Clin Oncol. 2016;34(24):2866–2873. | ||
Soria JC, Tan DS, Chiari R, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non-small-cell lung cancer (ASCEND-4): a randomised, open-label, phase 3 study. Lancet. 2017;389(10072):917–929. | ||
Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19(5):679–690. | ||
Larkins E, Blumenthal GM, Chen H, et al. FDA approval: alectinib for the treatment of metastatic, ALK-positive non-small cell lung cancer following crizotinib. Clin Cancer Res. 2016;22(21):5171–5176. | ||
Kodama T, Tsukaguchi T, Yoshida M, Kondoh O, Sakamoto H. Selective ALK inhibitor alectinib with potent antitumor activity in models of crizotinib resistance. Cancer Lett. 2014;351(2):215–221. | ||
Gainor JF, Dardaei L, Yoda S, et al. Molecular mechanisms of resistance to first- and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–1133. | ||
Kodama T, Hasegawa M, Takanashi K, Sakurai Y, Kondoh O, Sakamoto H. Antitumor activity of the selective ALK inhibitor alectinib in models of intracranial metastases. Cancer Chemother Pharmacol. 2014;74(5):1023–1028. | ||
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. | ||
Seto T, Kiura K, Nishio M, et al. CH5424802 (RO5424802) for patients with ALK-rearranged advanced non-small-cell lung cancer (AF-001JP study): a single-arm, open-label, phase 1–2 study. Lancet Oncol. 2013;14(7):590–598. | ||
Tamura T, Kiura K, Seto T, et al. Three-year follow-up of an alectinib phase I/II study in ALK-positive non-small-cell lung cancer: AF-001JP. J Clin Oncol. 2017;35(14):1515–1521. | ||
Shaw AT, Gandhi L, Gadgeel S, et al. Alectinib in ALK-positive, crizotinib-resistant, non-small-cell lung cancer: a single-group, multicentre, phase 2 trial. Lancet Oncol. 2016;17(2):234–242. | ||
Ou SH, Ahn JS, De Petris L, et al. Alectinib in crizotinib-refractory ALK-rearranged non-small-cell lung cancer: a phase II global study. J Clin Oncol. 2016;34(7):661–668. | ||
Muller IB, De Langen AJ, Honeywell RJ, Giovannetti E, Peters GJ. Overcoming crizotinib resistance in ALK-rearranged NSCLC with the second-generation ALK-inhibitor ceritinib. Expert Rev Anticancer Ther. 2016;16(2):147–157. | ||
Hida T, Nokihara H, Kondo M, et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): an open-label, randomised phase 3 trial. Lancet. 2017;390(10089):29–39. | ||
Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. Epub 2017 Jun 6. | ||
Markham A. Brigatinib: first global approval. Drugs. 2017;77(10):1131–1135. | ||
Khozin S, Blumenthal GM, Zhang L, et al. FDA approval: ceritinib for the treatment of metastatic anaplastic lymphoma kinase-positive non-small cell lung cancer. Clin Cancer Res. 2015;21(11):2436–2439. | ||
FDA Report Alectinib (revision date December 2015); 2015 [cited May 24, 2017]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208434s000lbl.pdf. Accessed August 12, 2017. | ||
Hamilton G, Rath B, Burghuber O. Pharmacokinetics of crizotinib in NSCLC patients. Expert Opin Drug Metab Toxicol. 2015;11(5):835–842. | ||
Shaw AT, Friboulet L, Leshchiner I, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. | ||
Kodityal S, Elvin JA, Squillace R, et al. A novel acquired ALK F1245C mutation confers resistance to crizotinib in ALK-positive NSCLC but is sensitive to ceritinib. Lung Cancer. 2016;92:19–21. | ||
Katayama R, Friboulet L, Koike S, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;20(22):5686–5696. | ||
Ou SH, Greenbowe J, Khan ZU, et al. I1171 missense mutation (particularly I1171N) is a common resistance mutation in ALK-positive NSCLC patients who have progressive disease while on alectinib and is sensitive to ceritinib. Lung Cancer. 2015;88(2):231–234. | ||
Toyokawa G, Inamasu E, Shimamatsu S, et al. Identification of a novel ALK G1123S mutation in a patient with ALK-rearranged non-small-cell lung cancer exhibiting resistance to ceritinib. J Thorac Oncol. 2015;10(7):e55–e57. | ||
Tchekmedyian N, Ali SM, Miller VA, Haura EB. Acquired ALK L1152R mutation confers resistance to ceritinib and predicts response to alectinib. J Thorac Oncol. 2016;11(7):e87–e88. | ||
Friboulet L, Li N, Katayama R, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–673. | ||
Debruyne DN, Bhatnagar N, Sharma B, et al. ALK inhibitor resistance in ALK(F1174L)-driven neuroblastoma is associated with AXL activation and induction of EMT. Oncogene. 2016;35(28):3681–3691. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.