")
Back to Journals » Journal of Asthma and Allergy » Volume 11
Anaphylaxis in the 21st century: phenotypes, endotypes, and biomarkers
Authors Jimenez-Rodriguez TW , Garcia-Neuer M , Alenazy LA, Castells M
Received 9 December 2017
Accepted for publication 21 February 2018
Published 20 June 2018 Volume 2018:11 Pages 121—142
DOI https://doi.org/10.2147/JAA.S159411
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Teodorikez Wilfox Jimenez-Rodriguez,1–3 Marlene Garcia-Neuer,1 Leila A Alenazy,1,4 Mariana Castells1
1Division of Rheumatology, Immunology and Allergy, Department of Medicine, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA; 2Allergy Section, Alicante General University Hospital, Alicante, Spain; 3PhD Program in Public Health, Medical and Surgical Sciences, Miguel Hernandez University, Alicante, Spain; 4Department of Medicine, College of Medicine, King Saud University, Riyadh, Saudi Arabia
Abstract: Anaphylaxis is the most serious of all allergic reactions and can be fatal. The diagnosis is frequently delayed, and misdiagnosis often occurs with asthma or urticaria. Biomarkers such as tryptase are not routinely checked, and appropriate treatment with epinephrine is not administered in a majority of cases, increasing the risk of poor outcomes. The objective of this review is to provide a better understanding of the pathophysiology of anaphylaxis with a description of phenotypes, endotypes, and biomarkers available in both the clinical and research settings. Expanding knowledge with regard to the presentation, causes, and triggers for anaphylaxis among health care providers will improve its diagnosis and management, increase patient safety, and decrease morbidity and mortality.
Keywords: anaphylaxis, hypersensitivity reactions, tryptase, autoimmune progesterone dermatitis, epinephrine, precision medicine
Defining anaphylaxis: history and consensus
In 1839, François Magendie was the first to describe the phenomenon of anaphylaxis experimentally when he found that rabbits that were able to tolerate a single injection of egg albumin often died after the second or third injection. He assumed these reactions were “toxic” because the injected albumin in these animals acted as a poison.1 In 1902, Charles Richet and Paul Portier coined the term anaphylaxis from ana (against) and phylaxia (protection) in Greek for the “property that has a poison to lower immunity rather than reinforce it.”2 Current expert consensus has defined anaphylaxis as a serious allergic reaction that is rapid in onset and can be fatal.3,4 The diagnosis is based on three possible clinical scenarios:
- Sudden onset of an illness (minutes to hours) with involvement of the skin, mucosal tissue (or both), and at least one of the following: a) respiratory compromise and b) reduced blood pressure or symptoms of end-organ dysfunction;
- Two or more of the following that occur after exposure to a likely allergen or other triggers (minutes to several hours): skin/mucosal symptoms and signs, respiratory compromise, reduced blood pressure or associated symptoms, and/or gastrointestinal symptoms (crampy abdominal pain or vomiting).
- Reduced blood pressure after exposure to a known allergen (minutes to hours).5
Critically, anaphylaxis involves at least two organ systems or sudden changes in vital signs. Skin and mucosal changes are usually but not always present, whereas hypotension and shock features are not mandatory for the diagnosis.6,7
Anaphylactoid reactions are clinically indistinguishable from anaphylaxis, with no demonstrable involvement of immunoglobulin E (IgE), and the term is no longer used, mainly to avoid unnecessary delays in diagnosis and treatment.3,8,9
Despite advances in the field of allergy, the symptoms of anaphylaxis continue to be under-recognized, diagnosis is often missed, treatment is often delayed (including the lack of epinephrine use), and the underlying causes are under-investigated worldwide.5,7,10
In 2003, to homogenize the nomenclature of allergy worldwide, the World Allergy Organization (WAO) proposed two classifications of anaphylaxis on the basis of the pathophysiological mechanism involved in the reaction. The term allergic anaphylaxis denotes reactions mediated by an immunologic mechanism – for example, IgE-, Ig, or an immune complex complement-related (corresponding to the classic hypersensitivity reactions [HSRs] described by Gell and Coombs) pathways. The term non-allergic anaphylaxis denotes reactions mediated by other mechanisms (eg, direct activation by bradykinin or complement), which are usually triggered by agents or events that induce sudden mast cell or basophil activation.11
Between 2004 and 2005, several organizations came together to update the definition and emphasize the use of epinephrine as a first-line treatment for anaphylaxis.6,12 Several studies were carried out to validate the criteria and to expand the scientific evidence with regard to the pathophysiology, triggers, and clinical management of anaphylaxis. The WAO provided a consensus document for the diagnosis and treatment of anaphylaxis. This report has been reviewed and updated to incorporate changes based on updated scientific evidence.3,4,8,9
Recently, a consensus document between the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology was published, which summarizes current knowledge in the field of allergy. They proposed a new approach based on precision medicine through phenotypes, which is associated with specific mechanisms that are defined as endotypes and the associated diagnostic biomarkers in food and drug allergies and anaphylaxis.13 This new classification encompasses the classic HSRs described by Gell and Coombs as well as reactions outside the classification.14
Phenotypes, endotypes, and biomarkers of anaphylaxis
Phenotypes are defined by clinical presentation, and endotypes refer to the cellular and molecular mechanisms of the HSRs defined by the diagnostic biomarkers (skin testing, tryptase, IgE, interleukin [IL]-6, and others).
Phenotypes of anaphylaxis are classified, by their clinical presentation, into Type I reactions, cytokine-release reactions (CRRs), mixed reactions, and, finally, bradykinin- and complement-like reactions. The corresponding endotypes underlying these phenotypes include IgE- and non–IgE-mediated mechanisms, cytokine-mediated mechanisms, mixed processes, and direct activation of immune cells by either complement or bradykinin14 (Figure 1).
 | Figure 1 Pathways of anaphylaxis. Note: Reprinted from the Journal of Allergy and Clinical Immunology, Volume 140/Edition 2, Castells M. Diagnosis and management of anaphylaxis in precision medicine, pages 321–333, Copyright 2017, with permission from Elsevier.14 Abbreviations: IgE, immunoglobulin E; IgG, immunoglobulin G; IL, interleukin; PAF, platelet-activating factor; TNF-α, tumor necrosis factor alpha. |
Type I
IgE-mediated anaphylaxis is the major mechanism underlying allergic anaphylaxis.1 After exposure to the allergen, a series of signals trigger the production of allergen-specific IgE by B cells (sensitization phenomenon). In subsequent exposures, the antigen–allergen-specific IgE complex binds to the Fc-epsilon-RI receptor on mast cells and/or basophils and, with adequate signaling, activates and degranulates these cells, thereby releasing preformed mediators, enzymes, and cytokines and facilitating the synthesis of de novo inflammatory mediators (eg, tryptase, histamine, leukotrienes, prostaglandins, platelet-activating factor [PAF], cytokines).15,16 The mediators cause allergic symptoms by directly acting on tissues. The reaction is propagated by recruiting and activating additional inflammatory cells – particularly eosinophils, which release more mediators, including lipid-derived mediators such as prostaglandin D2 and cysteinyl leukotrienes.16
In addition to the classical pathway mediated by IgE, other possible pathways have been described in animal models that are difficult to explore in humans.17 One of these alternative pathways is similar to the IgE-mediated pathway, but involves IgG antibodies. IgG-mediated reactions are mediated by IgG complexes that cross-link to the macrophage low-affinity receptor (FcgRIII) thus stimulating PAF (rather than histamine) release.18
PAF causes platelet aggregation and release of the potent vasoconstrictor thromboxane A2 and serotonin; acts directly on vascular endothelial cells to increase vascular permeability; decreases cardiac output, which can produce hypotension and cardiac dysfunctions; and increases smooth muscle contraction in the airways, gut, and uterus, among other effects.19,20
Although IgG-dependent anaphylaxis has not been demonstrated in humans, it has been hypothesized that IgG antibodies can mediate systemic anaphylaxis if there are large numbers of both IgG and antigen present.21 IgG receptors are capable of activating macrophages and neutrophils to secrete PAF and activate mast cells in vitro, which may contribute to human anaphylaxis.20–23 Chimeric IgG monoclonal antibodies (mAbs), such as rituximab, have been shown to induce anaphylaxis even in the absence of IgE, suggesting IgG-dependent anaphylaxis.24,25
Recent reports with regard to the direct activation of mast cells, independent of those mediated by IgE, indicate that the human G-protein–coupled receptor – MRGPRX2 – may be the receptor for many drugs and cationic proteins, such as quinolone antibiotics (eg, ciprofloxacin, levofloxacin), general anesthetics such as atracuronium and rocuronium, icatibant, and other drugs with tetrahydroisoquinoline (THIQ) motifs.26–29
The endotype for IgE-mediated reactions is mast cell and basophil mediator release that causes flushing, pruritus, hives, angioedema, shortness of breath, wheezing, nausea, vomiting, diarrhea, hypotension, oxygen desaturation, and cardiovascular collapse along with other symptoms.16,30 The common triggers for these reactions include foods, drugs, latex, Hymenoptera venoms, and environmental allergens.3,8–10,31,32 There are important geographic and age-related variations between countries; however, the most common food allergens are peanut, milk, eggs, nuts, shellfish, fruits and vegetables;33 antibiotics such as β-lactams, nonsteroidal anti-inflammatory drugs (NSAIDs), chemotherapeutic agents such as platins and taxanes, chimeric humanized human mAbs, general anesthetics, and immunotherapy allergens are other common allergens both in children and adults.34,35
CRRs
The CRR phenotype is caused by the release of proinflammatory mediators such as tumor necrosis factor alpha (TNF-α), IL-1B, and IL-6, and their target cells (endotype) include monocytes, macrophages, mast cells, and other immune cells with the Fc gamma receptor (FcgR) – an essential participant in many immune system effector functions, including the release of inflammatory mediators and antibody-dependent cellular cytotoxicity.
Triggers for these reactions include chimeric, humanized, and human mAbs and chemotherapeutic agents, including oxaliplatin. These drugs have not only been used to treat neoplastic, autoimmune, and inflammatory diseases but also to treat allergic disorders including allergic asthma, eosinophilic asthma, and chronic urticaria.36,37 HSRs to biologic agents are less common than standard infusion reactions, and they vary based on the biological agents involved.25 Phenotypic symptoms include chills, fever, and pain; these respond to ibuprofen and fluids and have been clinically correlated with IL-6.38 CRRs are typically not as severe as cytokine storm reactions.
Cytokine storm reactions are acute, severe, and potentially lethal systemic complications due to the production of large quantities of cytokines and chemokines, which play a pathological role in the development of systemic symptoms.39,40 IL-6 and other inflammatory cytokines such as IL-8, TNF-α, interferon gamma (IFN-γ), and IL-1β induce the inactivation of cadherin, which mediate cell adhesion, leading to vascular leakage by increased capillary permeability; moreover, this induces the formation of tissue factor (thromboplastin) on the cell surface of monocytes, with subsequent activation of the extrinsic coagulation pathway.41,42 The effects of inflammatory cytokines play a pathological role in the development of pain, tissue hypoxia, hypotension, myocardial dysfunction, and, eventually, disseminated intravascular coagulation (DIC) and multiorgan dysfunction. IL-6 is an excellent biomarker of cytokine storm reactions because of its correlation with the severity of the reaction and its longevity in blood serum.40
This phenotype is characterized by chills, fever, and generalized malaise followed by hypotension, desaturation, and cardiovascular collapse.14,25,37 Premedication with anti-inflammatory COX-1 inhibitors and corticosteroids can decrease the intensity of these symptoms but does not protect from severe reactions.14
Mixed reactions (Type I/CRRs)
Mixed reactions occur as a mixture of Type I and CRR phenotypes and, typically, are observed during chemotherapy and/or mAbs HSR, wherein symptoms of IgE-mediated reactions such as redness, pruritus, urticaria, angioedema, difficulty breathing, wheezing, nausea, vomiting, diarrhea, hypotension, desaturation, cardiovascular collapse, and life-threatening anaphylaxis – occurring secondary to the release of mast cell/basophil mediators (tryptase, histamine, leukotrienes, and prostaglandins) – overlap with symptoms secondary to the release of proinflammatory cytokines and chemokines (IL-1β, IL-6, and TNF-α) such as chills, fever, malaise, hypotension, desaturation, and cardiovascular collapse, thereby making it impossible to differentiate between mechanisms.
Complement/bradykinin-like reactions
Complement reactions involve direct activation of mast cells and other immune cells through complement activation as well as direct and indirect activation of the intrinsic coagulation pathway.16,22,43 Immune complexes can activate the complement system, generating anaphylatoxins such as C3a and C5a, which can bind to complement receptors resulting in release of histamine, leukotrienes, and prostaglandins that can induce flushing, hives, hypoxia, vasodilation, and hypotension.43,44 This mechanism has been described with drugs such as vancomycin,45 contrast media,46 dialysis membranes, and infusions of drugs that are suspended in certain lipid vehicles such as Cremophor EL, polysorbate 80, and polyethylene glycol.17,47 Notably, reports have also suggested that complement can have an important role in vespid-induced anaphylaxis, exacerbating the reaction due to the activation of complement by proteases present in the venom and adding to the IgE-mediated reaction.44,48
The molecular pathway of bradykinin reactions has been elucidated in animal models and involves an increase in heparin and Factor XII-driven contact system that results in the production of bradykinin and ultimately accounts for the increased vascular permeability (clinically, hypotension and desaturation).22 These symptoms have been associated with contamination of heparin with oversulfated chondroitin sulfate.49
Biomarkers for diagnosis of anaphylaxis
Skin tests (STs)
A careful history is important for the identification of culprit allergens. The ST is a highly specific test for Type I reactions, which is used to identify sensitization for numerous allergens including airborne, food-related, drugs (including platins), β-lactams, general anesthetics, and Hymenoptera venom triggers.31,50
Skin testing is considered safe for patients with a history of anaphylaxis and mastocytosis, provided comorbidities such as asthma are controlled. Medications such as β-blockers and angiotensin-converting enzyme (ACE) inhibitors may introduce greater risk for skin testing in case of a serious reaction; therefore, they should be discontinued before testing.51 Patients with CRRs and complement-activation reactions are likely to have negative ST results due to non-IgE mechanisms; however, the use of ST is still useful to identity patients who may convert from CRR to Type I reactions or those with mixed reactions.38,52
There should be a 4- to 6-week gap between anaphylactic reaction and ST due to the temporary loss of cutaneous activity following anaphylaxis; full restoration may take up to 6 weeks. However, due to treatment schedules, exceptions are made to expedite skin testing to 2 weeks post reaction, which should be sufficient to restore cutaneous reactivity; however, negative results may be inconclusive and demand further evaluations.53 For drug allergy, skin testing should be undertaken after resolution of clinical symptoms and clearance of culprit drug and rescue medication from the circulation.54 In case of Hymenoptera hypersensitivity, test reactivity may be falsely negative for approximately 6 weeks following such a reaction; therefore, STs should be performed later.55
Both ST and antigen challenge tests are reliable in vivo tests. However, not all allergenic compounds are present in the current skin testing extracts; therefore, the gold standard to clear an allergy after negative skin testing is a challenge. Patients with a convincing history of anaphylaxis and evidence of sensitization to a specific food should not undergo oral food challenges due to the high risk of anaphylaxis. For those with an equivocal history, low or moderate evidence of sensitization, or both, might benefit from a physician-monitored incremental oral food challenge. A positive (failed) challenge provides a sound basis for continued avoidance of the food. A negative (passed) challenge allows introduction or reintroduction of the specific food into the patient’s diet.56
For evaluation of penicillin allergy, skin testing of both major and minor determinants is required because patients are likely sensitized to minor determinants that are not currently commercially available in the US.57,58 For patients with a distant history of penicillin allergy and symptoms inconsistent with Type I reactions, a direct oral challenge with penicillin may be indicated.59–63 Reactions to beta-lactams can be by reactions to the beta-lactam ring or by reactions to its side-chain, so that there may be cross-reactivity dependent on each of these structures. It has been described patients capable of tolerating penicillin, who present anaphylactic reactions to other β-lactams that share the same side-chain, such as some aminopenicillins and cephalosporins.64,65 Efforts are currently underway to generate suitable ST reagents and establish the predictive value of cephalosporin STs.66
The specificity and sensitivity of mAb skin testing have not been defined because the allergenic components/epitopes are not yet defined and non–IgE-mediated mechanisms are believed to play a role. For chimeric mAbs, such as rituximab and infliximab, mouse epitopes are thought to be involved in the allergic response, and ST results are positive in 60–70% of patients with Type I and mixed reactions for rituximab; however, only 50% of patients with infliximab-induced Type I reactions have positive ST results.67 The negative predictive value for most mAbs is not known. Of 23 patients desensitized to trastuzumab, infliximab, or rituximab, 13 had positive ST results, although all had symptoms compatible with Type I or mixed reactions.67 Another study showed that 58/106 patients treated with mAbs had STs, and 41% of those were positive. Per phenotype, ST was positive in 44% of Type I reactions, 11% of CRRs, and 54% of the mixed reactions. A positive ST was associated with greater severity of initial reaction. These results indicate the value of ST for mAbs as well as the need for continued research.38
Serology and specific IgE
Many patients with allergic disorders (eg, atopic dermatitis, atopic asthma, allergic rhinitis) have elevated levels of total IgE.68 This elevation is not specific and low levels cannot be used to exclude the presence of atopic disease, because patients can still have local production of allergen-specific IgE in the tissues, and total IgE by itself is rarely adequate to diagnose allergic disease.69–71
Measurement of allergen-specific IgE is guided by the results of skin and/or serum testing.69 Specific IgE levels can be measured for food, environmental allergens, Hymenoptera venom, and drugs, including antibiotics and chemotherapy.72,73 They are not especially useful to identify and monitor food allergies because of their crude nature, lack of potency assessment, and variability among manufacturers and lots.74 Furthermore, skin testing may not be indicated due to diffuse skin diseases, significant dermatographism, inability to wean off medications interfering with the testing, or use of an extract believed to have a high probability of inducing a systemic reaction in the subject to be tested.69,75
A poor correlation between the IgE titer with ST result and the risk of anaphylaxis has been shown in previous studies; therefore, there is no evidence that either diagnostic test could accurately predict a severe allergic reaction in children or adults.76
Hymenoptera venom–specific IgE has high sensitivity and specificity, and components of the venoms are under recent evaluation, with a few components commercially available for clinical use. It has become apparent that patients treated with venom extracts may have severe anaphylaxis when re-stung in the field, which may be attributed to sensitization to minor determinants, such as Api m 10, which is absent in most current vaccines.77,78 Patients with specific IgE to Hymenoptera venoms who have been sensitized to major and minor determinants might not be protected after vaccination, and increasing the dose of venom has been recommended.79 The most effective test to determine the effectiveness of immunotherapy is the sting challenge test with live Hymenoptera; however, its safety is controversial.80–82
Penicillin-specific IgE has low sensitivity and is currently reserved for patients with near-fatal anaphylaxis, in whom skin testing is deemed unsafe.83 Measurement of specific IgE to other β-lactams, such as cephalosporins, has been used to assess cross-reactivity, but its clinical value has not been defined.84 Specific platin IgE has been shown to have lower sensitivity than skin testing, but has a higher specificity.85–87 A major advantage of specific IgE for platins is the ability to detect IgE antibodies shortly after the reactions and without the need to wait several weeks to determine ST reactivity, which can delay treatment to chemotherapy.88 Another advantage of specific IgE is that it can detect cross-reactivity with other similar drugs, for example, patients sensitized to oxaliplatin had specific IgE for other platins (carboplatin and cisplatin) without previous exposure.89
Component-resolved diagnostics
Advances in precision medicine have made it possible to identify and characterize some molecules present in the allergenic extracts, and this has resulted in component-resolved diagnosis which has changed our understanding of sensitization profiles and cross-reactivity90 as well as improved our ability to identify specific clinical phenotypes. In addition, it has allowed us to determine the relative risk of the severity of reactions in specific cases, such as soy, peanut, and hazelnut91 allergies, as well as predict the severity of allergic reaction92 – as in the case of Ara h 1, 2, and 3 associated with peanut-induced anaphylaxis, in contrast to patients with specific IgE to Ara h 8, 9, and 10 who might have oral allergy syndrome caused by cross-reactivity with tree pollen allergens and might not be at risk for anaphylaxis.93,94 Geographic variations have been seen – for instance, in the Mediterranean area, systemic reactions to hazelnut are generally mediated by Cor a 8, a lipid transfer protein (nsLTP),95–97 whereas reports from the US and Northern Europe have associated sensitization to Cor a 9 (an 11S globulin) and Cor a 14 (a 2S albumin) with severe hazelnut allergy in children.98,99 However, more studies are needed to validate the predictive values across different races, sex, and ethnic backgrounds.
Studies show that component-resolved diagnostics (CRD) offered greater specificity, but decreased sensitivity, in foods; therefore, its use is complementary and should not be used alone to rule out food allergy.100–102
In relation to pollen allergies, the utility of CRD focuses on the correct identification of species-specific markers (Bet v 1 can identify individuals allergic to the Betulaceae family; Ole e 1, those allergic to the Oleaceae family; Cup a 1, those allergic to the Cupressaceae family; and Pla a 1, those allergic to the Platanaceae family), especially in patients with multiple sensitization patterns secondary to cross-reactivity due to the presence of panallergens (polcalcin, profiling, or nsLTP).90 In cases of multiple pollen sensitization, responses are better to allergen-specific immunotherapy when it is prescribed on the basis of species-specific molecular markers with clinical relevance for each patient.103
In case of Hymenoptera allergy, CRD has become a very useful tool to distinguish between genuine sensitization and cross reactivity, because allergen sources contain potentially cross-reactive allergens between honey bee and wasp, such as Api m 2, Ves v 2 (hyaluronidases), Api m 5, Ves v 3 (dipetidylpeptidases), Api m 12, Ves v 6 (vitellogenins), and marker allergens that are specific for honey bee venom (Api m 1, Api m 3, Api m 4, and Api m 10) or yellow jacket venom (Ves v 1 and Ves v 5); these markers allow discrimination in clinical situations in which patients have a multiple sensitization profile.90
Basophil activation test
Basophils and mast cells originate from a common progenitor cell in bone marrow and share similar phenotypic/biochemical characteristics: they express high-affinity IgE receptors and contain special cytoplasmic granules. Basophils can be easily assessed because they are circulating granulocytes that have responded to allergic stimuli by migrating and accumulating at sites of allergic inflammation. Mast cells, unlike basophils, leave the bone marrow immature and complete their maturation in peripheral tissues, where they ultimately reside. Therefore, evaluation of human mast cells is limited to biopsy specimens from specific tissues.104,105
The basophil activation test (BAT) is a blood test used to diagnose IgE-mediated reactions to allergens such as food, environmental allergens, Hymenoptera venom, or drugs. BAT is a flow-cytometry–based functional assay that detects upregulation of the basophil activation surface markers CD63 and CD203c following exposure to an antigenic stimulus. Because both mast cells and basophils are sensitized with the same repertoire of IgE, BAT is considered a true reflection of sensitized mast cells.14
Currently, BAT is commercially available but has not been standardized or approved by the US Food and Drug Administration.
BAT has had success in evaluating immediate reactions to β-lactams, muscle relaxants, other drugs, as well as the diagnosis and monitoring of Hymenoptera venom allergies.13,106–108 BAT is not recommended for patients with a history of multiple reactions, NSAID hypersensitivity, or those with food allergies because of their increased histamine release at baseline.105
In a study with 15 patients allergic to platinum-containing compounds who were undergoing drug desensitization, BAT provided reliable information with regard to sensitization and upregulation of CD203c expression. Furthermore, reaction severity has been correlated with a higher expression of CD63, demonstrating that this technique can be used as a biomarker for desensitization.109
Tryptase
Tryptase is a serine protease and the most abundant secretory mediator formed and stored in human mast cells and basophils. The in vivo biologic activities of mature tryptase remain uncertain, but in vitro activities include generation of complement anaphylatoxins, inactivation of fibrinogen, and stimulation of a variety of different cell types. In addition to its release, it amplifies the allergic response.
The rise of tryptase levels can be detected in serum 30 minutes after the initial symptoms of anaphylaxis and peaks 1–2 hours post initiation of the reaction. Tryptase elevation is transient and typically resolves within 24–48 hours. Commercial immunoassays allow for detection of total (baseline release, reflecting mast cell and basophil burden) and mature tryptase (released only at the time of activation); however, they do not discriminate between the two. Increases above the normal range of 11.4 ng/mL are indicative of either acute mast cell/basophil activation or increase in total mast cell number;110 therefore, a baseline tryptase level is required 2 weeks post anaphylaxis. Tryptase levels ≥2 ng/mL + 1.2× baseline are considered significantly increased for patients with a low baseline tryptase.111,112
Total tryptase levels can be elevated (>20 ng/mL) in most patients with systemic mastocytosis (SM) – a clonal disorder associated with mutation of the membrane tyrosine kinase KIT; however, it is seen in other conditions such as hematologic disorders (acute myelocytic leukemia, myelodysplastic syndromes), immunologic disorders (hypereosinophilic syndrome), severe renal failure, or inherited variations such as familial tryptasemia – a recently characterized disease which is associated with multisystem complaints including cutaneous flushing and pruritus, dysautonomia, functional gastrointestinal symptoms, chronic pain, and connective tissue abnormalities, including joint hypermobility, due to the expression of more than two α-tryptase genes.113,114
Tryptase specificity is high whereas its sensitivity is low due to its release from different mast cell and basophil subsets, depending on the trigger. Tryptase levels are lower in mucosal mast cells than in cutaneous and perivascular mast cells, and anaphylactic reactions to intravenous drugs can elicit greater and more persistent increases than oral triggers, such as foods.
If anaphylaxis is suspected, blood sampling for tryptase measurement should be done between the first 30 minutes and 3 hours of reaction initiation to capture the maximum tryptase value.115 Elevations correlate with hypotension and support the diagnosis of anaphylaxis, although normal levels do not exclude the diagnosis.116,117
Other inflammatory mediators
Other mast cell/basophil mediators can be released during anaphylaxis including histamine, which is the only preformed granule mediator of these cells with vasoactive activity and direct spasmogenic action on the smooth muscle.
Elevation in plasma/urine histamine is consistent with anaphylaxis, although it is not exclusive, because other conditions can also have elevated levels without mast cell/basophil activation.115 Both histamine and its metabolite methylhistamine can be measured in 24-hour urine collection; however, the sensitivity is low because of the difficulties in timing 24-hour urine collections to symptom onset.
The contribution of PAF to anaphylaxis, which has been studied in mice, suggests a synergistic effect between histamine and PAF.16 A recent human study indicated an inverse correlation between serum PAF and acetylhydrolase levels (the enzyme that metabolizes and inactivates PAF) with the severity of anaphylaxis: low enzymatic levels were associated with high PAF levels, severe hypotension, and fatal and near-fatal anaphylaxis.118
Levels of other serum inflammatory mediators such as TNF-α, IL-6, and IL-1b can be elevated in patients with CRRs and anaphylaxis, but their sensitivity or specificity have not yet been validated.38 These inflammatory mediators provide insight into the phenoendotype of reactions and can help guide recommendations for desensitization protocols and premedications.40
Prostaglandin D2 and leukotrienes E4 and C4 can be measured in 24-hour urine collection. Despite high specificity, the sensitivity might be low due to the difficulties in timing 24-hour urine collections to symptom onset. However, prostaglandin D2 levels cannot be useful in patients receiving cyclooxygenase inhibitors.
Other mast cell proteases, such as chymase and carboxypeptidase, have been detected during anaphylaxis, although no commercial assays are available to date.
Patients at risk
Food-induced anaphylaxis and food-triggered exercise-induced anaphylaxis
Predictors for anaphylaxis risk have not been elucidated; one of the most important known risk factors associated with fatal anaphylaxis is asthma.119–121 Food-induced anaphylaxis affects both sexes equally, and fatal food-induced anaphylaxis occurs most often in adolescents and young adults who are unaware of their allergy, with the largest culprit being tree nuts.122
Anaphylaxis can be induced by many foods, the most common including cow’s milk, hen’s eggs, peanuts, tree nuts, fish, shellfish, soy and wheat, with variations depending on geography and local dietary patterns. Food-induced anaphylaxis is most common in children, but can occur in people of any age.123
In a study of 1,094 patients with peanut and tree nut allergy conducted in the UK, severe atopic disease was associated with the most severe reactions. Severe pharyngeal edema was associated with severe rhinitis; life-threatening bronchospasm was associated with severe asthma; and altered mental status was associated with severe eczema. These associations were independent of sex and age, although adults were 9 times more likely to have severe reactions than children and, of 122 patients in whom ACE and aminopeptidase levels were measured, patients with low levels had a greater number of severe reactions, highlighting the role of bradykinins in food-induced anaphylaxis as well as the need to discontinue ACE inhibitors in patients with food allergies.124
In a systematic review and meta-analysis that involved data from North America, Europe, and Australia, the estimated incidence of fatal food anaphylaxis in a food-allergic individual was 1.81 (95% CI 0.94–3.45) per million person-years, confirming that the incidence of fatal food-related anaphylaxis had little impact on the overall mortality risk in this group.125
In an Australian study, 4,453 infants with eczema were evaluated to characterize their risk of challenge-proven food allergy. They found that eczema was a strong risk factor for IgE-mediated food allergy, and the severity of eczema was associated with greater risk of food-induced allergies. They hypothesized that increased cutaneous exposure to food allergens in infants with weakened skin-barrier function may increase the risk of food sensitization.126
Recently, a multicenter study in the US evaluated allergic reactions in oral challenges for individuals with food allergies. Of the 6,377 oral provocations undertaken, the rate of allergic reactions was 14% (95% CI 13–16%) and the pooled estimate of anaphylaxis was 2% (95% CI 1–3%). Peanut was the most frequently involved allergenic food in the Northeast, Midwest, and West, whereas egg was the most frequent in the South. This showed that the likelihood of anaphylactic reaction in allergic individuals is low and geographical variations between trigger foods are present.127
Food-dependent exercise-induced anaphylaxis (FDEIA) is a disorder in which symptoms develop only if exertion takes place within a few hours of eating certain food(s) to which the patient is sensitized.128,129 This has been reported in patients of all ages, although most cases are described in adolescents and young adults, with wheat and omega-5 gliadin identified as the dominant triggers. The mechanisms are not fully understood; however, elimination of wheat before exercise reduces reactions.130–132
More recently, food ingestion associated with other cofactors, such as alcohol or the menstrual period, was found to induce anaphylaxis, expanding the understanding of FDEIA as a syndrome in which exercise might not be necessary.133
Mast cell activation disorders
Mast cell disorders are conditions in which mast cells are either increased in number (clonal mast cell disorders), hyper-reactive (non-clonal mast cell disorders), or both (Figure 2).134
 | Figure 2 Mast cell activation syndrome. Notes: Adapted from original provided by Dr Raied Talal Hufdi, Division of Rheumatology, Immunology and Allergy; Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA. Abbreviations: AHN, associated hematologic neoplasm; BM, bone marrow; EDS, Ehlers-Danlos Syndrome; GI, gastro-intestinal; IgE, immunoglobulin E; IgG, immunoglobulin G; IL, interleukin; MC, mast cell; MCAS, mast cell activation syndrome; MCL, mast cell leukemia; MMAS, monoclonal mast cell activation syndrome; NSAIDs, nonsteroidal anti-inflammatory drugs; PAF, platelet-activating factor; POTS, Postural Orthostatic Tachycardia Syndrome; RCM, radio contrast medium; SM, systematic mastocytosis; TMPE, telangiectasia macularis eruptiva perstans; TNF-α, tumor necrosis factor alpha; WHO, World Health Organization. |
Mast cells activate and accumulate in tissues due to a gain-of-function mutation, commonly in the KIT receptor gene that codes a tyrosine kinase transmembrane receptor involved in the regulation of crucial mast cell functions such as differentiation, activation, and survival. Upon receiving a trigger, they release mediators inappropriately, interfering with normal function, causing a variety of localized and systemic symptoms.134,135
Mast cell activation syndrome (MCAS) applies to all disorders (including anaphylaxis) that appear as a consequence of the release of vasoactive mediators by activated mast cells. Based on previous consensus, MCAS can be classified into three categories:112,136
- Primary MCAS, including SM and monoclonal MCAS (MMAS).
- Secondary MCAS.
- Idiopathic MCAS.
All of the criteria should be present to make a diagnosis of MCAS (Table 1).
 | Table 1 Proposed criteria for mast cell activation syndrome (all 3 must be present) Notes: *Documentation of a single meaningful increase in tryptase level is sufficient, whereas it is recommended to document at least two measurements of increased levels of other markers. Reproduced from Journal of Allergy and Clinical Immunology, Volume 140 (Edition 2), Akin C. Mast cell activation syndromes, pages 349–355, Copyright 2017, with permission from Elsevier.134 |
In primary MCAS (increased mast cells number), the presence of mast cell clones in the bone marrow of the affected individual must be demonstrated. In most patients (>95%) with SM and MMAS, mast cell clonality can be established by detecting KIT receptor-activating mutations. On the other hand, mast cells in secondary and idiopathic MCAS (increased activation) are normal, and symptoms of mast cell activation are related to other, secondary underlying conditions that activate these cells (allergies, neoplasms, inflammatory or autoimmune diseases, or unknown factors).137
Overall, 30% of patients with mastocytosis can present with anaphylaxis with known (including hymenoptera venom–induced anaphylaxis) or unknown triggers.138–140 Due to the lack of cutaneous manifestations (flushing, maculopapular rash in the form of urticaria pigmentosa, etc.), the diagnosis is often missed or delayed.141 Patients with syncope, hypotension, or cardiovascular collapse with severe anaphylaxis after Hymenoptera sting even with normal baseline tryptase levels should be evaluated for the KIT D816V mutation, which is present in >95% of patients with SM. In patients with positive KIT D816V mutation, a bone marrow biopsy is recommended.142–145 A tryptase level >20 ng/mL at baseline 4–6 weeks after an anaphylactic event is a minor criterion of SM and an indication for bone marrow biopsy.143 Other criteria for the diagnosis of SM are shown in Table 2. Systemic symptoms of mast cell mediator release such as osteoporosis, bone fractures, and gastrointestinal symptoms, including bloating and diarrhea, can be present for many years and are typically unrecognized.14
 | Table 2 WHO criteria for SM Note: Adapted with permission of American Society of Hematology, from Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts, Valent P, Akin C, Metcalfe DD, Colume 129(Edition 11), 2017; permission conveyed through Copyright Clearance Center, Inc.135 Abbreviations: MCs, mast cells; BM, bone marrow; SM, systemic mastocytosis; WHO, World Health Organization. |
Mastocytosis and MCAS can be identified as a cause of idiopathic anaphylaxis due to the fact that they can be controlled with tyrosine-kinase inhibitors.26
Treatment with venom immunotherapy (VIT) – if an IgE mechanism can be demonstrated, with or without omalizumab – provides a dramatic increase in patient safety when re-stung.143,146,147 Although there is no increase in the prevalence of drug allergy in patients with clonal mast cell disorders,148 reactions to NSAIDs are greater in these patients and can present as anaphylaxis.149
Drug allergy
The increase in anaphylaxis to drugs in the last 20 years is thought to be due to the increase in the variety and use of drugs. Some of these drugs have relatively high rates of HSRs including antibiotics, chemotherapeutic agents, mAbs, replacement factors, and biological agents. The pattern of presentation for HSRs continues to evolve, and new classifications have been created to encompass them.14,36,38
Medications are typically cited as among the most common triggers of fatal HSRs without age restriction and have been documented frequently in both pediatric and elderly populations.150 According to the European Anaphylaxis Registry, drugs were the third most frequent elicitor of anaphylaxis after food and insect venoms in the pediatric age group.151 Recently, a study from Turkey reported that 84 (15%) of 561 children were diagnosed with drug allergy; the most common drugs included β-lactam antibiotics (33%; penicillin, amoxicillin–clavulanic acid, ceftriaxone, cefixime, cefaclor, and piperacillin–tazobactam), NSAIDs (25%), chemotherapeutics (19%; asparaginase, oxaliplatin, etoposide, and docetaxel), biologic agents and enzymes (12%; rituximab, eculizumab, and enzymes like galsulfase and idursulfase), anesthetic and neuromuscular blocking agents (5%; propofol, midazolam, and rocuronium), and others (6%; ranitidine, triptorelin, cyclopentolate, iopromide, and Factor 9).34
On the other hand, it has been reported that older patients have both – higher rates of drug-induced anaphylaxis and an increased risk of severe reactions.152–154
Antibiotics are still the most common drug to cause anaphylaxis. IgE-mediated mechanisms are most commonly implicated in β-lactam allergy,63 and certain populations, such as patients with cystic fibrosis and patients treated with multiple courses of antibiotics, are at high risk.155
Patients presenting with anaphylaxis during anesthesia or after quinolone use for whom an IgE mechanism cannot be demonstrated by skin testing may have either increased protein levels or functional MRGPRX2 receptors that can bind and activate mast cells without IgE mediation; however, the participation of MRGPRX2 receptors has not been confirmed in human subjects.27–29
Due to the increased use of chemotherapies and targeted therapies, including mAbs, drug HSRs and anaphylaxis have increased dramatically worldwide, preventing the use of first-line therapies and affecting patient survival and quality of life.156 Some of the most immunogenic chemotherapies are carboplatin, cisplatin, oxaliplatin, and taxanes for ovarian, lung, breast, colon, and prostate cancers.157
Platins can elicit Type I reactions and typically require repeated exposures.85,88,158 Oxaliplatin is an exception to this, and first lifetime exposure reactions have been documented as well as CRRs and mixed reactions.159 Taxanes can induce similar reactions with atypical symptoms, such as back pain, through IgE- and non–IgE-mediated mechanisms. Cremophor and Polysorbate 80, used as diluents, are thought to be implicated in non–IgE-mediated reactions, which can occur at first or second lifetime exposures.156,158,160 In a recent study, STs conducted on 145/164 patients treated with taxane hypersensitivity were used to establish HSR risk and treatment protocol.161
Furthermore, mAbs are used in the treatment of neoplastic, autoimmune, and inflammatory diseases, and their clinical applications are becoming broader. Targets for these drugs include CD20, HER-2, epidermal growth factor receptor, IL-6 receptor, TNF-a, CD30, vascular endothelial growth factor A, programmed cell death protein 1/programmed death ligand L, IL-4, IL-5, IL-13, and IgE, among others.162 Although chimeric mAbs, such as rituximab and infliximab, present with the highest incidence of reaction, humanized and human mAbs – although less immunogenic – are engineered with mouse glycosylation patterns, which can result in allergenic determinants. Even with fully human mAbs, such as adalimumab and ofatumumab, anaphylaxis has been reported.18,163 Clinical presentation included a spectrum of atypical symptoms such as CRRs, infusion-related reactions, and mixed reactions that present with chills, fever, and generalized malaise followed by hypotension, desaturation, and cardiovascular collapse.106,164 These reactions can occur at first exposure but have also been seen after several exposures.67,163–165 Mixed reactions occur when these symptoms are associated with Type I symptoms and mast cells/basophils involvement or positive ST results, demonstrating an IgE mechanism in addition to the release of IL-6.89
Anaphylaxis at first exposure has been observed with cetuximab in patients with preformed carbohydrate galactose-a-1,3-galactose IgE antibodies due to tick exposure (Amblyomma americanum).166 Galactose-a-1,3-galactose is expressed on non-primate mammalian proteins and is present on the cetuximab heavy chain.167
Patients under treatment with β-blockers and ACE inhibitors
Evidence for β-blockers and ACE inhibitor treatment with anaphylaxis is sporadic and limited to retrospective data. There are no prospective trials to explain the relationship between these drugs and anaphylaxis.168 Theoretically, both β-blockers and ACE inhibitors may affect the patient’s likelihood of developing anaphylaxis and their ability to respond to treatment.169–171
The relationship between antihypertensive medication and the severity of anaphylaxis was associated with the conclusion that β-blocker, ACE-inhibitor, diuretic, and antihypertensive medication used in combination was significantly associated with grade 3 anaphylaxis (three or more organ/system involvement; OR, 2.8; 95% CI, 1.5–5.2; P = 0.0008), showing that the use of these drugs increased the severity of anaphylaxis.172
A telephonic survey conducted on German patients with a history of severe anaphylaxis had identified monotherapy with β-blockers or ACE inhibitors as a risk factor for severe anaphylaxis, which was more pronounced with combination therapy. In mouse models, the role of potential cofactors on anaphylactic responses confirmed that β-blocker and ACE inhibitors have modest anaphylaxis-promoting activities as single substances, but clearly display potentiating effects when administered in combination.173
The current recommendation is to avoid β-blockers and ACE inhibitors in patients at risk for anaphylaxis with no cardiovascular disease. Patients with cardiovascular disease should continue with β-blockers and ACE inhibitors, due to the proven decrease in mortality and increased life expectancy of this therapy.168
Latex allergy
Latex in gloves, condoms, and surgical materials has been shown to induce anaphylaxis,174 and cross-reactivity has been demonstrated with fruit allergens.175 In a recent publication, which looked at patients undergoing surgical procedures, latex was reported as the second most common cause of anaphylaxis (20%) after neuromuscular blocking agents (47%),176 and the risk increases with exposure.177
Although STs for latex have higher specificity (96–100%) and sensitivity (95–99%) than in vitro tests, they have been associated with anaphylactic events and are, therefore, discontinued in the US;178 diagnosis relies on serum IgE.
With the advent of latex-free facilities and the use of non-latex gloves, the incidence of latex-induced anaphylaxis has decreased.179
Hymenoptera venom
Epidemiological studies have estimated the prevalence of systemic reaction to Hymenoptera stings from 0.3 to 8.9%, and insect stings as 1.5–34.1% of all-cause anaphylaxis. Cases of fatal insect-sting anaphylaxis account for approximately 20% of fatal anaphylaxis from any cause.82
Risk factors for severe HSRs were determined from a European study of 962 patients with systemic reactions to Hymenoptera sting: elevated baseline tryptase levels (suggestive of MCAS), ACE inhibitor therapy, male, vespid sting, history of milder reactions, and systemic reactions to honey bee stings.180 Other factors determining the severity of reaction to Hymenoptera sting include advanced age as well as preexisting cardiovascular and respiratory disease.142 Mastocytosis may provoke fatal anaphylaxis despite VIT; however, so far, this has been observed only after discontinuation of treatment.82
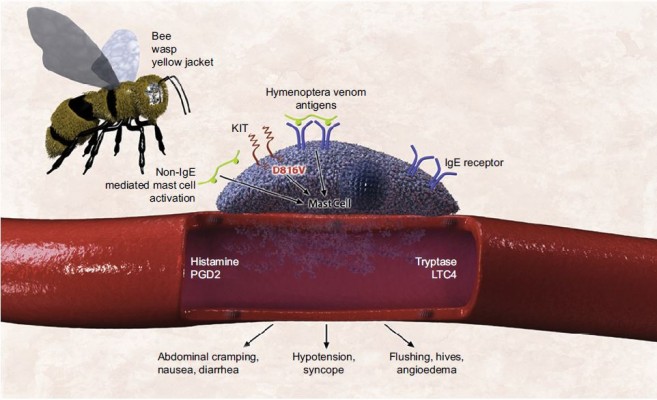
Hymenoptera anaphylaxis may be the presenting symptom of mastocytosis in an otherwise healthy individual.142 The mechanism of increased susceptibility to Hymenoptera venom anaphylaxis in mastocytosis has not been elucidated, but explanations include the following: 1) increased number of mast cells amplifying the severity of the reaction resulting from high mast cell mediator release; 2) perivascular location of the mast cells, providing direct access to the intravascular compartment; 3) D816V-mutant KIT amplifying the IgE-mediated reaction; and 4) additive direct (non–IgE-mediated) mast cell-activating properties of the Hymenoptera venom, including phospholipase A2 (Figure 3).142
 | Figure 3 Mast cell defects may collectively lead to increased risk for anaphylaxis in response to hymenoptera venom in patients with clonal mast cell disease. Note: Reproduced from The Journal of Allergy and Clinical Immunology: In Practice, Volume 3 (Edition 3), Castells MC, Hornick JL, Akin C. Anaphylaxis after hymenoptera sting: is it venom allergy, a clonal disorder, or both? Pages 350–355, Copyright 2015, with permission form Elsevier.142 Abbreviations: IgE, immunoglobulin E; LTC4, leukotriene C4; PGD2, prostaglandin D2. |
Furthermore, VIT with premedication has been suggested to increase VIT safety. Similarly, omalizumab has been shown to decrease the severity of allergic anaphylaxis following VIT administration in patients who had not previously tolerated immunotherapy,181 including patients with mastocytosis.146,147,182
Progestogen hypersensitivity
Progestogen hypersensitivity is a new terminology proposed for the already known autoimmune progesterone dermatitis – a rare and under-recognized disease that affects women of childbearing age.183 Symptoms can appear at any age, but most frequently appear in the third decade of life. A broader presentation of cyclical symptoms, including asthma and anaphylaxis, has been described during the progesterone surge.184,185 According to the cases previously reported, the onset of symptoms in 75% of patients occurs during the progesterone peak of the menstrual cycle183,186 (Figure 4). The lack of specific IgE against progesterone does not necessarily rule out the diagnosis.187 Endogenous and exogenous sources of progesterone can be allergenic triggers, and desensitization to progesterone has reversed infertility.183,186
 | Figure 4 Onset of symptoms in patients during the menstrual cycle in progesterone hypersensitivity. Note: Adapted from Fertility and Sterility, Volume 95 (Edition 3), Prieto-Garcia A, Sloane DE, Gargiulo AR, Feldweg AM, Castells M. Autoimmune progesterone dermatitis: clinical presentation and management with progesterone desensitization for successful in vitro fertilization, Pages 1121.e9–e13, Copyright 2011, with permission from Elsevier.186 |
One publication reports that four of six women with dermatitis or anaphylaxis related to the luteal phase of the menstrual cycle (some of them related to in vitro fertilization) and positive STs to progesterone were successfully desensitized to this hormone; this is the first evidence of hormonal desensitization that has culminated in successful pregnancies or good tolerance to oral contraceptives.186
More recently, a report based on 24 cases provided an algorithm for the classification, study, and management of these patients.183
Cardiovascular diseases
Kounis syndrome is diagnosed when an acute coronary syndrome co-incidentally occurred with a HSR following an allergenic event. It is thought to be caused by inflammatory mediators released by mast cells.188 The syndrome depends on the subtype of the coronary artery disease present, and symptoms reverse without sequelae within a few hours of the initial anaphylactic symptoms. Coronary damage can occur in severe episodes, but is rare. Use of intravenous epinephrine can have a detrimental role in patients with allergic angina, because of worsening myocardial ischemia, prolongation of the QTc interval, induction of coronary vasospasm, and arrhythmias; however, if necessary, intramuscular doses of epinephrine can be given.189
Takotsubo is defined as stress cardiomyopathy during anaphylaxis secondary to a rapid elevation of circulating catecholamines released in middle-aged women and is characterized by transient regional systolic and diastolic dysfunction of the left ventricle leading to a variety of wall motion abnormalities. This can lead to fatal cardiac arrhythmias with heart failure.190 Treatment is focused on stabilizing the acute onset and correct management of complications.191
Treatment and prevention
Epinephrine is the cornerstone in the acute treatment of anaphylaxis (Figure 5), and delayed administration is associated with increased mortality.3,7,14,32,35,192–198 The recommended dose of epinephrine is 0.01 mg/kg of a 1 mg/mL (1:1,000) dilution, to a maximum dose of 0.5 mg in an adult or 0.3 mg in a child, and can be repeated three times every 5–15 minutes. Patients should lie down with their legs elevated to avoid the empty inferior vena cava/empty ventricular syndrome and sudden death.4,32
 | Figure 5 Acute treatment of anaphylaxis. Abbreviations: ACE, angiotensin-converting enzyme; CV, cardiovascular; GI, gastrointestinal; IV, intravenous.
|
Epinephrine leads to vasoconstriction, increased peripheral vascular resistance, and decreased mucosal edema through the α1-adrenergic receptor, which is present in many tissues (including mast cells). It is the only drug with these effects among the medications used for the treatment of anaphylaxis, and clinically, it manifests as relief of airflow obstruction in the larynx as well as the lower airways, prevention of hypotension, and shock.199
In 2014, a review analyzed how the inadequate use of auto-injectable epinephrine could affect the management of anaphylaxis. Several critical points established by the authors included: problems in properly recognizing and diagnosing the disease; when it was recognized, it was treated with epinephrine, but even in these cases, the use of epinephrine was suboptimal; the patients did not carry auto-injectable epinephrine, or they were not trained for its use and there was no access to this drug in public spaces such as airports, schools, sport stadiums, and restaurants. Based on these findings, suggestions for approval included training health professionals and promoting the availability of epinephrine in public places to prevent cases of fatal anaphylaxis.196
If a patient presents with acute urticaria, antihistamines are recommended without the use of epinephrine unless another organ system is involved. In the case of urticaria with involvement of another organ system, the diagnosis of anaphylaxis should prevail, and epinephrine is the drug of choice.
Long-term treatment after an acute episode of anaphylaxis includes educating and training the patient and their family about the disease and the use of auto-injectable epinephrine32 (Table 3). Critically ill patients who have experienced an episode of anaphylaxis should be evaluated by an allergist, and triggers should be identified using allergen STs and/or measurement of allergen-specific IgE levels in serum.200 It is important to determine tryptase, both at baseline and during the reaction, to aid in a differential diagnosis.
 | Table 3 Preventive treatment of anaphylaxis Abbreviation: IgE, immunoglobulin E. |
Avoidance education based on the known trigger of anaphylaxis should be part of patient education. Anaphylaxis to stinging insects can be prevented by immunotherapy with the relevant insect venom(s), and anaphylaxis to certain medications can be overcome by desensitization.
For food allergy, the first step is to avoid the trigger and treat symptoms in case they appear. Oral immunotherapy for food-induced anaphylaxis is under investigation. In the US, results of a peanut protein oral immunotherapy (OIT) Phase III clinical trial in children aged 9–36 months demonstrated a good safety profile, with a decrease in specific IgE and increased tolerance, while achieving safe dietary reintroduction.201 A similar study reported sustained unresponsiveness following egg OIT, increasing the likelihood of tolerated unbaked eggs in diet,202 and a report from Europe using sublingual immunotherapy (SLIT) with Pru p3 (the primary sensitizer in fruits and responsible for severe HSRs in the Mediterranean area) also demonstrated good tolerance after treatment not only to peach, but also showed immunological changes in allergic patients to Ara h9 – an LTP from peanut.203
Immunotherapy should be considered in all patients who have had a history of anaphylaxis after an insect sting, and who have a positive ST or a positive result to an in vitro test for venom-specific IgE antibodies.204 Immunotherapy in patients with mastocytosis has been shown to significantly reduce the risk of anaphylaxis after a re-sting.80,205 VIT is recommended indefinitely in patients with mastocytosis and Hymenoptera allergy because mastocytosis is a chronic disorder with no curative options and systemic reactions and sting-related fatalities have been reported in patients who discontinued immunotherapy.142
Omalizumab – an anti-IgE mAb – has been shown to be a successful treatment for idiopathic anaphylaxis with IgE-mediated disease, effectively reducing the number of episodes and improving quality of life.206
Several cases have demonstrated the benefits of omalizumab as a pretreatment in patients who have not previously tolerated immunotherapy – both in healthy individuals and MCAS patients.146,181,207
Drug desensitization is a groundbreaking procedure for the management of immediate drug HSRs. It protects patients against anaphylaxis – maintaining patients on first-line therapy and thus representing an important advance in the treatment and prognosis of their medical condition.14
Desensitization to drugs has been used, for the last 15 years, in thousands of cases, with safety and great efficacy in patients with anaphylaxis to chemotherapeutic agents, mAbs, and antibiotics without any deaths. Powerful inhibitory mechanisms are initiated at low antigen doses, which can dominate the activator pathways and prevent anaphylaxis.118,208–210 Drug desensitization should be considered the standard of care when allergic patients require first-line therapy.
In a 2018 study, 526 desensitizations were carried out with 16 different mAbs in 104 patients, showing that reactions during the procedure are rare (23%) and the majority are mild (Grade I), with a decrease in HSR severity. STs may be useful as a predictor of severity when results are positive. The study demonstrated that desensitization provides a safe and effective re-treatment option to continue first-line therapy.38
The largest desensitization study worldwide has reported that 370 highly allergic patients received 2,177 successful desensitizations to 15 drugs. Most importantly, carboplatin-desensitized patients had a non-statistically significant lifespan advantage over non-allergic control subjects, indicating that the efficacy of carboplatin was not reduced in allergic patients and that rapid drug-desensitization protocols are as effective as standard infusions.211 Based on the results of the 2,177 desensitizations, 93% had no or mild reactions, whereas 7% had moderate-to-severe reactions that did not preclude the completion of the treatment; there were no deaths.211 Desensitization to mAbs and antibiotics has been successfully undertaken in targeted populations, such as patients with cystic fibrosis, with similar protocols. On the other hand, desensitization to aspirin for aspirin-exacerbated respiratory disease has induced an increased sense of smell, prevented the regrowth of polyps, and helped stabilize asthma symptoms.212–214
Current issues with available treatment modalities
Glucocorticoids and antihistamines should be used as complementary treatment in anaphylaxis. They are not life-saving (like epinephrine) and do not prevent or relieve upper airway obstruction, hypotension, or shock.32 Glucocorticoid and antihistamine administration should never delay the administration of epinephrine. The use of glucocorticoids and antihistamines alone as prophylactic agents cannot be recommended, as they do not prevent life-threatening reactions and their use has only been studied with radiocontrast HSRs.215
As mentioned previously, omalizumab has been demonstrated to be effective in patients who have episodes of food-induced, VIT-induced, and/or idiopathic anaphylaxis. In a selected group of patients, pretreatment with omalizumab is cost effective and allows the patient to be treated with fewer reactions.
Social aspects of treatment of anaphylaxis and quality of life
Quality of life of patients experiencing one or more episodes of food, medication, Hymenoptera venom, or exercise-induced anaphylaxis is a critical aspect of the patient’s treatment and management. It affects not only the patient but also their family, school, and workplace, as well as their traveling and other social interactions.216 Up to 12% of people who experience an anaphylactic shock can develop post-traumatic stress disorder, as well as long-term effects such as fear and anxiety that can dominate and restrict social, family, and professional interactions in adults and children.217–219 In particular, adolescents can present with high-risk behavior.220 Education of patients, health-care providers, and ED personnel can have a positive effect in all of these areas.221
Conclusion
Anaphylaxis should be considered when symptoms in two or more organs and/or sudden changes in vital signs occur in patients with or without evident exposure to allergens such as food, drugs including antibiotics, chemotherapy and mAbs, Hymenoptera stings, or environmental factors (ie, latex, animal dander, and pollen). Clinical manifestations do not always include skin and mucosal changes, and hypotension and shock may not be present. Non-typical symptoms such as pain, chills, rigors, and fever have been recognized as part of a new phenotypic expression of anaphylaxis during reaction to drugs such as chemotherapy and mAbs.
A new classification system based on precision medicine using phenotypes, endotypes, and biomarkers has expanded the Gell and Coombs classification to better identify and treat anaphylaxis.
Skin testing is a safe tool to identify sensitization to numerous allergens, even in high-risk patients, such as those with cancer, cystic fibrosis, and mastocytosis. Other biomarkers such as specific IgE, component-resolved diagnostics, and BAT are evolving as useful tools for the future.
The tryptase level – which can be detected from 30 minutes to 3 hours after the onset of a reaction – is the best biomarker at the present time which can also identify patients with clonal mast cell disorders.
Epinephrine is the cornerstone in the acute treatment of anaphylaxis, and delayed administration is associated with increased mortality.
Patients with drug-induced anaphylaxis should be considered candidates for desensitization if in need of first-line therapies. Desensitization can increase life expectancy and quality of life of patients with drug-induced anaphylaxis.
Allergists should educate patients and provide them with action plans and tools, including auto-injectable epinephrine. Education of all specialists and health-care providers in the symptoms, presentation, and acute management of anaphylaxis is key to increasing awareness of anaphylaxis.
Precision medicine requires further research on new biomarkers and exploration of new treatment modalities such as anti-IgE.
Acknowledgment
The authors did not receive compensation nor was the content of the article influenced in any way. Adamis Pharmaceuticals paid publication fees for the articles in this special issue on anaphylaxis.
Disclosure
Dr Teodorikez Wilfox Jimenez-Rodriguez has received grants from the Spanish Society of Allergy and Clinical Immunology, the Valencian Association of Allergy and Clinical Immunology, and the Medical Association of Alicante, Spain.
Marlene Garcia-Neuer reports no conflicts of interest in this work.
Leila A Alenazy has been sponsored by the College of Medicine Research Center, Deanship of Scientific Research, King Saud University.
Dr Mariana Castells has received consultancy fees from Sanofi, Genentech, Lytix Biopharma, Bentham Science, and UpToDate. The authors report no other conflicts of interest in this work.
References
Ring J, Behrendt H, de Weck A. History and classification of anaphylaxis. Chem Immunol Allergy. 2010;95:1–11. | ||
Rojido GM. Cien años de anafilaxia. Alergol Inmunol Clin. 2001;16:364–368. | ||
Simons FE. Anaphylaxis. J Allergy Clin Immunol. 2010;125(2 Suppl 2): S161–S181. | ||
Simons FE, Ardusso LR, Bilò MB, et al. International consensus on (ICON) anaphylaxis. World Allergy Organ J. 2014;7(1):9. | ||
Simons FE, Ardusso LR, Bilò MB, et al; World Allergy Organization. 2012 Update: World Allergy Organization Guidelines for the assessment and management of anaphylaxis. Curr Opin Allergy Clin Immunol. 2012;12(4):389–399. | ||
Sampson HA, Muñoz-Furlong A, Campbell RL, et al. Second symposium on the definition and management of anaphylaxis: summary report – second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Ann Emerg Med. 2006;47(4):373–380. | ||
Muraro A, Roberts G, Worm M, et al; EAACI Food Allergy and Anaphylaxis Guidelines Group. Anaphylaxis: guidelines from the European Academy of Allergy and Clinical Immunology. Allergy. 2014;69(8):1026–1045. | ||
Simons FE. Anaphylaxis: recent advances in assessment and treatment. J Allergy Clin Immunol. 2009;124(4):625–636; quiz 637–638. | ||
Simons FE. Anaphylaxis. J Allergy Clin Immunol. 2008;121(2 Suppl):S402–S407; quiz S420. | ||
Clark S, Camargo CA Jr. Epidemiology of anaphylaxis. Immunol Allergy Clin North Am. 2007;27(2):145–163, v. | ||
Johansson SG, Bieber T, Dahl R, et al. Revised nomenclature for allergy for global use: report of the Nomenclature Review Committee of the World Allergy Organization, October 2003. J Allergy Clin Immunol. 2004;113(5):832–836. | ||
Sampson HA, Muñoz-Furlong A, Bock SA, et al. Symposium on the definition and management of anaphylaxis: summary report. J Allergy Clin Immunol. 2005;115(3):584–591. | ||
Muraro A, Lemanske RF Jr, Castells M, et al. Precision medicine in allergic disease-food allergy, drug allergy, and anaphylaxis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma and Immunology. Allergy. 2017;72(7):1006–1021. | ||
Castells M. Diagnosis and management of anaphylaxis in precision medicine. J Allergy Clin Immunol. 2017;140(2):321–333. | ||
Dombrowicz D, Brini AT, Flamand V, et al. Anaphylaxis mediated through a humanized high affinity IgE receptor. J Immunol. 1996;157(4):1645–1651. | ||
Reber LL, Hernandez JD, Galli SJ. The pathophysiology of anaphylaxis. J Allergy Clin Immunol. 2017;140(2):335–348. | ||
Finkelman FD, Khodoun MV, Strait R. Human IgE-independent systemic anaphylaxis. J Allergy Clin Immunol. 2016;137(6):1674–1680. | ||
Gillis CM, Jönsson F, Mancardi DA, et al. Mechanisms of anaphylaxis in human low-affinity IgG receptor locus knock-in mice. J Allergy Clin Immunol. 2017;139(4):1253–1265.e14. | ||
Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80(4):1669–1699. | ||
Gill P, Jindal NL, Jagdis A, Vadas P. Platelets in the immune response: revisiting platelet-activating factor in anaphylaxis. J Allergy Clin Immunol. 2015;135(6):1424–1432. | ||
Finkelman FD. Anaphylaxis: lessons from mouse models. J Allergy Clin Immunol. 2007;120(3):506–515; quiz 516–517. | ||
Sala-Cunill A, Björkqvist J, Senter R, et al. Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J Allergy Clin Immunol. 2015;135(4):1031–1043.e6. | ||
Kajiwara N, Sasaki T, Bradding P, et al. Activation of human mast cells through the platelet-activating factor receptor. J Allergy Clin Immunol. 2010;125(5):1137–1145.e6. | ||
Vultaggio A, Matucci A, Nencini F, et al. Anti-infliximab IgE and non-IgE antibodies and induction of infusion-related severe anaphylactic reactions. Allergy. 2010;65(5):657–661. | ||
Castells MC. Anaphylaxis to chemotherapy and monoclonal antibodies. Immunol Allergy Clin North Am. 2015;35(2):335–348. | ||
Lieberman P, Garvey LH. Mast cells and anaphylaxis. Curr Allergy Asthma Rep. 2016;16(3):20. | ||
Subramanian H, Gupta K, Ali H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J Allergy Clin Immunol. 2016;138(3):700–710. | ||
Spoerl D, Nigolian H, Czarnetzki C, Harr T. Reclassifying anaphylaxis to neuromuscular blocking agents based on the presumed patho-mechanism: IgE-mediated, pharmacological adverse reaction or “innate hypersensitivity”? Int J Mol Sci. 2017;18(6). pii: E1223. | ||
Zhang T, Che D, Liu R, et al. Typical antimicrobials induce mast cell degranulation and anaphylactoid reactions via MRGPRX2 and its murine homologue MRGPRB2. Eur J Immunol. 2017;47(11):1949–1958. | ||
Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med. 2012;18(5):693–704. | ||
Altman AM, Camargo CA Jr, Simons FE, et al. Anaphylaxis in America: a national physician survey. J Allergy Clin Immunol. 2015;135(3):830–833. | ||
Simons FE, Ardusso LR, Dimov V, et al; World Allergy Organization. World Allergy Organization anaphylaxis guidelines: 2013 update of the evidence base. Int Arch Allergy Immunol. 2013;162(3):193–204. | ||
Mostmans Y, Blykers M, Mols P, Gutermuth J, Grosber M, Naeije N. Anaphylaxis in an urban Belgian emergency department: epidemiology and aetiology. Acta Clin Belg. 2016;71(2):99–106. | ||
Cavkaytar O, Karaatmaca B, Cetinkaya PG, et al. Characteristics of drug-induced anaphylaxis in children and adolescents. Allergy Asthma Proc. 2017;38(5):56–63. | ||
Turner PJ, Jerschow E, Umasunthar T, Lin R, Campbell DE, Boyle RJ. Fatal anaphylaxis: mortality rate and risk factors. J Allergy Clin Immunol Pract. 2017;5(5):1169–1178. | ||
Galvão VR, Castells MC. Hypersensitivity to biological agents-updated diagnosis, management, and treatment. J Allergy Clin Immunol Pract. 2015;3(2):175–185; quiz 186. | ||
Khan DA. Hypersensitivity and immunologic reactions to biologics: opportunities for the allergist. Ann Allergy Asthma Immunol. 2016;117(2):115–120. | ||
Isabwe G, Garcia Neuer M, de las Vecillas Sánchez L, Lynch DM, Marquis K, Castells M. Novel evidence-based phenotypes and endotypes of hypersensitivity reactions to 16 monoclonal antibodies: management with 526 desensitizations. J Allergy Clin Immunol. In press 2017. | ||
Schulert GS, Grom AA. Macrophage activation syndrome and cytokine-directed therapies. Best Pract Res Clin Rheumatol. 2014;28(2):277–292. | ||
Tanaka T, Narazaki M, Kishimoto T. Immunotherapeutic implications of IL-6 blockade for cytokine storm. Immunotherapy. 2016;8(8):959–970. | ||
Gomez-Salinero JM, Rafii S. Plasmin regulation of acute cytokine storm. Blood. 2017;130(1):5–6. | ||
Neumann FJ, Ott I, Marx N, et al. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler Thromb Vasc Biol. 1997;17(12):3399–3405. | ||
Muñoz-Cano RM, Picado C, Valero A, Bartra J. Mechanisms of anaphylaxis beyond IgE. J Investig Allergol Clin Immunol. 2016;26(2):73–82; quiz 2p following 83. | ||
Fregonese L, Swan FJ, van Schadewijk A, et al. Expression of the anaphylatoxin receptors C3aR and C5aR is increased in fatal asthma. J Allergy Clin Immunol. 2005;115(6):1148–1154. | ||
Chopra N, Oppenheimer J, Derimanov GS, Fine PL. Vancomycin anaphylaxis and successful desensitization in a patient with end stage renal disease on hemodialysis by maintaining steady antibiotic levels. Ann Allergy Asthma Immunol. 2000;84(6):633–635. | ||
Simon RA, Schatz M, Stevenson DD, et al. Radiographic contrast media infusions. Measurement of histamine, complement, and fibrin split products and correlation with clinical parameters. J Allergy Clin Immunol. 1979;63(4):281–288. | ||
Weiszhár Z, Czúcz J, Révész C, Rosivall L, Szebeni J, Rozsnyay Z. Complement activation by polyethoxylated pharmaceutical surfactants: Cremophor-EL, Tween-80 and Tween-20. Eur J Pharm Sci. 2012;45(4):492–498. | ||
van der Linden PW, Hack CE, Kerckhaert JA, Struyvenberg A, van der Zwan JC. Preliminary report: complement activation in wasp-sting anaphylaxis. Lancet. 1990;336(8720):904–906. | ||
Kishimoto TK, Viswanathan K, Ganguly T, et al. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med. 2008;358(23):2457–2467. | ||
Heinzerling L, Mari A, Bergmann KC, et al. The skin prick test – European standards. Clin Transl Allergy. 2013;3(1):3. | ||
Joint Task Force on Practice Parameters; American Academy of Allergy, Asthma and Immunology; American College of Allergy, Asthma and Immunology; Joint Council of Allergy, Asthma and Immunology. The diagnosis and management of anaphylaxis: an updated practice parameter. J Allergy Clin Immunol. 2005;115(3 Suppl 2):S483–S523. | ||
Sloane D, Govindarajulu U, Harrow-Mortelliti J, et al. Safety, costs, and efficacy of rapid drug desensitizations to chemotherapy and monoclonal antibodies. J Allergy Clin Immunol Pract. 2016;4(3):497–504. | ||
Pascal D, Jean Bousquet P, Romano A. Antibiotic-induced anaphylaxis. In: Castells MC, editor. Anaphylaxis and Hypersensitivity Reactions. Boston, MA: Humana Press; 2011:171–182. | ||
Brockow K, Romano A, Blanca M, Ring J, Pichler W, Demoly P. General considerations for skin test procedures in the diagnosis of drug hypersensitivity. Allergy. 2002;57(1):45–51. | ||
Goldberg A, Confino-Cohen R. Timing of venom skin tests and IgE determinations after insect sting anaphylaxis. J Allergy Clin Immunol. 1997;100(2):182–184. | ||
Nowak-Wegrzyn A, Assa’ad AH, Bahna SL, Bock SA, Sicherer SH, Teuber SS; Adverse Reactions to Food Committee of American Academy of Allergy, Asthma & Immunology. Work Group report: oral food challenge testing. J Allergy Clin Immunol. 2009;123(6 Suppl):S365–S383. | ||
Lin E, Saxon A, Riedl M. Penicillin allergy: value of including amoxicillin as a determinant in penicillin skin testing. Int Arch Allergy Immunol. 2010;152(4):313–318. | ||
Fernández J, Torres MJ, Campos J, Arribas-Poves F, Blanca M; DAP-Diater Group. Prospective, multicenter clinical trial to validate new products for skin tests in the diagnosis of allergy to penicillin. J Investig Allergol Clin Immunol. 2013;23(6):398–408. | ||
Blumenthal KG, Shenoy ES, Varughese CA, Hurwitz S, Hooper DC, Banerji A. Impact of a clinical guideline for prescribing antibiotics to inpatients reporting penicillin or cephalosporin allergy. Ann Allergy Asthma Immunol. 2015;115(4):294–300.e2. | ||
Demoly P, Adkinson NF, Brockow K, et al. International Consensus on drug allergy. Allergy Eur J Allergy Clin Immunol. 2014;69(4):420-437. | ||
Fox S, Park MA. Penicillin skin testing in the evaluation and management of penicillin allergy. Ann Allergy Asthma Immunol. 2011;106(1):1–7. | ||
Macy E, Schatz M, Lin C, Poon KY. The falling rate of positive penicillin skin tests from 1995 to 2007. Perm J. 2009;13(2):12–18. | ||
Macy E, Romano A, Khan D. Practical management of antibiotic hypersensitivity in 2017. J Allergy Clin Immunol Pract. 2017;5(3):577–586. | ||
Antúnez C, Martín E, Cornejo-García JA, et al. Immediate hypersensitivity reactions to penicillins and other betalactams. Curr Pharm Des. 2006;12(26):3327–3333. | ||
Antunez C, Blanca-Lopez N, Torres MJ, et al. Immediate allergic reactions to cephalosporins: evaluation of cross-reactivity with a panel of penicillins and cephalosporins. J Allergy Clin Immunol. 2006;117(2):404–410. | ||
Wheatley LM, Plaut M, Schwaninger JM, et al. Report from the National Institute of Allergy and Infectious Diseases workshop on drug allergy. J Allergy Clin Immunol. 2015;136(2):262–271.e2. | ||
Brennan PJ, Rodriguez Bouza T, Hsu FI, Sloane DE, Castells MC. Hypersensitivity reactions to mAbs: 105 desensitizations in 23 patients, from evaluation to treatment. J Allergy Clin Immunol. 2009;124(6):1259–1266. | ||
Gould HJ, Sutton BJ, Beavil AJ, et al. The biology of IGE and the basis of allergic disease. Annu Rev Immunol. 2003;21(1):579–628. | ||
Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol. 2010;125(2 Suppl 2): S73–S80. | ||
Smith JK, Krishnaswamy GH, Dykes R, Reynolds S, Berk SL. Clinical manifestations of IgE hypogammaglobulinemia. Ann Allergy Asthma Immunol. 1997;78(3):313–318. | ||
Wittig HJ, Belloit J, De Fillippi I, Royal G. Age-related serum immunoglobulin E levels in healthy subjects and in patients with allergic disease. J Allergy Clin Immunol. 1980;66(4):305–313. | ||
Paganelli R, Ansotegui IJ, Sastre J, et al. Specific IgE antibodies in the diagnosis of atopic disease. Clinical evaluation of a new in vitro test system, UniCAP (TM), in six European allergy clinics. Allergy. 1998;53(8):763–768. | ||
Sampson HA. Utility of food-specific IgE concentrations in predicting symptomatic food allergy. J Allergy Clin Immunol. 2001;107(5):891–896. | ||
Johnson TL, McCleskey PE, Rathkopf M, Meffert JJ, Hagan LL. Pathergy response to skin prick testing. J Allergy Clin Immunol. 2007;119(5):1270–1272. | ||
Krzysztof K, Lawrence D. Overview of in vitro allergy tests. UpToDate Online. 2016. Available form: https://www-uptodate-com.sangva.a17.csinet.es/contents/overview-of-in-vitro-allergy-tests?search=Overview%20of%20in%20vitro%20allergy%20tests.&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1. Accessed March 26, 2018. | ||
Harleman L, Sie A. History, blood tests or skin prick testing? Is it possible to predict the severity of allergic reactions in children with IgE-mediated food allergy? Arch Dis Child. 2015;100(6):594–598. | ||
Köhler J, Blank S, Müller S, et al. Component resolution reveals additional major allergens in patients with honeybee venom allergy. J Allergy Clin Immunol. 2014;133(5):1383–1389, 1389.e1–e6. | ||
Cifuentes L, Vosseler S, Blank S, et al. Identification of hymenoptera venom-allergic patients with negative specific IgE to venom extract by using recombinant allergens. J Allergy Clin Immunol. 2014;133(3):909–910. | ||
Nurmatov U, Dhami S, Arasi S, et al. Allergen immunotherapy for IgE-mediated food allergy: a systematic review and meta-analysis. Allergy. 2017;72(8):1133–1147. | ||
Ruëff F, Wenderoth A, Przybilla B. Patients still reacting to a sting challenge while receiving conventional hymenoptera venom immunotherapy are protected by increased venom doses. J Allergy Clin Immunol. 2001;108(6):1027–1032. | ||
Alfaya Arias T, Soriano Gómis V, Soto Mera T, et al; Hymenoptera Allergy Committee of the SEAIC. Key issues in hymenoptera venom allergy: an update. J Investig Allergol Clin Immunol. 2017;27(1):19–31. | ||
Bilò MB. Anaphylaxis caused by Hymenoptera stings: from epidemiology to treatment. Allergy. 2011;66(Suppl 95):35–37. | ||
Zhao Y, Qiao H. Detection of specific IgE antibodies to major and minor antigenic determinants in sera of penicillin allergic patients. Chin Med J (Engl). 2003;116(12):1904–1910. | ||
Romano A, Gaeta F, Valluzzi RL, Zaffiro A, Caruso C, Quaratino D. Natural evolution of skin-test sensitivity in patients with IgE-mediated hypersensitivity to cephalosporins. Allergy. 2014;69(6):806–809. | ||
Caiado J, Venemalm L, Pereira-Santos MC, Costa L, Barbosa MP, Castells M. Carboplatin-, oxaliplatin-, and cisplatin–specific IgE: cross-reactivity and value in the diagnosis of carboplatin and oxaliplatin allergy. J Allergy Clin Immunol Pract. 2013;1(5):494–500. | ||
Caiado J, Picard M. Diagnostic tools for hypersensitivity to platinum drugs and taxanes: skin testing, specific IgE, and mast cell/basophil mediators. Curr Allergy Asthma Rep. 2014;14(8):451. | ||
Tham EH, Cheng YK, Tay MH, Alcasabas AP, Shek LP. Evaluation and management of hypersensitivity reactions to chemotherapy agents. Postgrad Med J. 2015;91(1073):145–150. | ||
Hesterberg PE, Banerji A, Oren E, et al. Risk stratification for desensitization of patients with carboplatin hypersensitivity: clinical presentation and management. J Allergy Clin Immunol. 2009;123(6):1262–1267.e1. | ||
Caiado J, Castells M. Presentation and diagnosis of hypersensitivity to platinum drugs. Curr Allergy Asthma Rep. 2015;15(4):15. | ||
Matricardi PM, Kleine-Tebbe J, Hoffmann HJ, et al. EAACI molecular allergology user’s guide. Pediatr Allergy Immunol. 2016;27(Suppl 23):1–250. | ||
Sicherer SH, Dhillon G, Laughery KA, Hamilton RG, Wood RA. Caution: the Phadia hazelnut ImmunoCAP (f17) has been supplemented with recombinant Cor a 1 and now detects Bet v 1-specific IgE, which leads to elevated values for persons with birch pollen allergy. J Allergy Clin Immunol. 2008;122(2):413–414, 414.e2. | ||
Tuano KS, Davis CM. Utility of component-resolved diagnostics in food allergy. Curr Allergy Asthma Rep. 2015;15(6):32. | ||
Nicolaou N, Poorafshar M, Murray C, et al. Allergy or tolerance in children sensitized to peanut: prevalence and differentiation using component-resolved diagnostics. J Allergy Clin Immunol. 2010;125(1):191–197.e1–e13. | ||
Eigenmann PA, Lack G, Mazon A, et al. Managing nut allergy: a remaining clinical challenge. J Allergy Clin Immunol Pract. 2017;5(2):296–300. | ||
Ortolani C, Ballmer-Weber BK, Hansen KS, et al. Hazelnut allergy: a double-blind, placebo-controlled food challenge multicenter study. J Allergy Clin Immunol. 2000;105(3):577–581. | ||
Flinterman AE, Akkerdaas JH, den Hartog Jager CF, et al. Lipid transfer protein-linked hazelnut allergy in children from a non-Mediterranean birch-endemic area. J Allergy Clin Immunol. 2008;121(2):423–428.e2. | ||
Kattan JD, Sicherer SH. Optimizing the diagnosis of food allergy. Immunol Allergy Clin North Am. 2015;35(1):61–76. | ||
De Knop KJ, Verweij MM, Grimmelikhuijsen M, et al. Age-related sensitization profiles for hazelnut (Corylus avellana) in a birch-endemic region. Pediatr Allergy Immunol. 2011;22(1 Pt 2):e139–e149. | ||
Beyer K, Grishina G, Bardina L, Grishin A, Sampson HA. Identification of an 11S globulin as a major hazelnut food allergen in hazelnut-induced systemic reactions. J Allergy Clin Immunol. 2002;110(3):517–523. | ||
D’Urbano LE, Pellegrino K, Artesani MC, et al. Performance of a component-based allergen-microarray in the diagnosis of cow’s milk and hen’s egg allergy. Clin Exp Allergy. 2010;40(10):1561–1570. | ||
Gámez C, Sánchez-García S, Ibáñez MD, et al. Tropomyosin IgE-positive results are a good predictor of shrimp allergy. Allergy. 2011;66(10):1375–1383. | ||
Alessandri C, Zennaro D, Scala E, et al. Ovomucoid (Gal d 1) specific IgE detected by microarray system predict tolerability to boiled hen’s egg and an increased risk to progress to multiple environmental allergen sensitisation. Clin Exp Allergy. 2012;42(3):441–450. | ||
Douladiris N, Savvatianos S, Roumpedaki I, Skevaki C, Mitsias D, Papadopoulos NG. A molecular diagnostic algorithm to guide pollen immunotherapy in southern Europe: towards component-resolved management of allergic diseases. Int Arch Allergy Immunol. 2013;162(2):163–172. | ||
Ocmant A, Mulier S, Hanssens L, et al. Basophil activation tests for the diagnosis of food allergy in children. Clin Exp Allergy. 2009;39(8):1234–1245. | ||
McGowan EC, Saini S. Update on the performance and application of basophil activation tests. Curr Allergy Asthma Rep. 2013;13(1):101–109. | ||
Sanz ML, Gamboa PM, Mayorga C. Basophil activation tests in the evaluation of immediate drug hypersensitivity. Curr Opin Allergy Clin Immunol. 2009;9(4):298–304. | ||
Hausmann OV, Gentinetta T, Bridts CH, Ebo DG. The basophil activation test in immediate-type drug allergy. Immunol Allergy Clin North Am. 2009;29(3):555–566. | ||
Armstrong N, Wolff R, van Mastrigt G, et al. A systematic review and cost-effectiveness analysis of specialist services and adrenaline auto-injectors in anaphylaxis. Heal Technol Assess. 2013;17(17):1–117, v–vi. | ||
Giavina-Bianchi P, Galvão VR, Picard M, Caiado J, Castells MC. Basophil activation test is a relevant biomarker of the outcome of rapid desensitization in platinum compounds-allergy. J Allergy Clin Immunol Pract. 2017;5(3):728–736. | ||
Schwartz LB, Metcalfe DD, Miller JS, Earl H, Sullivan T. Tryptase levels as an indicator of mast-cell activation in systemic anaphylaxis and mastocytosis. N Engl J Med. 1987;316(26):1622–1626. | ||
Akin C, Soto D, Brittain E, et al. Tryptase haplotype in mastocytosis: relationship to disease variant and diagnostic utility of total tryptase levels. Clin Immunol. 2007;123(3):268–271. | ||
Valent P, Akin C, Arock M, et al. Definitions, criteria and global classification of mast cell disorders with special reference to mast cell activation syndromes: a consensus proposal. Int Arch Allergy Immunol. 2012;157(3):215–225. | ||
McNeil HP, Adachi R, Stevens RL. Mast cell-restricted tryptases: structure and function in inflammation and pathogen defense. J Biol Chem. 2007;282(29):20785–20789. | ||
Lyons JJ, Yu X, Hughes JD, et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat Genet. 2016;48(12):1564–1569. | ||
Schwartz LB. [webpage on the Internet] Laboratory tests to support the clinical diagnosis of anaphylaxis. UpToDate Online. 2016. Available from: https://www.uptodate.com/contents/laboratory-tests-to-support-the-clinical-diagnosis-of-anaphylaxis?source=search_result&search=Laboratory tests to support the clinical diagnosis of anaphylaxis&selectedTitle=1~150. Accessed November 27, 2017. | ||
Sampson HA, Mendelson L, Rosen JP. Fatal and near-fatal anaphylactic reactions to food in children and adolescents. N Engl J Med. 1992;327(6):380–384. | ||
van der Linden PW, Struyvenberg A, Kraaijenhagen RJ, Hack CE, van der Zwan JK. Anaphylactic shock after insect-sting challenge in 138 persons with a previous insect-sting reaction. Ann Intern Med. 1993;118(3):161–168. | ||
Vadas P, Gold M, Perelman B, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N Engl J Med. 2008;358(1):28–35. | ||
Louro E, Romano-Lieber NS, Ribeiro E. [Adverse events to antibiotics in inpatients of a university hospital]. Rev Saude Publica. 2007;41(6):1042–1048. Portuguese [with English abstract]. | ||
Bird JA, Burks AW. Food allergy and asthma. Prim Care Respir J. 2009;18(4):258–265. | ||
Foong RX, du Toit G, Fox AT. Asthma, food allergy, and how they relate to each other. Front Pediatr. 2017;5:89. | ||
Bock SA, Muoz-Furlong A, Sampson HA. Fatalities due to anaphylactic reactions to foods. J Allergy Clin Immunol. 2001;107(1):191–193. | ||
Dhami S, Sheikh A. Anaphylaxis: epidemiology, aetiology and relevance for the clinic. Expert Rev Clin Immunol. 2017;13(9):889–895. | ||
Summers CW, Pumphrey RS, Woods CN, McDowell G, Pemberton PW, Arkwright PD. Factors predicting anaphylaxis to peanuts and tree nuts in patients referred to a specialist center. J Allergy Clin Immunol. 2008;121(3):632–638.e2. | ||
Umasunthar T, Leonardi-Bee J, Turner PJ, et al. Incidence of food anaphylaxis in people with food allergy: a systematic review and meta-analysis. Clin Exp Allergy. 2015;45(11):1621–1636. | ||
Martin PE, Eckert JK, Koplin JJ, et al; HealthNuts Study Investigators. Which infants with eczema are at risk of food allergy? Results from a population-based cohort. Clin Exp Allergy. 2015;45(1):255–264. | ||
Akuete K, Guffey D, Israelsen RB, et al. Multicenter prevalence of anaphylaxis in clinic-based oral food challenges. Ann Allergy Asthma Immunol. 2017;119(4):339–348.e1. | ||
Feldweg AM. Exercise-induced anaphylaxis. Immunol Allergy Clin North Am. 2015;35(2):261–275. | ||
Sheffer AL, Austen KF. Exercise-induced anaphylaxis. J Allergy Clin Immunol. 1980;66(2):106–111. | ||
Shadick NA, Liang MH, Partridge AJ, et al. The natural history of exercise-induced anaphylaxis: survey results from a 10-year follow-up study. J Allergy Clin Immunol. 1999;104(1):123–127. | ||
Castells MC, Horan RF, Sheffer AL. Exercise-induced anaphylaxis. Curr Allergy Asthma Rep. 2003;3(1):15–21. | ||
Matsuo H, Kohno K, Morita E. Molecular cloning, recombinant expression and IgE-binding epitope of omega-5 gliadin, a major allergen in wheat-dependent exercise-induced anaphylaxis. FEBS J. 2005;272(17):4431–4438. | ||
Feldweg AM. Food-dependent, exercise-induced anaphylaxis: diagnosis and management in the outpatient setting. J Allergy Clin Immunol Pract. 2017;5(2):283–288. | ||
Akin C. Mast cell activation syndromes. J Allergy Clin Immunol. 2017;140(2):349–355. | ||
Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017;129(11):1420–1428. | ||
Akin C, Valent P, Metcalfe DD. Mast cell activation syndrome: proposed diagnostic criteria. J Allergy Clin Immunol. 2010;126(6):1099–1104.e4. | ||
González-de-Olano D, Álvarez-Twose I. Insights in anaphylaxis and clonal mast cell disorders. Front Immunol. 2017;8:792. | ||
González De Olano D, De La Hoz Caballer B, Núñez López R, et al. Prevalence of allergy and anaphylactic symptoms in 210 adult and pediatric patients with mastocytosis in Spain: a study of the Spanish network on mastocytosis (REMA). Clin Exp Allergy. 2007;37(10):1547–1555. | ||
Zanotti R, Lombardo C, Passalacqua G, et al. Clonal mast cell disorders in patients with severe Hymenoptera venom allergy and normal serum tryptase levels. J Allergy Clin Immunol. 2015;136(1):135–139. | ||
Brockow K, Jofer C, Behrendt H, Ring J. Anaphylaxis in patients with mastocytosis: a study on history, clinical features and risk factors in 120 patients. Allergy. 2008;63(2):226–232. | ||
Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; The American Academy of Allergy, Asthma & Immunology; And the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137(1):35–45. | ||
Castells MC, Hornick JL, Akin C. Anaphylaxis after hymenoptera sting: is it venom allergy, a clonal disorder, or both? J Allergy Clin Immunol Pract. 2015;3(3):350–355. | ||
Valent P, Escribano L, Broesby-Olsen S, et al; European Competence Network on Mastocytosis. Proposed diagnostic algorithm for patients with suspected mastocytosis: a proposal of the European Competence Network on Mastocytosis. Allergy. 2014;69(10):1267–1274. | ||
Escribano L, Álvarez-Twose I, Sánchez-Muñoz L, et al. Prognosis in adult indolent systemic mastocytosis: a long-term study of the Spanish Network on Mastocytosis in a series of 145 patients. J Allergy Clin Immunol. 2009;124(3):514–521. | ||
Álvarez-Twose I, González de Olano D, Sánchez-Muñoz L, et al. Clinical, biological, and molecular characteristics of clonal mast cell disorders presenting with systemic mast cell activation symptoms. J Allergy Clin Immunol. 2010;125(6):1269–1278.e2. | ||
Carter M, Robyn JA, Bressler PB, Walker JC, Shapiro GG, Metcalfe DD. Omalizumab for the treatment of unprovoked anaphylaxis in patients with systemic mastocytosis. J Allergy Clin Immunol. 2007;119(6):1550–1551. | ||
González de Olano D, Álvarez-Twose I, Esteban-López MI, et al. Safety and effectiveness of immunotherapy in patients with indolent systemic mastocytosis presenting with Hymenoptera venom anaphylaxis. J Allergy Clin Immunol. 2008;121(2):519–526. | ||
Bonadonna P, Pagani M, Aberer W, et al. Drug hypersensitivity in clonal mast cell disorders: ENDA/EAACI position paper. Allergy. 2015;70(7):755–763. | ||
Murali MR, Castells MC, Song JY, Dudzinski DM, Hasserjian RP. Case records of the Massachusetts General Hospital. Case 9-2011. A 37-year-old man with flushing and hypotension. N Engl J Med. 2011;364(12):1155–1165. | ||
Kuruvilla M, Khan DA. Anaphylaxis to drugs. Immunol Allergy Clin North Am. 2015;35(2):303–319. | ||
Grabenhenrich LB, Dolle S, Moneret-Vautrin A, et al. Anaphylaxis in children and adolescents: The European Anaphylaxis Registry. J Allergy Clin Immunol. 2016;137(4):1128–1137.e1. | ||
Clark S, Wei W, Rudders SA, Camargo CA Jr. Risk factors for severe anaphylaxis in patients receiving anaphylaxis treatment in US emergency departments and hospitals. J Allergy Clin Immunol. 2014;134(5):1125–1130. | ||
Jerschow E, Lin RY, Scaperotti MM, McGinn AP. Fatal anaphylaxis in the United States, 1999-2010: temporal patterns and demographic associations. J Allergy Clin Immunol. 2014;134(6):1318–1328.e7. | ||
Brown SG, Stone SF, Fatovich DM, et al. Anaphylaxis: clinical patterns, mediator release, and severity. J Allergy Clin Immunol. 2013;132(5):1141–1149.e5. | ||
Parmar JS, Nasser S. Antibiotic allergy in cystic fibrosis. Thorax. 2005;60(6):517–520. | ||
Bonamichi-Santos R, Castells M. Diagnoses and management of drug hypersensitivity and anaphylaxis in cancer and chronic inflammatory diseases: reactions to taxanes and monoclonal antibodies. Clin Rev Allergy Immunol. Epub 2016 Jun 8. | ||
Banerji A, Lax T, Guyer A, Hurwitz S, Camargo CA Jr, Long AA. Management of hypersensitivity reactions to carboplatin and paclitaxel in an outpatient oncology infusion center: a 5-year review. J Allergy Clin Immunol Pract. 2014;2(4):428–433. | ||
Castells MC. Hypersensitivity to antineoplastic agents. Curr Pharm Des. 2008;14(27):2892–2901. | ||
Wong JT, Ling M, Patil S, Banerji A, Long A. Oxaliplatin hypersensitivity: evaluation, implications of skin testing, and desensitization. J Allergy Clin Immunol Pract. 2014;2(1):40–45. | ||
Boulanger J, Boursiquot JN, Cournoyer G, et al; Comité de l’évolution des pratiques en oncologie. Management of hypersensitivity to platinum- and taxane-based chemotherapy: CEPO review and clinical recommendations. Curr Oncol. 2014;21(4):e630–e641. | ||
Picard M, Castells MC. Re-visiting hypersensitivity reactions to taxanes: a comprehensive review. Clin Rev Allergy Immunol. 2015;49(2):177–191. | ||
Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8:130. | ||
Puxeddu I, Giori L, Rocchi V, et al. Hypersensitivity reactions during treatment with infliximab, etanercept, and adalimumab. Ann Allergy Asthma Immunol. 2012;108(2):123–124. | ||
Vultaggio A, Castells MC. Hypersensitivity reactions to biologic agents. Immunol Allergy Clin North Am. 2014;34(3):615–632. | ||
Warrington RJ, Lee KR, McPhillips S. The value of skin testing for penicillin allergy in an inpatient population: analysis of the subsequent patient management. Allergy Asthma Proc. 2000;21(5):297–299. | ||
Chung CH, Mirakhur B, Chan E, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008;358(11):1109–1117. | ||
Commins SP, James HR, Kelly LA, et al. The relevance of tick bites to the production of IgE antibodies to the mammalian oligosaccharide galactose-α-1,3-galactose. J Allergy Clin Immunol. 2011;127(5):1286–1293.e6. | ||
Coop CA, Schapira RS, Freeman TM. Are ACE inhibitors and beta-blockers dangerous in patients at risk for anaphylaxis? J Allergy Clin Immunol Pract. 2017;5(5):1207–1211. | ||
TenBrook JA Jr, Wolf MP, Hoffman SN, et al. Should beta-blockers be given to patients with heart disease and peanut-induced anaphylaxis? A decision analysis. J Allergy Clin Immunol. 2004;113(5):977–982. | ||
Caviglia AG, Passalacqua G, Senna G. Risk of severe anaphylaxis for patients with Hymenoptera venom allergy: are angiotensin-receptor blockers comparable to angiotensin-converting enzyme inhibitors? J Allergy Clin Immunol. 2010;125(5):1171; author reply 1171–1172. | ||
Moneret-Vautrin DA, Latarche C. [Drugs as risk factors of food anaphylaxis in adults: a case-control study]. Bull Acad Natl Med. 2009;193(2):351–362; discussion 362–363. French [with English abstract]. | ||
Lee S, Hess EP, Nestler DM, et al. Antihypertensive medication use is associated with increased organ system involvement and hospitalization in emergency department patients with anaphylaxis. J Allergy Clin Immunol. 2013;131(4):1103–1108. | ||
Nassiri M, Babina M, Dölle S, Edenharter G, Ruëff F, Worm M. Ramipril and metoprolol intake aggravate human and murine anaphylaxis: evidence for direct mast cell priming. J Allergy Clin Immunol. 2015;135(2):491–499. | ||
Bousquet J, Flahault A, Vandenplas O, et al. Natural rubber latex allergy among health care workers: a systematic review of the evidence. J Allergy Clin Immunol. 2006;118(2):447–454. | ||
Crespo JF, Rodríguez J, James JM, Daroca P, Reaño M, Vives R. Reactivity to potential cross-reactive foods in fruit-allergic patients: implications for prescribing food avoidance. Allergy. 2002;57(10):946–949. | ||
Yunker NS, Wagner BJ. A pharmacologic review of anaphylaxis. Plast Surg Nurs. 2016;36(4):173–179. | ||
Kahn SL, Podjasek JO, Dimitropoulos VA, Brown CW Jr. Natural rubber latex allergy. Dis Mon. 2016;62(1):5–17. | ||
Kelly KJ, Sussman G. Latex allergy: where are we now and how did we get there? J Allergy Clin Immunol Pract. 2017;5(5):1212–1216. | ||
Brown RH, Taenkhum K, Buckley TJ, Hamilton RG. Different latex aeroallergen size distributions between powdered surgical and examination gloves: Significance for environmental avoidance. J Allergy Clin Immunol. 2004;114(2):358–363. | ||
Ruëff F, Przybilla B, Biló MB, et al. Predictors of severe systemic anaphylactic reactions in patients with Hymenoptera venom allergy: importance of baseline serum tryptase-a study of the European Academy of Allergology and Clinical Immunology Interest Group on Insect Venom Hypersensitivity. J Allergy Clin Immunol. 2009;124(5):1047–1054. | ||
Ricciardi L. Omalizumab: a useful tool for inducing tolerance to bee venom immunotherapy. Int J Immunopathol Pharmacol. 2016;29(4):726–728. | ||
Sokol KC, Ghazi A, Kelly BC, Grant JA. Omalizumab as a desensitizing agent and treatment in mastocytosis: a review of the literature and case report. J Allergy Clin Immunol Pract. 2014;2(3):266–270. | ||
Foer D, Buchheit KM, Gargiulo AR, Lynch DM, Castells M, Wickner PG. Progestogen hypersensitivity in 24 cases: diagnosis, management, and proposed renaming and classification. J Allergy Clin Immunol Pract. 2015;4(4):723–729. | ||
Bernstein IL, Bernstein DI, Lummus ZL, Bernstein JA. A case of progesterone-induced anaphylaxis, cyclic urticaria/angioedema, and autoimmune dermatitis. J Womens Health (Larchmt). 2011;20(4):643–648. | ||
Barr RG, Wentowski CC, Grodstein F, et al. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch Intern Med. 2004;164(4):379–386. | ||
Prieto-Garcia A, Sloane DE, Gargiulo AR, Feldweg AM, Castells M. Autoimmune progesterone dermatitis: clinical presentation and management with progesterone desensitization for successful in vitro fertilization. Fertil Steril. 2011;95(3):1121.e9–e13. | ||
Lieberman P. Catamenial anaphylaxis. J Allergy Clin Immunol Pract. 2014;2(3):358–359. | ||
Fassio F, Losappio L, Antolin-Amerigo D, et al. Kounis syndrome: A concise review with focus on management. Eur J Intern Med. 2016;30:7–10. | ||
Lieberman P, Simons FE. Anaphylaxis and cardiovascular disease: therapeutic dilemmas. Clin Exp Allergy. 2015;45(8):1288–1295. | ||
Kotsiou OS, Xirogiannis KI, Gourgoulianis KI. Kounis syndrome: is it really a takotsubo-like syndrome? J Investig Allergol Clin Immunol. 2017;27(3):198–200. | ||
Y-Hassan S, Tornvall P. Epidemiology, pathogenesis, and management of takotsubo syndrome. Clin Auton Res. 2018;28(1):53–65. | ||
Simons FE, Ardusso LR, Bilò MB, et al; World Allergy Organization. 2012 Update: World Allergy Organization Guidelines for the assessment and management of anaphylaxis. Curr Opin Allergy Clin Immunol. 2012;12(4):389–399. | ||
Sheikh A, Simons FE, Barbour V, Worth A. Adrenaline auto-injectors for the treatment of anaphylaxis with and without cardiovascular collapse in the community. Cochrane Database Syst Rev. 2012;(8):CD008935. | ||
Mostmans Y, Grosber M, Blykers M, Mols P, Naeije N, Gutermuth J. Adrenaline in anaphylaxis treatment and self-administration: experience from an inner city emergency department. Allergy. 2017;72(3):492–497. | ||
Simons FE. Anaphylaxis in infants: can recognition and management be improved? J Allergy Clin Immunol. 2007;120(3):537–540. | ||
Song TT, Worm M, Lieberman P. Anaphylaxis treatment: current barriers to adrenaline auto-injector use. Allergy. 2014;69(8):983–991. | ||
Camargo C, Simmons F. [webpage on the Internet] Anaphylaxis: rapid recognition and treatment. UpToDate Online. 2013:1–41. Available from: http://www.uptodate.com/contents/anaphylaxis-rapid- recognition-and-treatment. Accessed November 27, 2017. | ||
Greenberger PA. Fatal and near-fatal anaphylaxis: factors that can worsen or contribute to fatal outcomes. Immunol Allergy Clin North Am. 2015;35(2):375–386. | ||
Simons FE. First-aid treatment of anaphylaxis to food: focus on epinephrine. J Allergy Clin Immunol. 2004;113(5):837–844. | ||
Lieberman P, Nicklas RA, Oppenheimer J, et al. The diagnosis and management of anaphylaxis practice parameter: 2010 update. J Allergy Clin Immunol. 2010;126(3):477–480.e1–e42. | ||
Vickery BP, Berglund JP, Burk CM, et al. Early oral immunotherapy in peanut-allergic preschool children is safe and highly effective. J Allergy Clin Immunol. 2017;139(1):173–181.e8. | ||
Jones SM, Burks AW, Keet C, et al. Long-term treatment with egg oral immunotherapy enhances sustained unresponsiveness that persists after cessation of therapy. J Allergy Clin Immunol. 2017;137(4):1117–1127. | ||
Gomez F, Bogas G, Gonzalez M, et al. The clinical and immunological effects of Pru p 3 sublingual immunotherapy on peach and peanut allergy in patients with systemic reactions. Clin Exp Allergy. 2017;47(3):339–350. | ||
Casale TB, Burks AW. Hymenoptera-sting hypersensitivity. N Engl J Med. 2014;370(15):1432–1439. | ||
Bonadonna P, Gonzalez-de-Olano D, Zanotti R, et al. Venom immunotherapy in patients with clonal mast cell disorders: efficacy, safety, and practical considerations. J Allergy Clin Immunol Pract. 2013;1(5):474–478. | ||
Warrier P, Casale TB. Omalizumab in Idiopathic Anaphylaxis. Ann Allergy Asthma Immunol. 2009;102(3):257–258. | ||
Jagdis A, Vadas P. Omalizumab for a case of monoclonal mast cell activation syndrome with recurrent anaphylaxis. J Allergy Clin Immunol. 2014;113(2):AB27. | ||
Prieto-Garcia A, Zheng D, Adachi R, et al. Mast cell restricted mouse and human tryptase-heparin complexes hinder thrombin-induced coagulation of plasma and the generation of fibrin by proteolytically destroying fibrinogen. J Biol Chem. 2012;287(11):7834–7844. | ||
Castells M. Mast cell mediators in allergic inflammation and mastocytosis. Immunol Allergy Clin North Am. 2006;26(3):465–485. | ||
de Las Vecillas Sánchez L, Alenazy LA, Garcia-Neuer M, Castells MC. Drug hypersensitivity and desensitizations: mechanisms and new approaches. Int J Mol Sci. 2017;18(6). pii: E1316. | ||
Romano A, Torres MJ, Castells M, Sanz ML, Blanca M. Diagnosis and management of drug hypersensitivity reactions. J Allergy Clin Immunol. 2011;127(3 Suppl):S67–S73. | ||
Laidlaw TM, Boyce JA. Pathogenesis of Aspirin-Exacerbated Respiratory Disease and Reactions. Immunol Allergy Clin North Am. 2013;33(2):195–210. | ||
Cahill KN, Laidlaw TM. Pathogenesis of aspirin-induced reactions in aspirin-exacerbated respiratory disease. Immunol Allergy Clin North Am. 2016;36(4):681–691. | ||
Macy E, Bernstein JA, Castells MC, et al; Aspirin Desensitization Joint Task Force. Aspirin challenge and desensitization for aspirin-exacerbated respiratory disease: a practice paper. Ann Allergy Asthma Immunol. 2007;98(2):172–174. | ||
Tramer MR, von Elm E, Loubeyre P, Hauser C. Pharmacological prevention of serious anaphylactic reactions due to iodinated contrast media: systematic review. BMJ. 2006;333(7570):675. | ||
Warren CM, Otto AK, Walkner MM, Gupta RS. Quality of life among food allergic patients and their caregivers. Curr Allergy Asthma Rep. 2016;16(5):38. | ||
Chung MC, Walsh A, Dennis I. Trauma exposure characteristics, past traumatic life events, coping strategies, posttraumatic stress disorder, and psychiatric comorbidity among people with anaphylactic shock experience. Compr Psychiatry. 2011;52(4):394–404. | ||
Chen H, Huang N, Li WJ, et al. Clinical and laboratory features, and quality of life assessment in wheat dependent exercise-induced anaphylaxis patients from central China. J Huazhong Univ Sci Technolog Med Sci. 2016;36(3):410–415. | ||
Oude Elberink JN, Dubois AE. Quality of life in insect venom allergic patients. Curr Opin Allergy Clin Immunol. 2003;3(4):287–293. | ||
Warren CM, Dyer AA, Otto AK, et al. Food allergy-related risk-taking and management behaviors among adolescents and young adults. J Allergy Clin Immunol Pract. 2016;5(2):381–390.e13. | ||
Muraro A, Roberts G, Clark A, et al; EAACI Task Force on Anaphylaxis in Children. The management of anaphylaxis in childhood: position paper of the European Academy of Allergology and Clinical Immunology. Allergy. 2007;62(8):857–871. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.