")
Back to Journals » OncoTargets and Therapy » Volume 12
Analysis of long non-coding RNAs in glioblastoma for prognosis prediction using weighted gene co-expression network analysis, Cox regression, and L1-LASSO penalization
Authors Liang R, Zhi YQ, Zheng G, Zhang B, Zhu H, Wang M
Received 22 April 2018
Accepted for publication 4 September 2018
Published 21 December 2018 Volume 2019:12 Pages 157—168
DOI https://doi.org/10.2147/OTT.S171957
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Ruqing Liang,1,* Yaqin Zhi,2,* Guizhi Zheng,3 Bin Zhang,2 Hua Zhu,2 Meng Wang2
1Department of Neurology, Affiliated Hospital of Jining Medical University, Jining, Shandong Province 272000, China; 2Department of Oncology, Jining No 1 People’s Hospital, Jining, Shandong Province 272000, China; 3College of Integrated Chinese and Western Medicine, Jining Medical College, Jining, Shangdong 272067, China
*These authors contributed equally to this work
Purpose: This study focused on identification of long non-coding RNAs (lncRNAs) for prognosis prediction of glioblastoma (GBM) through weighted gene co-expression network analysis (WGCNA) and L1-penalized least absolute shrinkage and selection operator (LASSO) Cox proportional hazards (PH) model.
Materials and methods: WGCNA was performed based on RNA expression profiles of GBM from Chinese Glioma Genome Atlas (CGGA), National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), and the European Bioinformatics Institute ArrayExpress for the identification of GBM-related modules. Subsequently, prognostic lncRNAs were determined using LASSO Cox PH model, followed by constructing a risk scoring model based on these lncRNAs. The risk score was used to divide patients into high- and low-risk groups. Difference in survival between groups was analyzed using Kaplan–Meier survival analysis. IncRNA-mRNA networks were built for the prognostic lncRNAs, followed by pathway enrichment analysis for these networks.
Results: This study identified eight preserved GBM-related modules, including 188 lncRNAs. Consequently, C20orf166-AS1, LINC00645, LBX2-AS1, LINC00565, LINC00641, and PRRT3-AS1 were identified by LASSO Cox PH model. A risk scoring model based on the lncRNAs was constructed that could divide patients into different risk groups with significantly different survival rates. Prognostic value of this six-lncRNA signature was validated in two independent sets. C20orf166-AS1 was associated with antigen processing and presentation and cell adhesion molecule pathways, involving nine common genes. LBX2-AS1, LINC00641, PRRT3-AS1, and LINC00565 were related to focal adhesion, extracellular matrix receptor interaction, and mitogen-activated protein kinase signaling pathways, which shared 12 common genes.
Conclusion: This prognostic six-lncRNA signature may improve prognosis prediction of GBM. This study reveals many pathways and genes involved in the mechanisms behind these lncRNAs.
Keywords: lncRNA, risk score, WGCNA, network, pathway
Introduction
Glioblastoma (GBM), grade IV glioma, is the most common and aggressive type of brain cancer characterized by high morbidity and mortality and dismal prognosis.1,2 Reportedly, the median survival of patients with newly diagnosed GBM is approximately 15 months.3 Despite the development of medical interventions such as surgical resection, radiological therapy, and chemotherapeutic therapy, the survival rate remains largely unchanged over the past years.4 A deep understanding on the pathogenesis of GBM and the discovery of molecular biomarkers likely contribute to the improvement of GBM survival.
Long non-coding RNAs (lncRNAs) are defined as transcripts greater than 200 nucleotides that do not code proteins.5 With the development of genome-wide expression profiling, a huge amount of novel lncRNAs have been discovered. These lncRNAs are known to play key roles in a broad range of biological processes such as cell differentiation, human diseases, and tumorigenesis.6 Unraveling potential roles of lncRNAs in GBM has emerged as a leading edge of GBM research.7 For instance, Han et al8 revealed that ASLNC22381 and ASLNC2081 may engage in recurrence and progression of GBM through conducting lncRNA and mRNA profiling. In addition, Zhang et al9 reported a set of lncRNAs that have prognostic value for GBM by lncRNAs bioinformatics analysis in The Cancer Genome Atlas (TCGA). Moreover, a recent study identifies an immune-related lncRNA signature for prognostic prediction based on TCGA data of GBM patients.10 Despite these valuable findings, the majority of lncRNAs in GBM remains poorly understood.
In comparison with previous studies that identified prognostic lncRNA signatures based on the limited microarray data from TCGA,9,10 we carried out a comprehensive analysis on all publicly available gene expression data of GBM from Chinese Glioma Genome Atlas (CGGA), National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO), and the European Bioinformatics Institute (EBI) ArrayExpress repositories through a series of bioinformatics approaches. We searched for GBM-related key modules through constructing a weighted gene co-expression network analysis (WGCNA). Based on the lncRNAs contained in these key modules, we acquired a panel of lncRNAs as prognostic biomarkers by univariate Cox regression analysis, in combination with Cox proportional hazards (PH) model based on the L1-penalized least absolute shrinkage and selection operator (LASSO) estimation. Subsequently, a prognostic scoring system was constructed based on these prognostic lncRNAs to evaluate the death risk due to GBM. In addition, lncRNA alterations in GBM compared to normal samples were analyzed using metaDE method. Furthermore, pathway enrichment analysis using Gene Set Enrichment Analysis (GSEA) was conducted to give some insights into the underlying mechanisms of these predictive lncRNAs.
Materials and methods
Data resource
The data sets in this study were derived from three sources.
First, the gene expression data of 325 glioma samples, named “Part D”,11 was downloaded from the CGGA (http://cgga.org.cn/), including 144 GBM samples that were selected as the training set in this study (platform: Illumina HiSeq 2000 RNA Sequencing). Survival information was available for 138 patients with GBM, of whom, 92 were dead, while 46 were alive with a median survival time of 13.22±11.44 months.
Second, the data sets were searched in the NCBI GEO (http://www.ncbi.nlm.nih.gov/geo/) and the EBI ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) repositories for publication of human GBM with no less than 40 samples. As a result, three data sets of GSE51062, GSE36245, and E-TABM-898 were obtained, including 52 samples, 46 samples, and 56 samples, separately. The platform for all the three data sets was Affymetrix-GPL570. We also searched for human GBM data sets that had no less than 50 samples and simultaneously available survival information in NCBI GEO and EBI ArrayExpress. Two data sets, the GSE74187 (n=60) and GSE83300 (n=50), meeting the criteria were included in this study. The platform for both of them was Agilent-014850. In addition, we needed human GBM gene expression data sets that had both GBM samples and paired normal tissue samples, with the total number of samples greater than 40. Through exploring NCBI GEO and EBI ArrayExpress, the GSE22866 (including 40 GBM samples and six normal samples; platform: Affymetrix-GPL570), GSE50161 (including 34 GBM samples and 13 normal samples; platform: Affymetrix-GPL570), and GSE4290 (including 77 GBM samples and 23 normal samples; platform: Agilent-014850) were acquired.
Third, RNA-seq data set comprising 154 GBM samples and 18 normal samples was downloaded from the TCGA (https://gdc-portal.nci.nih.gov/). There were 152 samples available with survival information, including 102 dead and 50 live samples.
Data preprocessing
For the data sets downloaded from Affymetrix-GPL570 platform, raw data (CEL files) were background corrected and normalized12 using the oligo package (version 1.41.1, http://www.bioconductor.org/packages/release/bioc/html/oligo.html) in R language (version 3.4.1). With respect to the data sets from Agilent-014850 platform, raw data (TXT files) underwent log2 transformation to yield approximately normal distribution with the limma13 software (version 3.34.0, https://bioconductor.org/packages/release/bioc/html/limma.html), followed by standardization using the median method. CGGA and TCGA data were subject to quantile normalization using the preprocessCore package14 (version1.40.0, http://bioconductor.org/packages/release/bioc/html/preprocessCore.html) in R language (version 3.4.1).
Next, according to platform annotation files, the probes in all data sets that had RefSeq transcript ID and annotation information as non-coding RNA in the Refseq database were chosen. Moreover, the platform sequencing data were aligned to human genome (GRCh38 version) by using the Clustal2 (http://www.clustal.org/clustal2/).15 The acquired lncRNAs combining with the annotated lncRNAs in the Refseq database16 were extracted for further analysis.
WGCNA
The WGCNA (version 1.61, https://cran.r-project.org/web/packages/WGCNA/index.html)17 was applied to build a WGCNA to mine GBM-related preserved modules. For this network analysis, the CGGA data were referred to as the training set, while the GSE51062, GSE36245, and E-TABM-898 as validation sets. Initially, comparability between the four sets was analyzed using correlation analysis. A WGCNA was constructed in accordance with a previous study.18 Briefly, using scale-free topology criterion, the soft threshold power of β was established, through which the weighted adjacency matrix was developed. The modules with size ≥150 and minimum cut height of 0.99 were selected using dynamic tree cut algorithm, and the preserved modules were determined using the module preservation function of WGCNA package. In addition, the possible biological functions of the significantly preserved modules were studied using userListEnrichment function of WGCNA package.
Selection of prognosis-related lncRNAs
Based on the lncRNAs in the preselected preserved WGCNA modules and the corresponding survival information, univariate Cox regression analysis was used to identify the lncRNAs that were significantly correlated with prognosis (logrank P<0.05) by using survival package (version 2.4, https://cran.r-project.org/web/packages/survival/index.html) in R language (version 3.4.1).19
Construction of prognosis scoring model based on lncRNAs
The identified prognosis-related lncRNAs were used to fit a Cox PH model based on the LASSO estimation20 to select the optimal panel of prognostic lncRNAs. The optimal value for penalization coefficient lambda was selected by running cross-validation likelihood (cvl) 1,000 times. Subsequently, the Cox PH coefficients and expression levels of these prognostic lncRNAs were extracted to calculate the risk score as a measure of survival risk for each patient using the following formula:
|
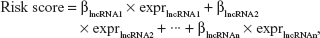
where βlncRNAn represents Cox PH coefficient of lncRNAn and exprlncRNA represents expression level of lncRNAn.
All samples in the CGGA set were dichotomized into high- and low-risk groups by risk score, with median risk score as the threshold. Then, three independent sets with concomitant survival information (TCGA set, GSE74187, and GSE83300) were utilized to evaluate the effectiveness and robustness of the abovementioned risk scoring model. As mentioned above, the three data sets contained all available GBM data with survival information in TCGA, NCBI GEO, and EBI ArrayExpress. In the same manner, samples in each set were categorized by risk score into predicted high- and low-risk groups. Survival difference between different risk groups in each set was analyzed using the Kaplan–Meier curve in combination with the Wilcoxon logrank test.
Analysis of consensus differentially expressed RNAs (DERs)
GSE22866, GSE50161, and GSE4290 contained both GBM samples and normal control samples. We screened the overlapped DERs between GBM and normal samples across the three data sets using MetaDE package (https://cran.r-project.org/web/packages/MetaDE/),21,22 under the thresholds of tau2=0, Qpval>0.05, P<0.05 and false discovery rate (FDR) <0.05. Of them, tau2 and Qpval were measures of heterogeneity for the heterogeneity test. When tau2=0 and Qpval>0.05, the gene was homogeneous and unbiased.
Pathway enrichment analysis
We built lncRNA-mRNA networks with the selected prognostic lncRNAs and their correlated mRNAs in WGCNA modules. GSEA is a powerful approach for annotating gene expression data that are characterized by focusing on gene set with common biological function, chromosomal location, or regulation (http://software.broadinstitute.org/gsea/index.jsp).23 We performed pathway enrichment analysis for the lncRNA-mRNA networks using GSEA. Pathways with nominal (NOM) P-value <0.05 were considered significant. GSEA-enriched results were shown by normalized enrichment score (NES) that was calculated as previously described.24
Results
WGCNA co-expression network construction and module mining
After preprocessing, 609 lncRNAs and 14,948 mRNAs were overlapped in CGGA data set, GSE51062, GSE36245, and E-TABM-898. By using WGCNA, correlation analysis between any two sets of the four data sets, CGGA data set (training set), GSE51062, GSE36245, and E-TABM-898 (validation sets), were performed. As shown in Figure 1, the correlation coefficients ranged from 0.5 to 1 (P-values<1e–200), suggesting that the expression of common RNAs among the four data sets was coincident.
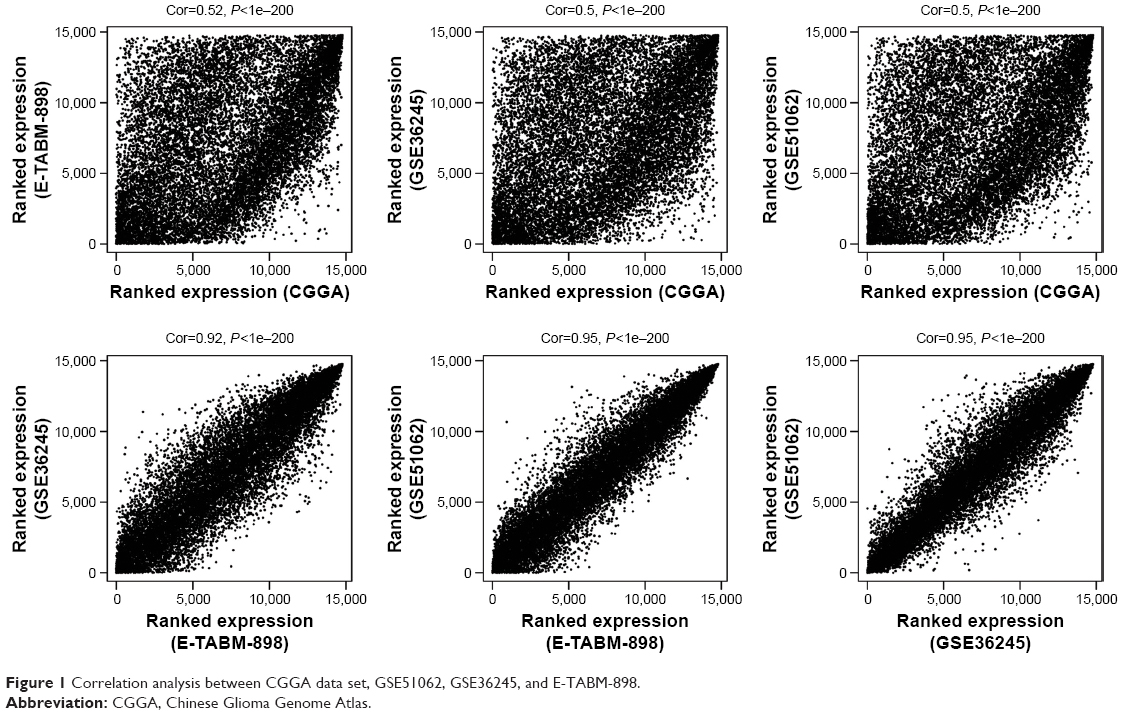 | Figure 1 Correlation analysis between CGGA data set, GSE51062, GSE36245, and E-TABM-898. |
Initially, WGCNA of RNAs was built for the training set (CGGA set). According to the scale-free topology criterion, the soft threshold power of β was set as 5 when scale-free topology model-fit R2=0.9. The phylogenetic tree mined nine co-expression modules (module size, ≥50; cut height, ≥0.99) in the WGCNA (Figure 2A). As shown by the color bands underneath the phylogenetic tree, nine modules were represented by branches of different colors (M1, black; M2, blue; M3, brown; M4, green; M5, gray; M6, pink; M7, red; M8, turquoise; M9, yellow). Moreover, these modules were validated in E-TABM-898, GSE51062, and GSE36245 (Figure 2B and D). In the three validation sets, genes were colored in the same manner as in TCGA set.
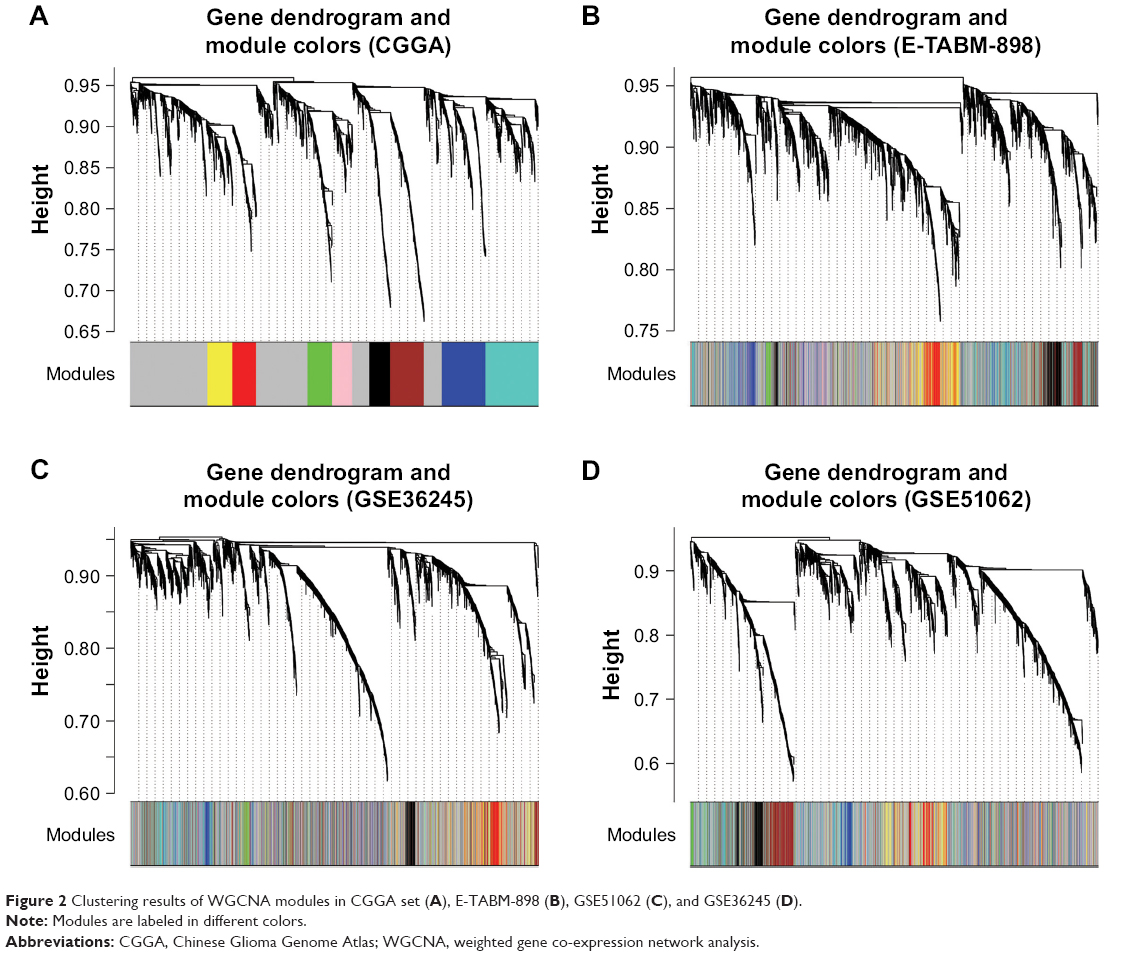 | Figure 2 Clustering results of WGCNA modules in CGGA set (A), E-TABM-898 (B), GSE51062 (C), and GSE36245 (D). |
As can be seen from a multidimensional scaling (MDS) for gene expression data of the nine modules (Figure 3A), genes in yellow and red modules showed similar expression and genes in brown and black modules exhibit similar expression. Hierarchical clustering analysis of modules found that the yellow and red modules were on the same branch (Figure 3B). These observations illustrate that the yellow and red modules possess similar gene expression patterns.
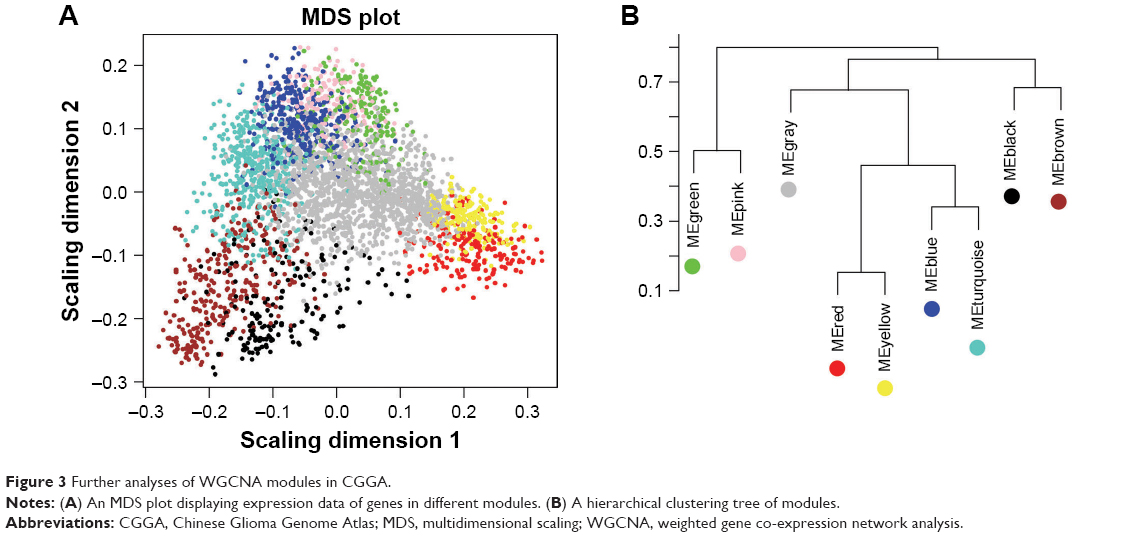 | Figure 3 Further analyses of WGCNA modules in CGGA. |
Module preservation analysis found that among the nine nodules, eight modules had Z-score >5 (Table 1). The eight modules were ranked in a descending order of Z-score. Top three modules were yellow module (Z-score=34.5011), red module (Z-score=34.3040), and black module (Z-score=24.5504), which were highly overlapped across all datasets. This observation indicates that the three modules may provide important information concerning the pathological mechanisms of GBM. With regard to functional annotation, the yellow module (84 lncRNAs) was related to biological adhesion, the red module (26 lncRNAs) was associated with immune response, and the brown module (eight lncRNAs) was possibly involved in synaptic transmission (Table 1).
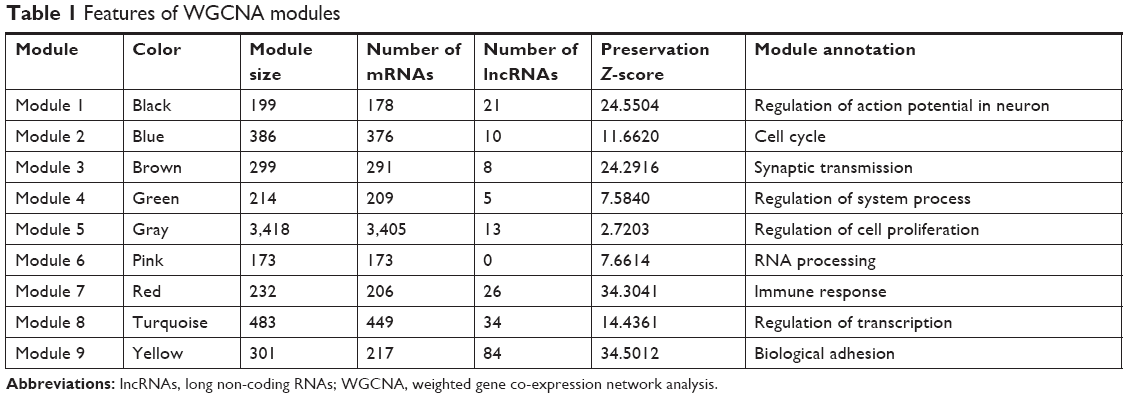 | Table 1 Features of WGCNA modules |
Identification of prognosis-related lncRNAs
There were 188 lncRNAs in the eight overlapped WGCNA modules. Based on the survival information of CGGA set, 32 lncRNAs were identified to be significant prognosis-related lncRNAs by univariate Cox regression analysis. As shown in Figure 4, among the 32 prognosis-related lncRNAs, 11 were in the yellow module, eight in the red module, and eight in the turquoise module. As aforementioned, the yellow and red modules had similar gene expression patterns. Moreover, the two modules were functionally related to biological adhesion and immune response, which were critical for GBM pathogenesis.25,26 Therefore, the 19 lncRNAs in the yellow and red modules were selected for further analysis.
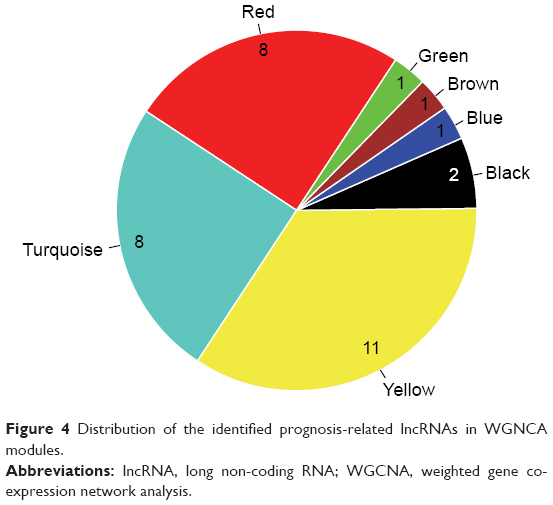 | Figure 4 Distribution of the identified prognosis-related lncRNAs in WGNCA modules. |
Development of a six-lncRNA prognostic scoring system
Expression of the 19 lncRNAs in the yellow and red modules were used as input for LASSO Cox PH model. When the cvl was maximized to be −466.2711, the optimal lambda value was 18.0151. As a result, a panel of six lncRNAs was selected as predictive factors for survival, including C20orf166-AS1, LINC00645, LBX2-AS1, LINC00565, LINC00641, and PRRT3-AS1 (Table 2). For predicting each individual patient’s survival probability, risk score was calculated for each patient with the following formula:
|
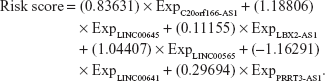
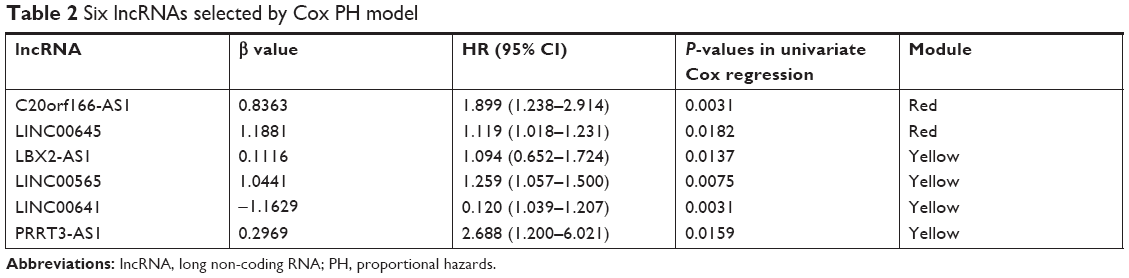 | Table 2 Six lncRNAs selected by Cox PH model |
Prediction of overall survival (OS) of GBM patients
The aforementioned lncRNA-based risk scoring system was applied to the CGGA set. With the median risk score as cutoff, all patients in the CGGA set were categorized into a high-risk group (n=69) and a low-risk group (n=69). The results showed that the low-risk group had significantly longer OS compared to the high-risk group (16.61±14.22 months vs 9.83±6.17 months, logrank, P=0.000127; Figure 5A).
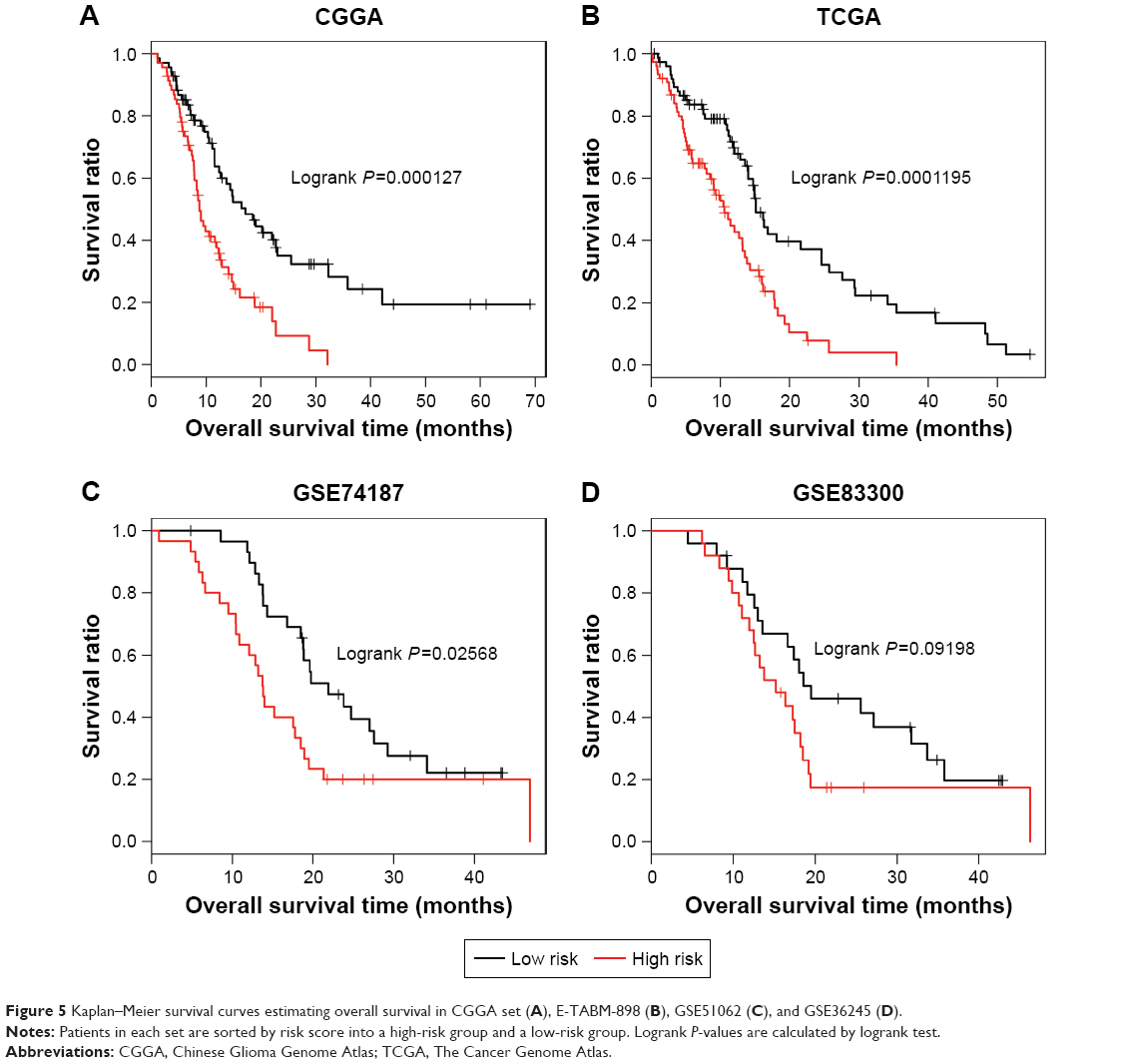 | Figure 5 Kaplan–Meier survival curves estimating overall survival in CGGA set (A), E-TABM-898 (B), GSE51062 (C), and GSE36245 (D). |
The predictive capability of this prognostic scoring system was tested in TCGA set, GSE74187, and GSE83300, and the risk score and risk group categories were similar for each of them. As shown in Figure 5B, for TCGA set (n=152), when compared to the high-risk group, a notably better survival was observed in the low-risk group (14.93±12.54 months vs 9.19±6.65 months, logrank P=0.0001195). Consistent results were also found for GSE74187 (n=60; 22.47±10.14 month vs 15.83±10.11 month, log-rank p=0.02568, Figure 5C). For GSE83300, the low-risk group had a longer OS compared to the high-risk group, with marginally significant difference (logrank P=0.09198; Figure 5D). It may be attributed to the relatively small sample size (n=50) of GSE83300. These findings offer strong evidence for the prognostic power of the six-lncRNA prognostic scoring system.
Identification of overlapped DERs in GSE22866, GSE50161, and GSE4290
GSE22866, GSE50161, and GSE4290 with both GBM and normal tissue samples were used in the present study to find overlapped DERs between GBM and normal samples. Totally, 3,989 overlapped DERs (tau2=0, Qpval>0.05, P-value<0.05, and FDR<0.05) were found; of which, 98 were lncRNAs. Notably, of the aforementioned six prognostic lncRNAs, LBX2-AS1, LINC00641, LINC00645, and LINC00565 were DERs. A heatmap for expression of these overlapped DERs demonstrates that the DERs in the GSE22866, GSE50161, and GSE4290 had similar expression patterns (Figure 6).
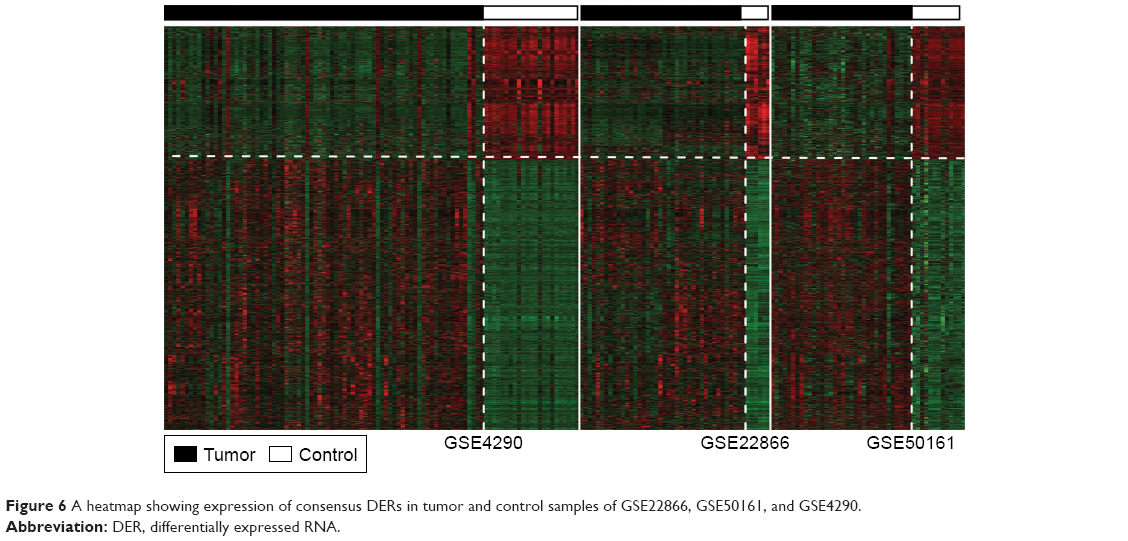 | Figure 6 A heatmap showing expression of consensus DERs in tumor and control samples of GSE22866, GSE50161, and GSE4290. |
Establishment of lncRNA-mRNA networks
To explore the relationships between the six prognostic lncRNAs and genes in the yellow and red modules, lncRNA-mRNA networks were constructed for the two modules, respectively (Figure 7A and B). For the red module, the lncRNA-mRNA network was composed of two lncRNAs (C20orf166-AS1 and LINC00645) and 206 genes, of which, five were downregulated DERs and 72 were upregulated (Figure 7A). For the yellow module, the lncRNA-mRNA network contained four lncRNAs (LBX2-AS1, LINC00641, PRRT3-AS1, and LINC00565) and 217 genes, of which, four were downregulated DERs and 97 were upregulated (Figure 7B).
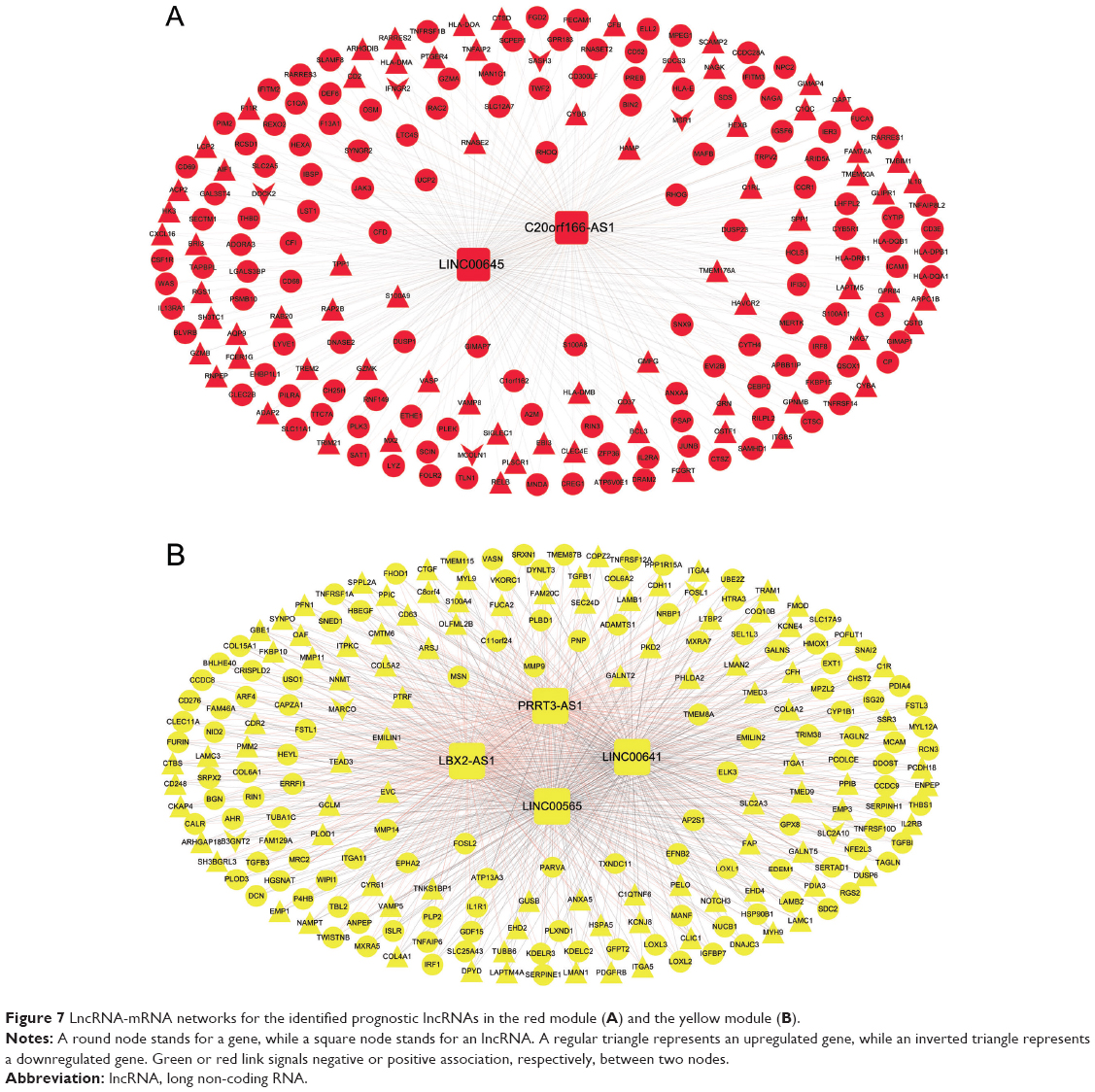 | Figure 7 LncRNA-mRNA networks for the identified prognostic lncRNAs in the red module (A) and the yellow module (B). |
Pathway analysis
We conducted pathway enrichment analysis using GSEA for the two lncRNA-mRNA networks. It was revealed that C20orf166-AS1 in the red module was significantly enriched in antigen processing and presentation and cell adhesion molecules (CAMs) pathways (Table 3). The two pathways involved nine common genes: major histocompatibility complex, class II, DM alpha (HLA-DMA), major histocompatibility complex, class II, DM beta (HLA-DMB), major histocompatibility complex, class II, DP beta 1 (HLA-DPB1), CD2, sialic acid-binding Ig-like lectin 1 (SIGLEC1), major histocompatibility complex, class II, DO alpha (HLA-DOA), major histocompatibility complex, class II, DQ alpha 1 (HLA-DQA1), major histocompatibility complex, class II, DR beta 1 (HLA-DRB1), and major histocompatibility complex, class II, DQ beta 1 (HLA-DQB1). As shown in Table 4, LBX2-AS1, LINC00641, PRRT3-AS1, and LINC00565 in the yellow module were significantly associated with cancer, focal adhesion, extracellular matrix (ECM) receptor interaction, and mitogen-activated protein kinase (MAPK) signaling pathways. Twelve common genes were involved in the four pathways, including laminin subunit beta (LAMB) 1, collagen type V alpha 2 chain (COL5A2), TGFB 1, integrin subunit alpha (ITGA) 5, platelet-derived growth factor receptor beta (PDGFRB), TNF receptor superfamily (TNFRSF) 12A, dual-specificity phosphatase (DUSP) 6, laminin subunit gamma (LAMC) 1, LAMC3, TNFRSF1A and myosin light chain (MYL) 9.
 | Table 3 Results of pathway enrichment analysis for the lncRNA-mRNA network of the red module |
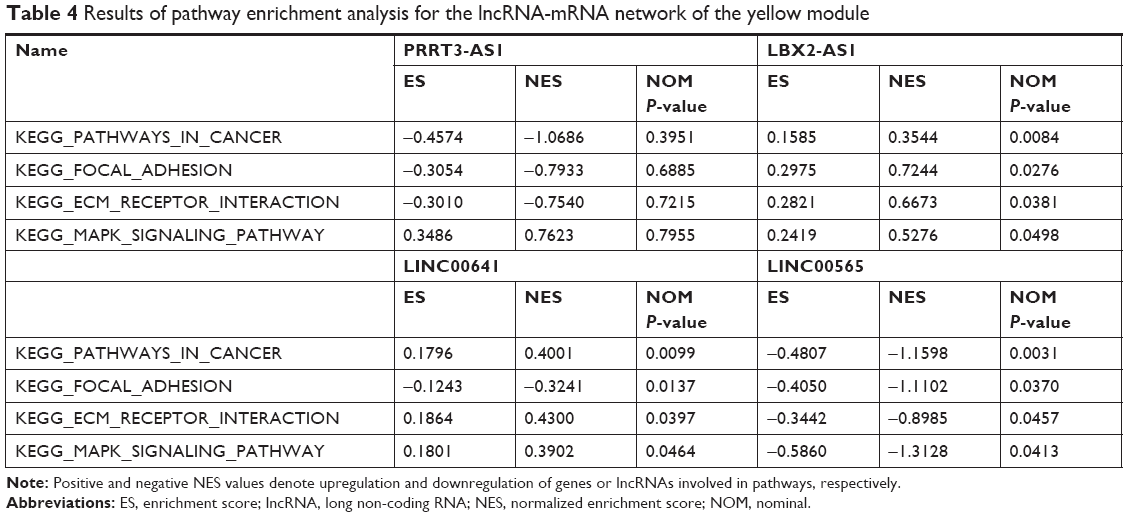 | Table 4 Results of pathway enrichment analysis for the lncRNA-mRNA network of the yellow module |
Discussion
Increasing evidence indicates that a growing number of lncRNAs are associated with various cancer types.27 This discovery leads to a growing interest in the study of lncRNAs in GBM. Based on the gene expression data of GBM from CGGA, NCBI GEO, and EBI ArrayExpress, we identified a prognostic signature of six lncRNAs (C20orf166-AS1, LINC00645, LBX2-AS1, LINC00565, LINC00641, and PRRT3-AS1) through a combination of WGCNA, univariate Cox regression analysis, and LASSO PH model. Moreover, a six-lncRNA-based risk scoring system was constructed and capable to classify GBM patients into two risk groups with significantly different survival rates. The prognostic performance of the risk scoring model was successfully validated in two independent sets. It indicates that the six lncRNAs are promising prognostic biomarkers for GBM and may play important roles in tumorigenesis of GBM.
LINC00645 is an endometrial cancer-specific lncRNA.28 Emerging studies have proved that C20orf166-AS1 is aberrantly expressed in prostate cancer and bladder cancer.42,43 However, the involvement of LINC00645 and C20orf166-AS1 in GBM has not been reported yet. In the present study, C20orf166-AS1 was identified as an important lncRNA of prognostic value for GBM. Moreover, pathway enrichment analysis showed that C20orf166-AS1 was significantly related to antigen processing and presentation and CAMs pathways, and both pathways shared HLA-DMA, HLA-DMB, HLA-DPB1, CD2, SIGLEC1, HLA-DOA, HLA-DQA1, HLA-DRB1, and HLA-DQB1. Among the nine common genes, HLA-DMA, HLA-DMB, HLA-DPB1, HLA-DOA, HLA-DQA, HLA-DRB1, and HLA-DQB1 are major histocompatibility complex class II molecules that are mainly expressed on antigen-presenting cells and play an important role in immune response. The protein encoded by CD2 gene is a CAM located on the surface of T cells and NK cells, and it acts as a specific marker for these cells.29 SIGLEC1 protein is a member of siglecs that are predominately expressed on the surface of immune cells and bind to glycans enclosing sialic acids.30 Interactions between siglecs and glycans are implicated in cell adhesion and cell signaling. These findings reveal that C20orf166-AS1 might participate in immune response and cell adhesion in GBM through the regulation of these genes in antigen processing and presentation and CAM pathways.
Recent studies report that upregulation of LBX2-AS1 has been observed in lung cancer.31,32 Interestingly, LBX2-AS1 is significantly upregulated with the increase of tumor grade in GBM,33 suggesting that this lncRNA probably has an important regulatory role in GBM prognosis. Alterations of LINC00641, PRRT3-AS1, and LINC00565 in cancer have been scarcely reported. The current study provided evidence that the four lncRNAs had predictive value for survival of GBM patients. Notably, the study uncovered that they were significantly linked to focal adhesion, ECM receptor interaction, and MAPK signaling pathways. These pathways involved 12 common genes, including LAMB1, COL5A2, TGFB1, ITGA5, PDGFRB, TNFRSF12A, DUSP6, LAMC1, LAMC3, TNFRSF1A, and MYL9. Increasing evidence has established that MAPK pathway is involved in regulating GBM cell migration and proliferation.34,35 LAMB1, LAMC1, and LAMC3 are members of ECM glycoproteins. COL5A2 encodes an alpha chain for fibrillar collagen, a major component of ECM proteins.36 TGF-β1 is a member of TGF β superfamily and plays a role in the regulation of growth, proliferation, and differentiation of glioma cells.37 Integrin alpha-5 protein encoded by ITGA5 belongs to the integrin alpha chain family that is critical for cell adhesion.38 Recently, it is found that PDGFRB is elevated in GBM microvascular proliferation compared to GBM tumor cells and selectively expressed PDGFRB protein in pericytes.39 DUSP6 protein belongs to the dual-specificity protein phosphatase subfamily that acts as a negative regulator over MAPK members.40 Besides, it has been found that DUSP6 is upregulated in GBM and promotes the development of GBM.41 These results imply that the involvement of LBX2-AS1, LINC00641, PRRT3-AS1, and LINC00565 in GBM may be involved in focal adhesion, ECM receptor interaction, and MAPK signaling pathways. These common genes might be potential therapeutic targets for GBM.
Conclusion
Based on the comprehensive analysis of publicly accessible GBM data in CGGA, NCBI GEO, and EBI ArrayExpress, this study identifies a novel six-lncRNA signature for GBM prognostic prediction. This study also highlights the pathways and genes involved in the regulatory mechanisms underlying these prognostic lncRNAs. Further studies are warranted prior to the application of this lncRNA signature in clinical practice.
Availability of data and material
The raw data were collected and analyzed by the authors, and they are not ready to share their data because the data have not been published.
Author contributions
RL and YQZ participated in the design of this study, and they both performed the statistical analysis. GZ and BZ carried out the study and collected important background information. HZ and MW drafted the manuscript. All authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015;129(6):829–848. | ||
Kesari S. Understanding glioblastoma tumor biology: the potential to improve current diagnosis and treatments. Semin Oncol. 2011;38 (Suppl 4):S2–S10. | ||
Alifieris C, Trafalis DT. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol Ther. 2015;152:63–82. | ||
Bleeker FE, Molenaar RJ, Leenstra S. Recent advances in the molecular understanding of glioblastoma. J Neurooncol. 2012;108(1):11–27. | ||
Johnsson P, Lipovich L, Grandér D, Morris KV. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta. 2014;1840(3):1063–1071. | ||
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21. | ||
Haemmerle M, Gutschner T. Long non-coding RNAs in cancer and development: where do we go from here? Int J Mol Sci. 2015;16(1):1395–1405. | ||
Han L, Zhang K, Shi Z, et al. LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int J Oncol. 2012;40(6):2004–2012. | ||
Zhang XQ, Sun S, Lam KF, et al. A long non-coding RNA signature in glioblastoma multiforme predicts survival. Neurobiol Dis. 2013;58(10):123–131. | ||
Zhou M, Zhang Z, Zhao H, Bao S, Cheng L, Sun J. An Immune-Related Six-lncRNA Signature to Improve Prognosis Prediction of Glioblastoma Multiforme. Mol Neurobiol. 2018;55(5):3684–3697. | ||
Bao ZS, Chen HM, Yang MY, et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014;24(11):1765–1773. | ||
Parrish RS, Spencer HJ 3rd. Effect of normalization on significance testing for oligonucleotide microarrays. J Biopharm Stat. 2004;14(3):575–589. | ||
Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. | ||
Bolstad BM, Irizarry RA, Åstrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. | ||
Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. | ||
Sun C, Jiang H, Sun Z, Gui Y, Xia H. Identification of long non-coding RNAs biomarkers for early diagnosis of myocardial infarction from the dysregulated coding-non-coding co-expression network. Oncotarget. 2016;7(45):73541–73551. | ||
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):559. | ||
Zhai X, Xue Q, Liu Q, Guo Y, Chen Z. Colon cancer recurrence-associated genes revealed by WGCNA co-expression network analysis. Mol Med Rep. 2017;16(5):6499–6505. | ||
Wang W, Zhang L, Wang Z, et al. A three-gene signature for prognosis in patients with MGMT promoter-methylated glioblastoma. Oncotarget. 2016;7(43):69991–69999. | ||
Tibshirani R. The lasso method for variable selection in the Cox model. Stat Med. 1997;16(4):385–395. | ||
Wang X, Kang DD, Shen K, et al. An R package suite for microarray meta-analysis in quality control, differentially expressed gene analysis and pathway enrichment detection. Bioinformatics. 2012;28(19):2534–2536. | ||
Qi C, Hong L, Cheng Z, Yin Q. Identification of metastasis-associated genes in colorectal cancer using metaDE and survival analysis. Oncol Lett. 2016;11(1):568–574. | ||
Tilford CA, Siemers NO. Gene set enrichment analysis. Methods Mol Biol. 2009;563(23):99. | ||
White-Al Habeeb NM, Ho LT, Olkhov-Mitsel E, et al. Integrated analysis of epigenomic and genomic changes by DNA methylation dependent mechanisms provides potential novel biomarkers for prostate cancer. Oncotarget. 2014;5(17):7858–7869. | ||
Moura-Neto V, Campanati L, Matias D, et al. Glioblastomas and the Special Role of Adhesion Molecules in Their Invasion. Glioma Cell Biol. 2014:293–315. | ||
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. | ||
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–1261. | ||
Chen BJ, Byrne FL, Takenaka K, et al. Transcriptome landscape of long intergenic non-coding RNAs in endometrial cancer. Gynecol Oncol. 2017;147(3):654–662. | ||
Jining L, Makagiansar I, Yusuf-Makagiansar H, Chow VT, Siahaan TJ, Jois SD. Design, structure and biological activity of beta-turn peptides of CD2 protein for inhibition of T-cell adhesion. Eur J Biochem. 2004;271(14):2873–2886. | ||
Zheng Q, Hou J, Zhou Y, Yang Y, Xie B, Cao X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015;25(10):1121–1136. | ||
Tian Z, Wen S, Zhang Y, et al. Identification of dysregulated long non-coding RNAs/microRNAs/mRNAs in TNM I stage lung adenocarcinoma. Oncotarget. 2017;8(31):51703–51718. | ||
Bao L, Zhang Y, Wang J, et al. Variations of chromosome 2 gene expressions among patients with lung cancer or non-cancer. Cell Biol Toxicol. 2016;32(5):419–435. | ||
Wu F, Zhao Z, Chai R, et al. Expression profile analysis of antisense long non-coding RNA identifies WDFY3-AS2 as a prognostic biomarker in diffuse glioma. Cancer Cell Int. 2018;18(1):107. | ||
Zohrabian VM, Forzani B, Chau Z, Murali R, Jhanwar-Uniyal M. Rho/ROCK and MAPK signaling pathways are involved in glioblastoma cell migration and proliferation. Anticancer Res. 2009;29(1):119–123. | ||
Mao H, Lebrun DG, Yang J, Zhu VF, Li M. Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Invest. 2012;30(1):48–56. | ||
Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326(5957):1216–1219. | ||
Kaminska B, Kocyk M, Kijewska M. TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol. 2013;986(986):171–187. | ||
Calderwood DA, Shattil SJ, Ginsberg MH. Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J Biol Chem. 2000;275(30):22607–22610. | ||
Xu G, Li JY. Differential expression of PDGFRB and EGFR in microvascular proliferation in glioblastoma. Tumour Biol. 2016;37(8):10577–10586. | ||
Owens DM, Keyse SM. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 2007;26(22):3203–3213. | ||
Messina S, Frati L, Leonetti C, et al. Dual-specificity phosphatase DUSP6 has tumor-promoting properties in human glioblastomas. Oncogene. 2011;30(35):3813–3820. | ||
Eeles RA, Olama AA, Benlloch S, et al. Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet. 2013;45(4):385–391, 391e1–2. | ||
Wang H, Niu L, Jiang S, et al. Comprehensive analysis of aberrantly expressed profiles of lncRNAs and miRNAs with associated ceRNA network in muscle-invasive bladder cancer. Oncotarget. 2016;7(52):86174–86185. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.