")
Back to Journals » Cancer Management and Research » Volume 12
Analyses of Potential Driver and Passenger Bacteria in Human Colorectal Cancer
Authors Wang Y , Zhang C , Hou S, Wu X, Liu J, Wan X
Received 3 August 2020
Accepted for publication 25 October 2020
Published 12 November 2020 Volume 2020:12 Pages 11553—11561
DOI https://doi.org/10.2147/CMAR.S275316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Chien-Feng Li
Yijia Wang,1,* Chunze Zhang,2,* Shaobin Hou,3 Xiaojing Wu,1 Jun Liu,4 Xuehua Wan5
1Laboratory of Oncologic Molecular Medicine, Tianjin Union Medical Center, Nankai University, Tianjin, People’s Republic of China; 2Department of Colorectal Surgery, Tianjin Union Medical Center, Nankai University, Tianjin, People’s Republic of China; 3Advanced Studies in Genomics, Proteomics, and Bioinformatics, University of Hawaii at Manoa, Honolulu, HI, USA; 4Department of Radiology, Tianjin Union Medical Center, Nankai University, Tianjin, People’s Republic of China; 5TEDA Institute of Biological Sciences and Biotechnology, Nankai University, TEDA, Tianjin, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun Liu
Tianjin Union Medical Center, No. 190, Jieyuan Road, Hongqiao District, Tianjin City, People’s Republic of China
Email [email protected]
Xuehua Wan
Nankai University, No. 94, Weijin Road, Nankai District, Tianjin City, People’s Republic of China
Email [email protected]
Introduction: Besides genetic and epigenetic alterations that lead to carcinogenesis and development of colorectal cancer (CRC), intestinal microbiomes are recently recognized to play a critical role in CRC progression. The abundant species associated with human CRC have been proposed for their roles in promoting tumorigenesis. However, a recent “driver-passenger” model suggests that these CRC-associated species with high relative abundances may be passenger bacteria that take advantage of the tumor environment instead of initiating CRC, whereas the driver species that initiate CRC have been replaced by passenger bacteria due to the alteration of the intestinal niche.
Methods: Here, to reveal potential driver and passenger bacteria during CRC progression, we compare the gut mucosal microbiomes of 75 triplet-paired CRC samples collected from on-tumor site, adjacent-tumor site, and off-tumor site, and 26 healthy controls.
Results: Our analyses revealed potential driver bacteria in four genera and two families, and potential passenger bacteria in 14 genera or families. Bacillus, Bradyrhizobium, Methylobacterium, Streptomyces, Intrasporangiaceae and Sinobacteraceae were predicted to be potential driver bacteria. Moreover, 14 potential passenger bacteria were identified and divided into five groups. Group I passenger bacteria contain Fusobacterium, Campylobacter, Streptococcus, Schwartzia, and Parvimonas. Group II passenger bacteria contain Dethiosulfatibacter, Selenomonas, Peptostreptococus, Leptotrichia. Group III passenger bacteria contain Granulicatella. Group IV passenger bacteria contain Shewanella, Mogibacterium, and Eikenella. Group V passenger bacteria contain Anaerococus. Co-occurrence network analysis reveals a low correlation relationship between driver and passenger bacteria in CRC patients compared with healthy controls.
Discussion: These driver and passenger species may serve as bio-marker species for screening cohorts with high risk to initiate CRC or patients with CRC, respectively. Further functional studies will help understand the roles of driver and passenger bacteria in CRC initiation and development.
Keywords: colorectal cancer, driver-passenger model, microbiota
Introduction
Although it has been well accepted that genetic and epigenetic alterations are driving forces in carcinogenesis and progression of colorectal cancer (CRC), accumulated evidences suggest that specific microbial compositions in the intestine environment are frequently associated with CRC. For decades, development of high-throughput sequencing technologies facilitates delineating the catalogue of intestinal microbiota. More than 1,000 microbial species from the three domains of life have been identified in the human gastrointestinal tract, based on the analyses of their small subunit ribosomal RNA gene sequences.1,2 Currently, the most detailed report to date suggests that we are only approximately 47% human on average by cell count.3 Thus, such enormously diversified microbiota with enormous numbers comprise a complex ecosystem that contributes to the host metabolism in the gastrointestinal tract as well as host health and disease. It has been proposed that intestinal microbiota may play key roles in driving CRC. However, dominant bacterial taxa associated with CRC and their roles and potential mechanisms in CRC evolution remain to be completely understood.
Tjalsma et al4 proposed a bacterial driver-passenger model for CRC, in which pathogenic bacteria initiating CRC development may be transiently colonized and replaced by other bacterial species with competitive growth advantage in the tumor niche.4 The former is defined as driver bacterium, and the latter is defined as passenger bacterium.4 Candidate driver bacteria show pro-carcinogenic features such as production of DNA-damaging compounds, interruption of tumor suppressor protein function, and induction of host inflammatory response.5,6 These bacteria may colonize with low relative abundance but persistently interact with host cells to induce asymptomatic but chronic change in normal intestinal epithelium to initiate CRC.7 Candidate passenger bacteria may show low relative abundances at the normal intestinal epithelium or the early stage of CRC, but competitively proliferate in the latter tumor niche.8 They take advantage of altered metabolism of tumor cells and outcompete the abundances of driver bacteria during development of CRC. These bacteria are hard to colonize on healthy colon tissue and cannot breach the intact colon wall. But they can easily invade a broken colon wall of adenoma or carcinoma.4 For example, Fusobacterium spp. appear to be the most common passenger bacteria in CRC.9 These passenger bacteria may continue contributing to the development of CRC. Survey of driver and passenger bacteria may serve as taxonomic bio-markers for determination of cohort with high risk to initiate CRC or patients with CRC, respectively.
In this work, to identify potential driver and passenger bacteria in CRC evolution, we compare the gut mucosal microbiomes of 75 triplet-paired CRC samples collected from on-tumor site, adjacent-tumor site, and off-tumor site, and 26 healthy controls, which were obtained in Tianjin Union Medical Center. Because tumorigenesis is a chronic process in a localized position, off-tumor sites may represent the normal intestinal epithelium before the patients got CRC. Thus, highly abundant bacterial taxa associated with off-tumor sites may function as potential driver bacteria, which is presumed to have higher abundance in patients before they got CRC than healthy controls. On the other hand, highly abundant bacteria taxa associated with on-tumor sites may function as passenger bacteria. Using these criteria, we identify four genera and two families as potential driver bacteria, and 14 genera or families as potential passenger bacteria.
Materials and Methods
Sample Collection
We analyzed intestinal microbiome samples collected from 75 patients and 26 healthy controls in Tianjin Union Medical Center. The patient information about age and sex is listed in Table 1. Written informed consent was obtained from all participants prior to their inclusion in the study. All protocols and procedures of this study were approved by the Medical Ethics Board of Tianjin Union Medical Center. Three locations in the same removed tissue of a cancer patient were collected, including on-tumor site (T), adjacent-tumor site (P) and off-tumor site (N). Cotton swabs were used to dip into the intestinal surface of removed colorectal cancer tissues to collect the tumor and adjacent flora. All off-tumor sites were above the tumor to avoid contamination. Furthermore, intestinal microflora samples on intestinal mucosa of healthy people were also collected by colonoscopy as healthy samples (H). Bacterial DNA of these samples was extracted using ZR Fungal/Bacterial DNA Kit according to the manufacturer’s instructions.
 |
Table 1 The Age and Gender Information of Patients in the Study |
16S rRNA Sequence Analysis
The 16S ribosomal RNA amplicon libraries were constructed according to the Illumina manufacturer's manual. Briefly, the following primers were used to amplify the V3-V4 region of 16S rRNA gene: forward primer (PCR1_Forward), 5ʹTCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG and reverse primer (PCR1_Reverse), 5ʹGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC. Amplified DNA library was subsequently purified using AMPure XP beads and quantified using Quant-iT PicoGreen dsDNA assay kit (Invitrogen). The paired-end sequencing reads were generated on Illumina MiSeq platform. Quality control and filtering of raw sequences were carried out using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The filtered paired-end reads were assembled using PandaSeq.10 The assembled sequences were loaded on QIIME11 for OTU picking, taxonomic assignment, and diversity analyses. Rarefaction curves were analyzed using alpha_rarefaction.py command from the QIIME pipeline (Supplementary Figure 1). The 16S rRNA sequencing reads have been submitted to the NCBI SRA database under accession number PRJNA606879. All the data are provided in this paper and supplementary Figure 1.
Determination of Potential Driver and Passenger Bacteria
Relative abundance of bacterial taxa was determined using arcsine square root.12 Candidate driver bacteria were determined when the relative abundance at an off-tumor site (N-site) was significantly higher than those at an on-tumor site (T-site). Candidate passenger bacteria were determined when its relative abundance at an on-tumor site (T-site) was significantly higher than those at an off-tumor site (N-site) and healthy controls (H). The statistical significances of multiple samples comparisons were calculated using one-way ANOVA with Kruskal–Wallis test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Correlation Network Analysis
To calculate Spearman rank correlation values between bacterial taxa, taxa summary from QIIME output was loaded in R package Hmisc (Harrell, Vanderbilt University School of Medicine, Nashville, TN, USA). Correlation network analysis of the Spearman rank correlation values was carried out using Cytoscape 3.7.2.13
Statistical Analysis
Statistical analyses were performed using statistical analysis in social science program (SSPS). The statistical significances of multiple sample comparisons were calculated using one-way ANOVA with Kruskal–Wallis test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Ethics Statement
Research was conducted according to the principles expressed in the Declaration of Helsinki. The study was approved by the Ethics Committee of Tianjin Union Medical Center. Patients provided written informed consent for the collection of samples and subsequent analysis when required.
Results
Determination of Potential Driver Bacteria in CRC
To determine potential driver bacteria in CRC initiation, we surveyed relative abundances of bacterial taxa at genus level at an on-tumor site (T), an adjacent-tumor site (P), an off-tumor site (N), and in healthy controls (H) (Figure 1). We identified Bacillus, Bradyrhizobium, and Methylobacterium, with high levels of relative abundance (average > 1%) as potential driver bacteria, whose relative abundance at off-tumor sites (N) were significantly higher than those at on-tumor sites (T) and in healthy controls (H) (Figure 2A–C). Moreover, relative abundances of these three genera at adjacent-tumor site (P) are significantly higher than those at on-tumor sites (T), respectively (Figure 2A–C). Particularly, the relative abundance of Bradyrhizobium at adjacent-tumor sites (P) was similar to that at off-tumor sites (N) and significantly higher than that in healthy controls (H) (Figure 2B). These data indicate that these bacteria have a competitive growth advantage and outcompete other species in colonizing the intestinal niche of patients and may contribute to CRC initiation.
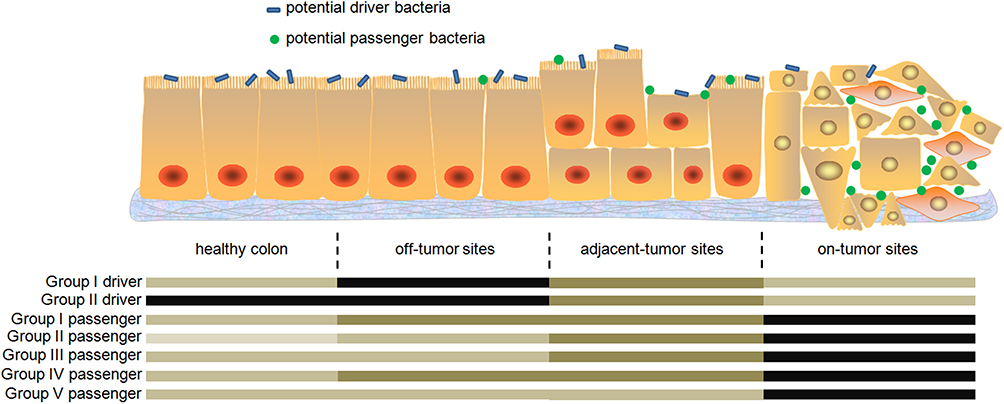 |
Figure 1 Schematic showing the off-tumor site, adjacent-tumor site, and on-tumor site in the intestinal tract of a CRC patient. Driver bacteria but not passenger bacteria can easily attach to intact colon wall at off-tumor sites. The latter are good colonizers at on-tumor sites and can invade distorted structure of carcinoma tissue. Relative abundances of each group are shown below the image, in which deep color indicates high abundance and light color indicates low abundance. |
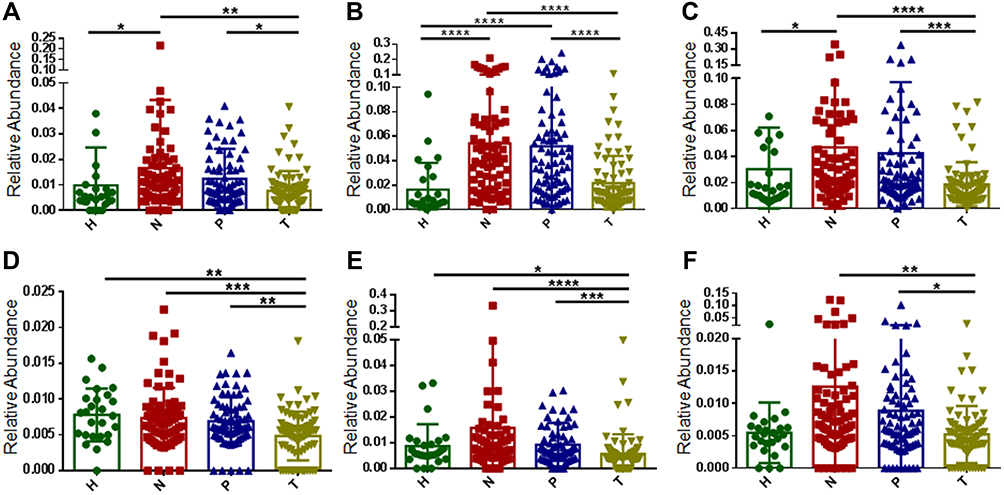 |
Figure 2 Identification of potential driver bacteria associated with CRC. Bacillus (A), Bradyrhizobium (B), Methylobacterium (C), Streptomyces (D), Intrasporangiaceae (E), and Sinobacteraceae (F) as potential driver bacteria. H: healthy control, N: off-tumor site, P: adjacent-tumor site, T: on-tumor site. The statistical significances of multiple sample comparisons were calculated using one-way ANOVA with Kruskal–Wallis test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
We also identified Streptomyces and two families, Intrasporangiaceae and Sinobacteraceae, with high levels of relative abundance, whose relative abundance at off-tumor sites (N) and adjacent-tumor sites (P) were both significantly higher than those at on-tumor sites (T) (Figure 2D–F). Their average relative abundance at off-tumor sites (N) were higher than those from healthy controls (H) but without statistical significance (Figure 2D–F). Moreover, the relative abundance of Streptomyces and Intrasporangiaceae in healthy controls (H) were significantly higher than those at on-tumor sites (T) (Figure 2D and F). We also considered these bacterial taxa as potential driver bacteria.
Determination of Potential Group I Passenger Bacteria in CRC
We next investigated potential passenger bacteria in CRC by filtering the relative abundance at on-tumor sites (T) which were higher than those at off-tumor sites (N). Based on the differential relative abundances of bacterial taxa at different sites, we divided the potential passenger bacteria into five groups. Group I passenger bacteria contained Fusobacterium, Campylobacter, Streptococcus, Schwartzia, and Parvimonas, whose relative abundances at on-tumor sites (T) were higher than those at off-tumor sites (N) and healthy controls (H) (Figure 3A–E). Relative abundances of Campylobacter and Parvimonas at off-tumor sites (N) and adjacent-tumor sites (P) were significantly higher than those from healthy controls (H) (Figure 3B and E), suggesting that the whole intestinal niche of patients is different from that of healthy people and can be considered for screening passenger bacteria by comparing their abundance distributions at on- and off-tumor sites. Particularly, the relative abundances of Fusobacterium, Campylobacter, and Schwartzia at adjacent-tumor sites (P) were significantly lower than those at on-tumor sites (T) (Figure 3A, B and D).
 |
Figure 3 Identification of potential group I passenger bacteria associated with CRC. Fusobacterium (A), Campylobacter (B), Streptococcus (C), Schwartzia (D), and Parvimonas (E) as potential group I passenger bacteria. H: healthy control, N: off-tumor site, P: adjacent-tumor site, T: on-tumor site. The statistical significances of multiple sample comparisons were calculated using one-way ANOVA with Kruskal–Wallis test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
Determination of Potential Group II, III, IV, and V Passenger Bacteria in CRC
Groups II, III, IV and V passenger bacteria did not meet the criteria for group I, but their relative abundances at on-tumor sites (T) were significantly higher than those at either off-tumor sites (N) or in healthy controls (H) (Figure 4A–I). Group II passenger bacteria contained Dethiosulfatibacter, Selenomonas, Peptostreptococus, and Leptotrichia, whose relative abundances in healthy controls (H) were significantly lower than those at off-tumor sites (N), adjacent-tumor sites (P), and on-tumor sites (T), suggesting that these bacteria may be inhibited by probiotics or other factors in heathy intestinal environments (Figure 4A–D). Group III passenger bacteria contain Granulicatella, whose relative abundance in healthy controls (H) was significantly lower than that at adjacent-tumor sites (P) and on-tumor sites (T), suggesting that the integrity of the colon wall may be the main factor affecting the growth of these bacteria (Figure 4E). Group IV passenger bacteria contained Shewanella, Mogibacterium, and Eikenella, whose relative abundances in healthy controls (H) were lower than those at on-tumor sites (T) (Figure 4F–H), indicating that these bacteria were enriched at on-tumor sites. Group V passenger bacteria contained Anaerococus, whose relative abundance at on-tumor sites (T) was significantly higher than that at off-tumor sites (P) (Figure 4I), suggesting that these bacteria were prone to colonize on interrupted colon walls. Taken together, the niche of on-tumor sites is the most suitable environment for these groups of passenger bacteria to proliferate.
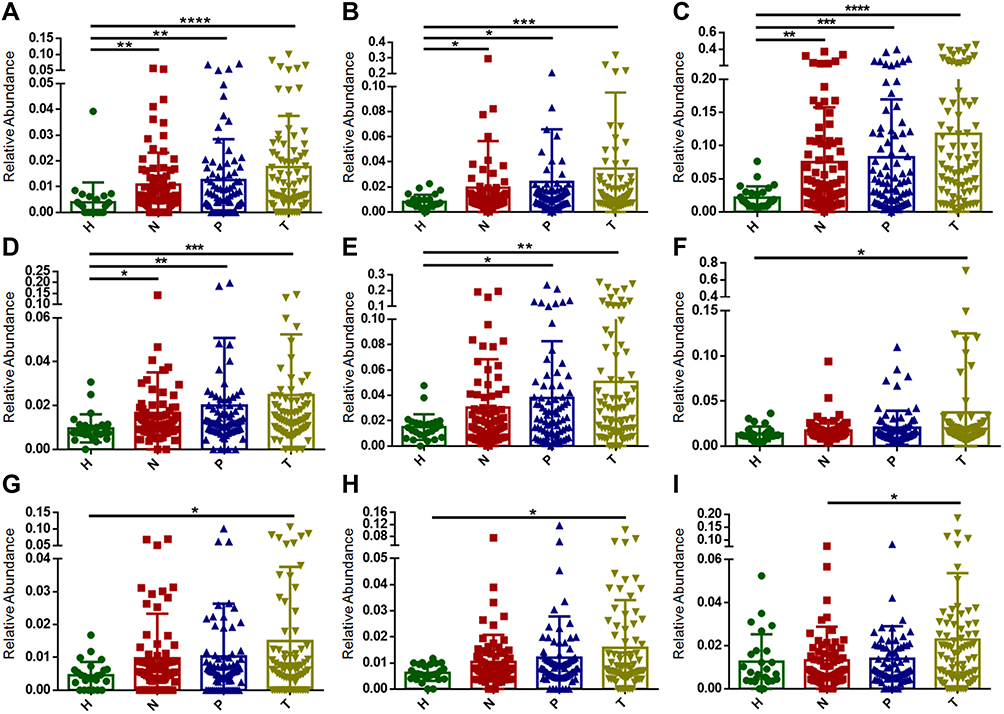 |
Figure 4 Identification of potential group II (A–D), III (E), IV (F–H), and V (I) passenger bacteria associated with CRC. Dethiosulfatibacter (A), Selenomonas (B), Peptostreptococus (C), and Leptotrichia (D) as potential group II passenger bacteria. Granulicatella (E) as a potential group III passenger bacteria. Shewanella (F), Mogibacterium (G), and Eikenella (H) as potential group IV passenger bacteria. Anaerococus (I) as a potential group V passenger bacteria. H: healthy control, N: off-tumor site, P: adjacent-tumor site, T: on-tumor site. The statistical significances of multiple sample comparisons were calculated using one-way ANOVA with Kruskal–Wallis test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. |
Co-Occurrence Network of Potential Driver and Passenger Bacteria in CRC
Bacteria in mixed-species populations co-exist to function in the progression of CRC. We next preformed correlation analysis of the potential driver and passenger bacteria identified above. Spearman rank correlation was carried out using relative abundances of bacterial taxa in healthy controls (H), off-tumor sites (N), adjacent-tumor sites (P), and on-tumor sites (T), respectively. All the correlation relationships were positive, while none of the negative correlation relationships was found between any potential driver and passenger bacteria. Interestingly, the potential driver bacteria defined above are highly correlated in all types of samples, respectively (Figure 5A–D). By contrast, merely a few potential passenger bacteria were correlated, especially in off-tumor sites (N), adjacent-tumor sites (P), and on-tumor sites (T) (Figure 5B–D). In healthy controls, certain potential driver and passenger bacteria were positively correlated, suggesting that their abundances were balanced in healthy people. During initiation and development of CRC, abundance levels of most driver bacteria and passenger bacteria were differently correlated.
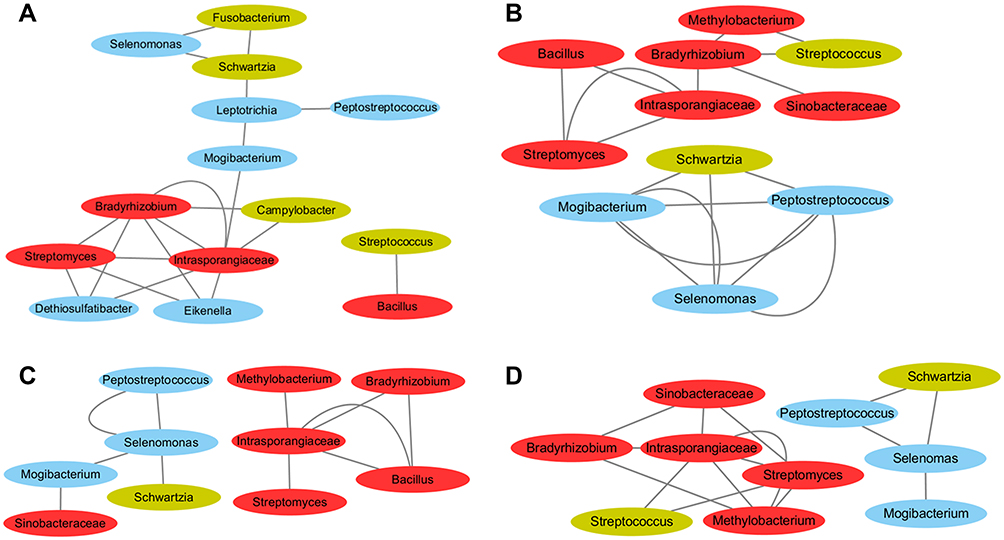 |
Figure 5 Correlation relationships among driver bacteria and passenger bacteria in healthy controls (A), off-tumor sites (B), adjacent-tumor sites (C), and on-tumor sites (D). Bacteria pairs with Spearman rank correlation above 0.50 were represented in the network. Edges represent correlation relationships between bacteria pairs. Red, potential driver bacteria. Green, potential group I passenger bacteria. Blue, potential group II, III, IV, and V passenger bacteria. |
Discussion
In this work, to reveal potential driver bacteria for CRC initiation and passenger bacteria for CRC development, we analyzed microbiomes of CRC patients and healthy controls in Tianjin Union Medical Center. We identified four genera including Bacillus, Bradyrhizobium, Methylobacterium, and Streptomyces, and two families including Intrasporangiaceae and Sinobacteraceae as potential driver bacteria. We identified 14 potential passenger bacteria and divided them into five groups. Group I passenger bacteria contained Fusobacterium, Campylobacter, Streptococcus, Schwartzia, and Parvimonas. Group II passenger bacteria contained Dethiosulfatibacter, Selenomonas, Peptostreptococus, Leptotrichia. Group III passenger bacteria contained Granulicatella. Group IV passenger bacteria contained Shewanella, Mogibacterium, and Eikenella. Group V passenger bacteria contained Anaerococus.
The potential driver bacteria of CRC defined in this study were found to be associated with other types of cancer as well. The analysis of 16S rRNA showed a higher relative abundance of Bacillus spp. in breast cancer patients compared with healthy samples.14 The relative abundance of Bradyrhizobium japonicum was increased in lung cancer patients with early-stage tumors compared with those in patients with advanced-stage tumors (I & II vs III & IV).15 The analysis of V1-V2 region of 16S rRNA showed that Methylobacteriaceae was abundant in malignant disease compared to benign disease in breast cancer patients.16 An intra-tumoral microbiome signature including Streptomyces and Bacillus clausii is highly predictive of long-term survivorship.17 Further physiological and functional studies of representative pathogenic bacteria among these potential driver bacteria will help understand their roles in CRC initiation.
The majority of the potential passenger bacteria defined here were also found to be associated with CRC in previous reports. Fusobacterium, Peptostreptococcus, Parvimonas, Leptotrichia, Campylobacter, Streptococcus, Selenomas, Shewanella, Mogibacterium, and Eikenella were detected to be highly associated with CRC.18–25 Due to their high relative abundances in CRC samples, they have received more attention than driver bacteria and some of these CRC-associated bacteria were under investigation for their physiological roles in CRC evolution. For example, Fusobacterium spp., anaerobic Gram-negative bacteria, are highly associated with CRC.8,18,26 Particularly, Fusobacterium nucleatum has been identified to be an invasive and proinflammatory agent for acute and chronic oral and gastrointestinal infections.18,27–31 In addition, Fusobacterium spp. may function as a vector for host tissue infection by co-adherent bacteria.18 Treatment of Fusobacterium with metronidazole (Fusobacterium-killing antibiotic) reduced tumor-growth in vivo,21 suggesting that Fusobacterium contributes to CRC growth and metastasis. Campylobacter jejuni produces a genotoxin, cytolethal distending toxin, which has DNAse activity and causes DNA double-strand breaks. C. jejuni promotes CRC and induces changes in microbial composition.32 Fusobacterium spp. have been observed in significant co-occurrence with Leptotrichia and Campylobacter species,18 or with Bacteroides, Selenomonas, and Prevotella species.21
Our co-occurrence network analysis of potential driver and passenger bacteria suggests bacterial compositions in different sample types show specific correlation patterns. Six driver bacteria taxa were highly correlated together in off-tumor sites. Sinobacteraceae was clustered with passenger bacteria in adjacent-tumor sites, while Bacillus was not correlated with either driver bacteria or passenger bacteria in on-tumor sites. In healthy samples, Bacillus, Bradyrhizobium, Streptomyces, and Intrasporangiaceae were correlated with passenger bacteria, while Methylobacterium and Sinobacteraceae were not correlated with any of them. Four passenger bacteria including Schwartzia in group I and Mogibacterium, Selenomonas, and Peptostreptococcus in groups II–V were highly correlated in off-tumor sites, adjacent-tumor sites, and on-tumor sites. Fusobacterium and Campylobacter were correlated with other driver and passenger bacteria in healthy samples, but none of them in off-tumor sites, adjacent-tumor sites, or on-tumor sites, suggesting that relative abundances of these potential driver and passenger bacteria were more balanced in healthy controls than CRC patients. Reduced bacterial composition in the intestine is the key feature for many intestinal and extra-intestinal disorders.33 It is suggested that a combination of alterations, rather than changed abundance of a particular strain in the intestinal microbiota promotes tumorigenesis.34 A high diversity with balanced driver and passenger bacteria might reflect a key feature of a healthy gut enabling a species-rich ecosystem to deal with environmental stresses that promote CRC. These mixed-species populations may interact, cooperate, or compete with each other in the intestinal ecosystem. These potential driver and passenger bacteria confirmed in this study are only based on bioinformatics analysis, so how microbial diversity protects against tumorigenesis and how driver bacteria initiate CRC and are replaced by passenger bacteria in the later stage remain to be further investigated. Furthermore, the precise role of these bacteria on the intestinal tract need to be revealed by further experiments in vitro and in vivo, especially for those predicted as passenger bacteria. Passenger bacteria have high relative abundances in tumor niches, but some of them may act as probiotics, which has the function of establishment of balanced intestinal microflora, synthesis of vitamins and inactivation of cancerogenic compounds.35,36
Taken together, this study reports potential driver and passenger bacteria identified in human CRC. Further retrospective study will help identify potential driver and passenger bacteria as well. In addition, it is important to investigate whether driver bacteria identified in off-tumor sites of CRC patients are also present in the fecal and colonoscopy samples of CRC patients. Only a few driver bacteria have been identified based on experimental data in vitro and in vivo,5 and many potential driver bacteria need to be found and identified. Furthermore, abundances of driver and passenger bacteria may serve as a reference biomarker in clinical diagnosis of CRC initiation risk and development. These potential driver and passenger bacteria found in our study provide a preliminary scope to carry out further investigations into the mechanisms involved.
Funding
This research was funded by the Natural Science Foundation of China Grant number 81972826, and the Fundamental Research Funds for the Central Universities, Nankai University Grant number 63191440, and Key R&D Projects in the Tianjin Science and Technology Pillar Program Grant number 19YFZCSY00420, and National key R&D Program of China Grant number 2017YFC1700604, and National key R&D Program of China Grant number 2017YFC1700606, and Tianjin 131 Innovative Talent Training Project in 2018, and the Research Foundation of Tianjin Union Medical Center number 2018YJ023.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Qin J, Li R, Raes J, et al. Europe PMC Funders Group Author Manuscripts. A human gut microbial gene catalog established by metagenomic sequencing. Nature. 2010;464:59–65. doi:10.1038/nature08821
2. Rajilić-Stojanović M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi:10.1111/1574-6976.12075
3. Knight R, Callewaert C, Marotz C, et al. The microbiome and human biology. Annu Rev Genomics Hum Genet. 2017;18:65–86. doi:10.1146/annurev-genom-083115-022438
4. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Micro. 2012;10:575–582. doi:10.1038/nrmicro2819
5. Rhee KJ, Wu SG, Wu XQ, et al. Induction of persistent colitis by a human commensal, enterotoxigenic bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77:1708–1718.
6. Kapral M, Węglarz L, Parfiniewicz B, et al. Quantitative evaluation of transcriptional activation of NF-κB p65 and p50 subunits and IκBα encoding genes in colon cancer cells by desulfovibrio desulfuricans endotoxin. Folia Microbiol. 2010;55:657–661. doi:10.1007/s12223-010-0106-6
7. Gao Z, Guo B, Gao R, et al. Microbiota disbiosis is associated with colorectal cancer. Front Microbiol. 2015;6:20. doi:10.3389/fmicb.2015.00020
8. Kostic AD, Gevers D, Pedamallu CS, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi:10.1101/gr.126573.111
9. Amitay EL, Werner S, Vital M, et al. Fusobacterium and colorectal cancer: causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis. 2017;38:781–788. doi:10.1093/carcin/bgx053
10. Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD. PANDAseq: paired-end assembler for illumina sequences. BMC Bioinformatics. 2012;13:31. doi:10.1186/1471-2105-13-31
11. Caporaso J, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi:10.1038/nmeth.f.303
12. Nakatsu G, Li X, Zhou H, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727. doi:10.1038/ncomms9727
13. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi:10.1101/gr.1239303
14. Eslami SZ, Majidzadeh AK, Halvaei S, et al. Microbiome and breast cancer: new role for an ancient population. Front Oncol. 2020;10:1–15. doi:10.3389/fonc.2020.00120
15. Jin J, Gan Y, Liu H, et al. Diminishing microbiome richness and distinction in the lower respiratory tract of lung cancer patients: a multiple comparative study design with independent validation. Lung Cancer. 2019;136:129–135.
16. Meng S, Chen B, Yang J, et al. Study of microbiomes in aseptically collected samples of human breast tissue using needle biopsy and the potential role of in situ tissue microbiomes for promoting malignancy. Front Oncol. 2018;8:318. doi:10.3389/fonc.2018.00318
17. Riquelme E, Zhang Y, Zhang L, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806. doi:10.1016/j.cell.2019.07.008
18. Warren RL, Freeman DJ, Pleasance S, et al. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome. 2013;1:1–12. doi:10.1186/2049-2618-1-16
19. Rajagopala SV, Vashee S, Oldfield LM, et al. The human microbiome and cancer. Cancer Prev Res. 2017;10:226–234. doi:10.1158/1940-6207.CAPR-16-0249
20. Bultman SJ. Emerging roles of the microbiome in cancer. Carcinogenesis. 2014;35:249–255. doi:10.1093/carcin/bgt392
21. Bullman S, Pedamallu CS, Sicinska E, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. 2017;358:1443–1448. doi:10.1126/science.aal5240
22. Flemer B, Lynch DB, Brown JMR, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer. Gut. 2017;66:633–643. doi:10.1136/gutjnl-2015-309595
23. Chen W, Liu F, Ling Z, et al. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi:10.1371/journal.pone.0039743
24. Hale VL, Chen J, Johnson S, et al. Shifts in the fecal microbiota associated with adenomatous polyps. Cancer Epidemiol Biomarkers Prev. 2017;26:85–94.
25. Allali I, Delgado S, Marron PI, et al. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes. 2015;6:161–172. doi:10.1080/19490976.2015.1039223
26. Castellarin M, Warren RL, Freeman JD, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306. doi:10.1101/gr.126516.111
27. Han YW, Shi W, Huang GT, et al. Interactions between periodontal bacteria and human oral epithelial cells: Fusobacterium nucleatum adheres to and invades epithelial cells. Infect Immun. 2000;68:3140–3146. doi:10.1128/IAI.68.6.3140-3146.2000
28. Krisanaprakornkit S, Kimball JR, Weinberg A, et al. Inducible expression of human beta-defensin 2 by Fusobacterium nucleatum in oral epithelial cells: multiple signaling pathways and role of commensal bacteria in innate immunity and the epithelial barrier. Infect Immun. 2000;68:2907–2915. doi:10.1128/IAI.68.5.2907-2915.2000
29. Peyret-Lacombe A, Brunel G, Watts M, et al. TLR2 sensing of F. nucleatum and S. sanguinis distinctly triggered gingival innate response. Cytokine. 2009;46:201–210. doi:10.1016/j.cyto.2009.01.006
30. Signat B, Roques C, Poulet P, et al. Fusobacterium nucleatum in periodontal health and disease. Curr Issues Mol Biol. 2011;13:25–36.
31. Swidsinski A, Dörffel Y, Loening-Baucke V, et al. Acute appendicitis is characterised by local invasion with Fusobacterium nucleatum/necrophorum. Gut. 2011;60:34–40. doi:10.1136/gut.2009.191320
32. He Z, Gharaibeh RZ, Newsome RC, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. 2019;68:289–300. doi:10.1136/gutjnl-2018-317200
33. Tilg H, Kaser A. Gut microbiome, obesity, and metabolic dysfunction. J Clin Invest. 2011;121:2126–2132. doi:10.1172/JCI58109
34. Tilg H, Adolph TE, Gerner RR, Moschen AR. The intestinal microbiota in colorectal cancer. Cancer Cell. 2018;33:954–964. doi:10.1016/j.ccell.2018.03.004
35. Uccello M, Malaguarnera G, Basile F, et al. Potential role of probiotics on colorectal cancer prevention. BMC Surg. 2012;12(Suppl 1):S35. doi:10.1186/1471-2482-12-S1-S35
36. MalaGuarnera G, leGGio F, Vacante M, et al. Probiotics in the gastrointestinal diseases of the elderly. J Nutr Health Aging. 2012;16:402–410. doi:10.1007/s12603-011-0357-1
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.