")
Back to Journals » Drug Design, Development and Therapy » Volume 15
An Update Review of Biosimilars of Adalimumab in Psoriasis – Bioequivalence and Interchangeability
Received 24 April 2021
Accepted for publication 16 June 2021
Published 8 July 2021 Volume 2021:15 Pages 2987—2998
DOI https://doi.org/10.2147/DDDT.S317382
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Tin Wui Wong
Xin Zhou,1 Zhuo Chen,2 Xinling Bi1
1Department of Dermatology, Changhai Hospital, Naval Medical University, Shanghai, 200433, People’s Republic of China; 2Department of Dermatology, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, 200127, People’s Republic of China
Correspondence: Xinling Bi
Department of Dermatology, Changhai Hospital, Naval Medical University, Shanghai, 200433, People’s Republic of China
Tel +86 13386278946
Email [email protected]
Abstract: Biologic drugs have revolutionized the treatment of psoriasis and other rheumatological diseases. In recent years, many biosimilar agents that are highly similar in structure and function to their originator products have been developed, including the tumor necrosis factor-alpha antagonist adalimumab. The considerably lower cost of these products has greatly cut the economic burden of the patients and increased the accessibility of biologic therapies worldwide. The US Food and Drug Administration and/or the European Medicines Agency have approved eight biosimilars of adalimumab (ABP 501/BI 695501/SB5/GP2017/FKB327/MSB11022/PF-06410293/CT-P17) for the treatment of psoriasis, and others are under review. Given that these agents showed pharmacokinetic, efficacy, safety, and immunogenicity profiles comparable to those of the originator, adalimumab biosimilars were licensed for all indications approved for reference adalimumab based on extrapolation; however, some of the equivalence studies were only conducted in one or two disease populations. This review discusses the bioequivalence of adalimumab biosimilars as demonstrated by various clinical trials, the extrapolation of indications, guidance and policies of the EU and US on interchangeability (nonmedical switching/automatic substitution) between biosimilars and originators, and the real-life practices of switching from reference adalimumab to the respective biosimilars. Further data from real-world studies and post-marketing analyses are needed better to address the efficacy and safety of the transition strategy.
Keywords: biosimilar, adalimumab, psoriasis, guidance of interchangeability, extrapolation of indications
Introduction
The occurrence of biologic medicines has brought a drastic change in the treatment regimens for psoriasis and other chronic rheumatic diseases over the past decades. Many biological agents have been licensed for treating chronic plaque psoriasis. Biologics targeting tumor necrosis factor-alpha (TNF-α) (infliximab, etanercept, and adalimumab), interleukin (IL)-12/23p40 (ustekinumab), IL-23p19 (guselkumab, tildrakizumab, and risankizumab), IL-17A (secukinumab and ixekizumab), and IL-17RA (brodalumab) were among the most commonly used medications in this class. However, the high expense often limits patient access to these medications.1
A biosimilar, as defined by the European Medicines Agency (EMA), is a biologic agent very similar to another already approved biological drug in the European Union (EU); although there might be minor differences from the originator, the biological properties and clinical performance in terms of pharmacokinetic (PK) and pharmacodynamic (PD) features, immunogenicity, efficacy, and safety should be comparable to the respective originator.2 The United States (US) Food and Drug Administration (FDA) defines a biosimilar as a biological medicine that has “no clinically meaningful differences” from an already licensed originator.3 The phrase “no clinically meaningful differences” means that the biosimilar should be comparable in terms of purity, safety, efficacy, and clinical immunogenicity to the reference drug. Biosimilars were created to reduce the financial expense of originators, thus allowing wider application of biologic treatment.4
Adalimumab (Humira, AbbVie Inc. North Chicago, Illinois, US) is a fully human, recombinant, IgG1 monoclonal antibody5 targeting TNF-α. After binding to TNF, adalimumab blocks the interaction of the cytokine with p55 and p75 cell surface TNF receptors, thus inhibiting TNF-related biological reactions.6 Results of the REVEAL6 study showed that 71% patients from adalimumab group achieved 75% improvement of Psoriasis Area and Severity Index (PASI) score at week 16 compared to that of placebo group (7%). Considering the results of the REVEAL6 and CHAMPION trials,7 it was approved by the FDA for treating adult psoriasis in 2008 and by the EMA in 2007. Since 2012, adalimumab has become the world’s top selling drug, with total sales of 2014 reaching as much as $12.89 billion.8 The tremendous commercial success worldwide makes adalimumab the most appealing target for biosimilar manufacturers. Upon expiration of the patents of Humira in the US in December 2016 and in Europe in October 2018,9 several biosimilars gained the approval of regulatory agencies and entered the market.
To date, through the years 2016–2020, the FDA and/or the EMA have approved eight adalimumab biosimilars (ABP 501: EMA 2017, FDA 2016; BI 695501: EMA 2017 (withdrawn 2019), FDA 2017; SB5: EMA 2017, FDA 2019; GP2017: EMA 2018, FDA 2018; FKB327: EMA 2018, FDA 2020; MSB11022: EMA 2019; PF-06410293: FDA 2019, EMA 2020;10 CT-P17: EMA2020 11), for treating chronic plaque psoriasis, and many others are in development (Table 1).
 |
Table 1 Adalimumab Biosimilars Approved or in Clinical Development for Psoriasis Treatment |
Some prior articles have reviewed adalimumab biosimilars. Olteanu et al12 reviewed published and ongoing studies relating to biosimilars targeting TNF-α. They listed three completed trials of adalimumab biosimilars (ABP 501 and BCD-057) and three ongoing trials of adalimumab biosimilars (SB5, M923, and GP2017). However, the results including safety, efficacy and immunogenicity of these trials were not available at the time. The authors concluded that the current situation is very unsatisfactory, which will give clinicians a certain degree of uncertainty in their treatment decisions. In addition, the review was written seven years ago, there were no adalimumab biosimilars approved by the FDA and the EMA, and the RCTs of adalimumab biosimilars were still ongoing at that time. Discussions about the guidance and policies of various countries on interchangeability, and the real-life practices of switching were missing. Reynolds et al10 reviewed biosimilars in the treatment of psoriasis, but mainly focused on the safety and efficacy. Extrapolation, interchangeability, and guidance of different societies and countries were not involved. The perspectives on the biosimilars have changed over years, so it is necessary to discuss about these problems that may influence clinicians in their clinical practice.
The purpose of this review is to present the most updated clinical trials’ outcomes of adalimumab biosimilars (ABP 501, BI 695501, SB5, GP2017, FKB327, MSB11022, PF-06410293 and CT-P17), extrapolation of indications, guidance and policies in the EU and USA on interchangeability (nonmedical switching/automatic substitution) between these biosimilars and their originators, and the real-life practices of switching from originator adalimumab to respective biosimilars through a narrative review of the existing literature. Additional adalimumab biosimilars that were approved in countries other than the US and the EU are also mentioned.
Methods
The literature review was conducted in the PubMed database to identify English articles related to Phase I PK studies and Phase III randomized clinical trials (RCTs) investigating the safety and effectiveness of adalimumab biosimilars in the treatment of psoriasis or other rheumatic diseases. Each phase III study recruited more than 400 patients from multiple centers, and the efficacy and safety results were analyzed by the full analysis set (FAS) and the safety analysis set (SAS) separately. We extracted data from the corresponding regulatory agencies’ approval documents, national registries of biologics or post-marketing surveillance analyses, and we also searched ClinicalTrials.gov to identify unpublished study results relating to adalimumab biosimilars. The search was performed by using the following key words: biosimilar adalimumab, ABP501, BI 695501, SB5, GP2017, FKB327, MSB11022, PF-06410293, CT-P17, psoriatic arthritis (PsA), psoriasis (PsO), rheumatoid arthritis (RA), ankylosing spondylitis (AS), immunogenicity, efficacy, safety, extrapolation of indication, interchangeability, switch, and substitution. The literature review was extended to Feb 28, 2021.
Efficacy, Safety and Bioequivalence Studies
ABP 501
The FDA and the EMA approved the first adalimumab biosimilar ABP 501 in 2016 and 2017, respectively.11,13 A phase I study enrolled healthy subjects to evaluate the PK similarity of ABP 501 to adalimumab.14 Healthy volunteers were randomized into three groups to be treated subcutaneously with 40 mg of ABP 501, US- or EU-sourced adalimumab. The mean serum concentration-time profiles after a single dose were similar across the three groups. The single doses of ABP501 and adalimumab were shown to be equivalent with respect to the area under the serum concentration-time curve from time 0 (AUC0) to infinity (AUCinf) and from AUC0 to terminal concentration (AUClast). The 90% confidence intervals (CIs) for the geometrical mean ratios (GMRs) of Cmax, AUCinf and AUClast were within the predefined range (0.80–1.25; primary endpoint), demonstrating the equivalence of PK between ABP501 and adalimumab.14
Papp et al conducted a multicenter, randomized, phase III study to compare the safety and efficacy of ABP 501 with adalimumab in psoriasis.15 Overall, 350 patients were enrolled, randomized 1:1 into two groups and included in the efficacy analysis; 347 patients (ABP 501 group, n = 174; adalimumab group, n = 173) were included within the safety analysis. At week 16, the percentage improvement of PASI score in the two groups was 80.9% (ABP 501) and 83.1% (adalimumab). Adverse events (AEs) (67.2% [ABP 501] vs 63.6% [adalimumab]) and antidrug antibody (ADA) incidence (55.2% [ABP 501] vs 63.6% [adalimumab]) were comparable between groups. Safety and immunogenicity remained similar across groups after a single transition at 20 weeks.15
Another phase III RCT was conducted in patients with RA.16 In this active comparator-controlled study, 526 patients were randomized, and 494 completed the study. At week 24, the American College of Rheumatology (ACR)-20 response rate was 74.6% in the ABP 501 group and 72.4% in the adalimumab group; the risk ratio (RR) was 1.039, and the 90% CI (0.954–1.133) fell within a predefined margin. The safety profiles were comparable between the two groups. No clinically significant differences were found in AEs or laboratory abnormalities. ADAs tested positive in 38.3% of the ABP501 group and in 38.2% of the adalimumab group.16 An open-label extension of this study evaluated patients from the former trial who switched to ABP501 at week 26 and those who maintained ABP 501 treatment throughout 68 weeks. The results confirmed the long-term safety and efficacy between groups.17
BI 695501
BI 695501 gained approval by both the FDA and the EMA in 2017.11,13 On 15 January 2019, the EMA withdrew the marketing authorization for BI 695501 in the EU at the request of the manufacturer for commercial reasons.18 Ninety percent CIs of all primary PK variables (BI 695501 to US-/EU-sourced adalimumab and US- to EU-sourced adalimumab) fell within the predefined margin in the phase I VOLTAIRE-PK study, confirming three-way PK equivalence. The similarity of BI 695501 to US-/EU-sourced adalimumab was further supported by comparison of secondary and additional PK parameters.19
The phase III VOLTAIRE-RA trial randomized active RA patients with concomitant methotrexate (MTX) to receive BI 695501 or Humira for 24 weeks. Before week 25, patients receiving adalimumab treatment in the first 24 weeks were rerandomized into the adalimumab maintenance group or BI 695501 transition group for another 24 weeks of treatment. The ACR-20 response rates were 67.0% (BI 695501) and 61.1% (adalimumab) at week 12 (90% CI −0.9 to 12.7) and 69.0% (BI 695501) and 64.5% (adalimumab) at week 24 (95% CI −3.4 to 12.5), confirming clinical similarity between the biosimilar BI 695501 and the adalimumab reference product (RP). The percentages of ACR20, ACR50, and ACR70 responders were comparable among the BI 695501 switching group, adalimumab maintenance group and BI 695501 maintenance group at week 48.20 The immunogenicity (ADAs, ADA titers and neutralizing antibodies), safety and tolerability data were similar among the treatment arms. VOLTAIRE-RAext21 was an open-label extension of the VOLTAIRE-RA study, which evaluated the long-term efficacy, safety and immunogenicity of BI 695501 in patients who had completed the VOLTAIRE-RA study. Patients from the abovementioned three groups received BI 695501 biweekly for another 48 weeks. The results showed comparable safety, efficacy, and immunogenicity between groups, without identifying any previously unknown side effects of adalimumab.
The preliminary results of a phase III study in patients with chronic moderate-to-severe plaque psoriasis showed comparable PASI75 response rates between groups (week 16: 68.2% [BI 695501] vs 70.4% [adalimumab], 95% CI −14.4 to 8.7; week 24: 75.3% [BI 695501] vs 72.4% [adalimumab], 95% CI −8.5 to 12.6).22 Multi-switching between adalimumab and BI695501 in patients with plaque psoriasis was conducted in another completed phase III study named VOLTAIRE-X; however, no results have been reported yet.23
SB5
SB5 was approved by the EMA in 2017 and by the FDA in 2019.11,13 A phase I PK clinical trial comparing SB5 with reference adalimumab in healthy subjects showed comparable results of mean PK parameters across the SB5, EU-adalimumab and US-adalimumab groups.24 The mean values of AUCinf, Cmax and AUClast were similar between groups, and the 90% CI fell within the prespecified equivalence margin.
The results from a randomized, double-blind, phase III clinical trial comparing the efficacy of SB5 with adalimumab RP in patients with moderate-to-severe RA showed that the ACR-20 response was equivalent between the SB5 group (72.4%) and the adalimumab group (72.2%) at week 24 (95% CI −7.83 to 8.13).25 Overall, the rates of treatment-emergent AEs (TEAEs) were 35.8% (SB5) and 40.7% (adalimumab) up to week 24. The incidence of ADA was also comparable between the SB5 (33.1%) and adalimumab (32.0%) groups.25 At 24 weeks, patients receiving adalimumab were rerandomized either to continue adalimumab treatment or to switch to SB5 until week 52, while patients receiving SB5 maintained their therapeutic drug throughout the 52 weeks of study. The results revealed comparable safety profiles, effectiveness and incidence of ADAs across groups after transition.26
Currently, no clinical trials have been performed to evaluate the efficacy and safety of this biosimilar in psoriasis populations.
GP2017
The FDA and the EMA approved GP2017 in 2018.11,13 A single-center, parallel group, three-arm PK similarity study was conducted in healthy male subjects. The GMRs for Cmax and AUC0-inf were 1.05 and 1.04 for GP2017/EU-adalimumab and 1.00 and 1.08 for GP2017/US-adalimumab, and the 90% CIs fell within the acceptance range, confirming the PK equivalence between GP2017 and adalimumab. The safety and immunogenicity data showed similarity across groups.27
In a phase III study, patients with psoriasis were randomized to receive GP2017 or adalimumab subcutaneously for 17 weeks. Then, PASI50 responders were rerandomized either to continue their former assigned treatment until week 35 or to shift between GP2017 and adalimumab every 6 weeks.28 At week 35, all patients received the originally assigned treatment at randomization until week 51. The response rates of PASI75 were comparable for GP2017 (66.8%) and adalimumab (65.0%); the 95% CI was −7.46 to 11.15. There was no impact on clinical efficacy after multiple switching between GP2017 and adalimumab. All the efficacy parameters, including PASI50, PASI75, PASI90 and PASI100, were similar across the transition groups and the maintenance groups over time. At 51 weeks, no significant differences with regard to efficacy, immunogenicity, or AEs were identified in switched groups compared to the continued groups.28
Another phase III trial named ADMYRA also revealed similar efficacy of GP2017 to the respective adalimumab originator in patients with methotrexate-resistant RA. There were no statistically significant differences regarding efficacy, safety and immunogenicity profiles among any of the groups.29
MSB11022
The EMA approved MSB11022 in April 2019.11 Three-way PK equivalence for all primary endpoints was demonstrated in a phase I study comparing MSB11022 with the adalimumab originator in healthy subjects. AEs were comparable across groups, with TEAE incidences of 64.1%, 57.5% and 62.0% among the MSB11022 and US-/EU-adalimumab groups, respectively.
In the phase III AURIEL-PsO trial, patients with moderate-to-severe plaque psoriasis were enrolled and assigned to receive MSB11022 or reference adalimumab subcutaneously every other week for 16 weeks, at which point patients receiving adalimumab were rerandomized either to continue adalimumab treatment or to switch to MSB11022 up to week 50, while patients receiving MSB11022 continued with MSB11022 until 50 weeks.30 The percentage of PASI75 responders was 89.7% (MSB11022 group) and 91.6% (adalimumab group) at week 16 (95% CI −7.82–4.07). Profiles of safety and immunogenicity were equivalent between the groups.30
In the multicenter, double-blind, parallel group, phase III AURIEL-RA study, patients with moderate-to-severe methotrexate-resistant RA were randomized into the MSB11022 group or the adalimumab group. At week 12, 79.6% of patients in the MSB11022 group achieved ACR20 and 80.9% of patients in the reference adalimumab group achieved ACR20 (95% CI −10.55–8.04), which was similar between the two groups.31 This similarity was maintained up to week 52. No significant differences were identified in efficacy, Dermatology Life Quality Index (DLQI), AE incidence, or immunogenicity among the treatment groups throughout the trial.31
FKB327
FKB327 gained approval in the EU in September 2018 and in the US in July 2020.11,13 A phase I three-way bioequivalence study confirmed PK similarity between FKB327 and EU- or US-sourced adalimumab.32
In a phase III equivalence study, patients with moderate-to-severe active RA were randomized to be treated with FKB327 or reference adalimumab for 24 weeks (period 1). Then, in period 2 (an open-label extension study), patients were rerandomized 2:1 to continue with the same study drug or to switch to the other up to week 54.33 The percentages of patients achieving ACR20 were 74.1% (FKB327 group) and 75.7% (adalimumab group) at week 24 (95% CI −7.9 to 4.7), which was maintained at over 70% of patients up to week 54 in all treatment arms. At week 24, the incidence of ADAs was similar between the groups (57.7% [FKB327] vs 55.5% [adalimumab]), and no notable difference in ADAs was identified between the maintenance and transition groups in the extension study.33 In period 3, all patients were given FKB327 for another 46 weeks until week 100.34 The results showed that the long-term (up to 2 years) effectiveness, safety and immunogenicity were comparable between FKB327 and adalimumab, and there was no impact on single- or double-switching treatment.
Currently, no clinical trials have been carried out to evaluate the efficacy of this agent in the psoriasis population.
PF‑06410293
PF‑06410293 was approved by the FDA in 2019 and by the EMA in 2020.11,13
The PK equivalence study REFLECTIONS B538-07, with 362 healthy volunteers, demonstrated similar PK profiles between PF-06410293 and both EU- and US-sourced adalimumab. The ratios and 90% CIs of all parameters (Cmax, AUC2wk, AUCt and AUC∞) fell within the acceptance range of 80–125% and included 100% of the sample.35
In a double-blind, randomized, phase III study (REFLECTIONS B538-02), patients with active RA were assigned to be treated with PF-06410293 or EU-adalimumab. The study was divided into 3 periods with each period lasting 26 weeks. At week 12 (primary endpoint), the ACR20 response rate was 68.7% in the PF-06410293 group and 72.7% in the EU-adalimumab group (95% CI −10.38 to 4.44). The safety, immunogenicity and PK profiles were comparable between groups through the first 26 weeks.36 The clinical comparability of PF-06410293 to EU-adalimumab was further supported by results from period 2.35 The ACR20 in patients who switched from reference adalimumab to PF-06410293 in period 2 was 86.6% at week 26 and 84.3% at week 52. Moreover, ACR20 remained similar throughout the first two periods in the PF-06410293 maintenance group (86.6% [week 26] and 82.7% [week 52]) and the adalimumab maintenance group (84.4% [week 26] and 79.3% [week 52]).35 The ACR20 response rate was consistent in all 3 treatment groups during treatment period 2.
CT-P17
CT-P17 was recently approved by the EMA in December 2020.11
PK equivalence was concluded by a phase I study with 312 healthy subjects. AUC0-inf, AUC0-last, and Cmax were similar among CT-P17, US- and EU-sourced adalimumab, with the ratios and 90% CIs of all parameters falling within the predefined acceptance range of 80–125%. The safety, efficacy and immunogenicity profiles were comparable across treatment arms.37
Kay et al conducted a 52-week multicenter, randomized, double-blind, phase III clinical trial (N=648) to evaluate the efficacy of CT-P17 compared with EU-adalimumab and reported results up to week 24.38 Moderate-to-severe RA patients were randomized to receive either CT-P17 or EU-adalimumab 40 mg injection every other week until week 24 (period 1). At week 26, patients receiving EU-adalimumab in period 1 were rerandomized either to transition to CT-P17 or to maintain EU-adalimumab treatment until 48 weeks (period 2). Patients receiving CT-P17 in period 1 continued CT-P17 treatment in period 2. The ACR20, ACR50 and ACR70 response rates at week 24 were 82.7%, 60.2% and 40.7% in the CT-P17 group and 82.7%, 63.6% and 44.4% in the EU-adalimumab group (95% CI −5.94 to 5.94),38 which showed similar therapeutic efficacy between CT-P17 and EU-adalimumab.
Others
Many other adalimumab biosimilars are in development or have already been approved in other countries worldwide (Table 1). Table 2 shows the results of some phase III studies, demonstrating comparable efficacy of these biosimilars to RP.
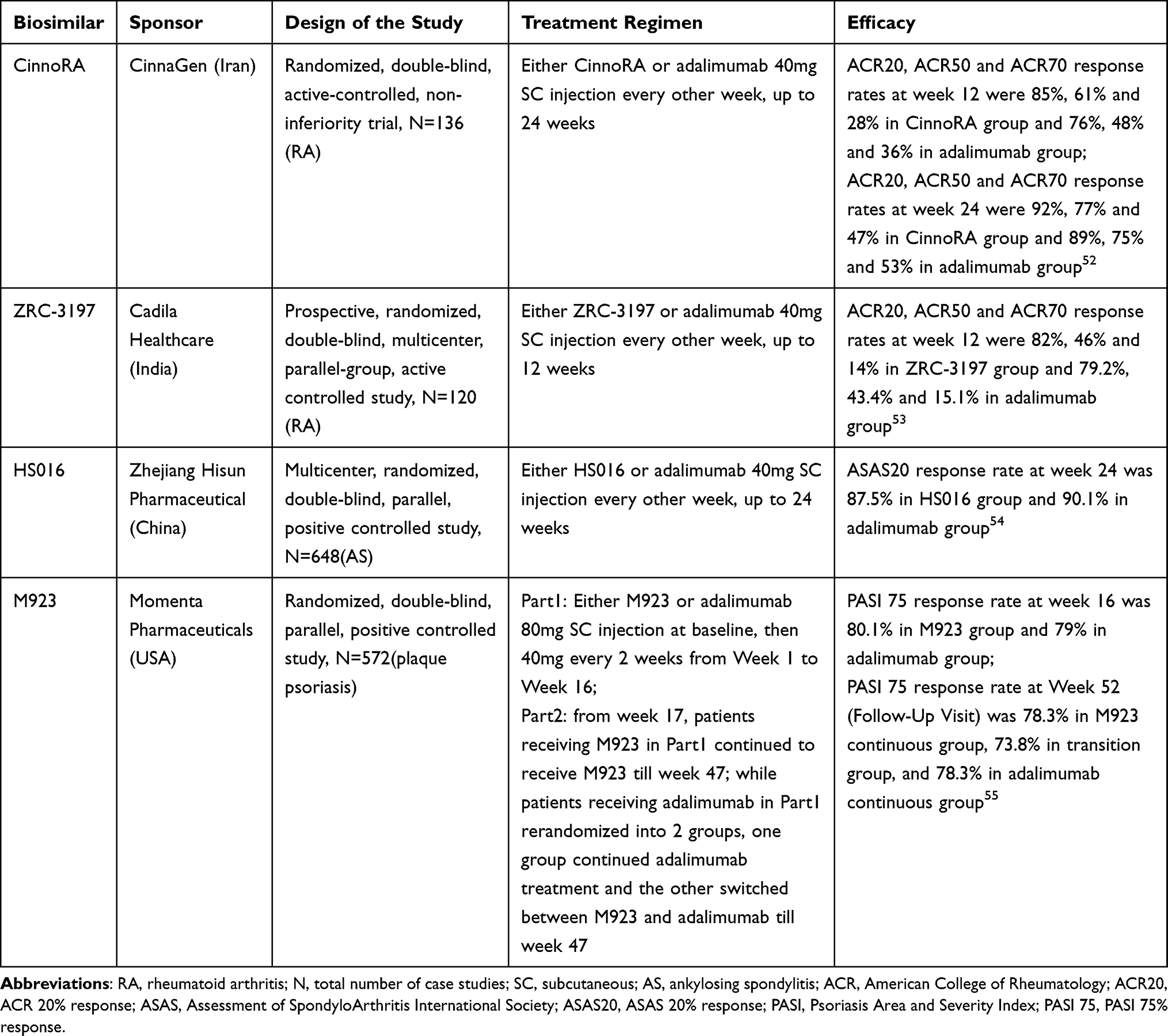 |
Table 2 Efficacy of Adalimumab Biosimilars in Some Phase III Clinical Trials |
Summary
The above clinical trials showed comparable PK, immunogenicity, efficacy and safety profiles between adalimumab biosimilars and their RP in psoriasis or other rheumatic diseases, which means that these biosimilars are very similar to the reference adalimumab and are promising alternatives. In some phase III clinical trials, there were multiple switches of drugs, the results of efficacy were similar among groups and the safety outcomes did not raise any concern, which allows doctors and patients to choose to use biosimilars with confidence in the real world.
Extrapolation of Indications
Provided that biosimilars are equivalent in structure and clinical performance to originators that have already been approved by regulatory agencies, the approval process for these agents is abbreviated compared to that for novel drugs. Extrapolation, as stated by the EMA, is “a well-established scientific principle which has been used for many years”.2 If a biosimilar is highly similar to an RP and has comparable PK, PD, immunogenicity, safety and efficacy in one indication, safety and efficacy data could be extrapolated to other indications approved for the RP, which means that clinical trials assessing equivalence can be carried out in a single disease, without trials in other relevant indications, the EMA would approve the product for all indications licensed for the RP.2 Based on this principle, adalimumab biosimilars were licensed for all indications approved for reference adalimumab although some of their equivalence studies were only conducted in rheumatic diseases.
This protocol may raise debate among dermatologists and affect their use of biosimilars in clinical practice. In a survey (2015) of US specialty physicians who already prescribe biologics, including dermatologists, gastroenterologists, medical oncologists, hematologist-oncologists, rheumatologists, and nephrologists, only 12% of respondents had positive perceptions of the concept of extrapolation and would use a biosimilar for all approved indications.39 In another survey conducted in the US from 2016 to 2017, rheumatologists, dermatologists and gastroenterologists were included. The majority of physicians (70%) knew that if a biosimilar showed that it treats one disease similarly to an originator biologic, the biosimilar may be extrapolated by regulators to treat all the diseases the originator biologic is approved to treat. However, half of the physicians were uncomfortable with biosimilars receiving approval by extrapolation.40
Interchangeability with Reference Adalimumab
Interchangeability is defined by the EMA as “replacing an RP with a biosimilar (or vice versa) or replacing one biosimilar with another”, which includes switching and substitution. Switching means the health provider decides to exchange one medicine for another, while substitution refers to replacement at the pharmacy level without intervention of the prescriber.2 If switching from an RP to a respective biosimilar (or switching conversely) or between biosimilars only means saving money, this process is also mentioned as nonmedical switching.41
The responsibilities of the regulation of switching and substitution practices and the designation of interchangeability fall within different EU member states. Healthcare providers should choose carefully before prescribing biosimilars to their patients, taking into account the legal framework, regulations, guidelines and advice in their areas of specialty and the patient’s perspective.2 In a 2017 consensus document of the joint task force comprising 25 experts from 8 European countries, the USA and Japan, in recommendation 6, it was considered effective and safe to switch from the originator to the respective biosimilar, but the patient’s perspective should be taken into consideration (evidence level 1b and degree of recommendation A).42 To date, national regulatory agencies and authorities have provided multiple guidelines about the practice of use and switching of biosimilars in real life. The Portuguese Society of Rheumatology states that switching from an RP to a more affordable biosimilar is desirable if it is done under a standardized protocol, the prescriber is involved, the patient is well informed and followed-up and the products’ traceability is guaranteed.43 The British Society for Rheumatology (BSR) states in a stricter way that switching from an RP to a biosimilar should be determined on a case-by-case basis until there are more data to support a safe transition. The statement of BSR says that strong monitoring is required in switching, decisions should be made jointly by clinicians and patients, and medications are traceable.44 Moreover, automatic substitution without information and consent of the prescriber is unacceptable.45
In the US, a designation pathway has been created to evaluate whether a biological product is interchangeable with an RP. According to the Biologics Price Competition and Innovation Act (BPCI Act), an interchangeable product must meet additional standards to demonstrate its interchangeability.45 It is expected to be shown that a proposed interchangeable product can produce the same clinical outcomes as RP in any indications. Switching studies should be carried out, and post-marketing surveillance (real-world observation) data are needed to support interchangeability.45 According to the Public Health Service Act (PHS Act) 351(k)(4), a prescribed biological RP may be substituted by an interchangeable biosimilar at the pharmacy level without consulting the health care provider. To date, the FDA has not considered any biosimilar as interchangeable.45
Although several phase III RCTs or open label extension studies on switching from reference adalimumab to the respective biosimilars or multiple switching between groups showed comparable safety, efficacy and immunogenicity,15,20,21,26 evidence on the nonmedical switching from reference adalimumab to the respective biosimilars in the real-world setting is limited and is mostly based on a few small-scale observational studies with a limited number of subjects, thus seemingly insufficient to recommend this switching procedure. Safety data from real-world practice are available only for SB5. Di Cesare et al46 reported a small cohort study of real-life PsO/PsA patients switching from reference adalimumab to SB5. Of 20 switched patients, two experienced loss of efficacy on cutaneous symptoms, with one case leading to SB5 discontinuation and switching to an IL-12/23 antagonist and the other case developing pustular psoriasis. No changes in PASI scores were noticed in 90% of patients who were shifted from adalimumab to SB5. In PsA patients, 9 out of 12 patients maintained PsA symptom remission, while the other 3 patients experienced axial disease flare as soon as 4 weeks, leading to an alternation of concomitant medication in 2 cases and a back-switch to adalimumab originator in the third. Bruni et al47 performed a retrospective real-world cohort of rheumatic joint diseases. Eighty-two patients with RA, PsA, juvenile idiopathic arthritis (JIA) and axial spondyloarthritis (axSpA) treated with adalimumab for at least 6 months and in stable condition were enrolled and switched from reference adalimumab to SB5. RA patients experienced stable disease condition, while PsA patients and axSpA patients showed mild disease flares at 3 months. There was minor adjustment in the concomitant medications, with values of all disease activity and disability measures greatly decreasing and being similar from baseline at 6 months. A total of 33.7% and 16.6% of patients reported AEs at 3 months and 6 months, respectively, mostly disease relapse and mild infections. Two patients discontinued SB5, and one of them back-switched to reference adalimumab. The authors concluded that their real-life data confirmed the safety of switching from reference adalimumab to SB5 in RA and the possibility of applying this procedure in axSpA and PsA patients, further supporting that switching to biosimilars is desirable in the treatment of inflammatory rheumatologic diseases. Another real-life practice48 with data collected from the DERMBIO registry in Denmark enrolled 43 psoriasis patients who were switched under surveillance from reference adalimumab to GP2017 and revealed that although there was no significantly notable impact on PASI scores and DLQI after switching, 39.5% of patients had increased AE rates, which were mainly pruritus, flares and headache.
Conclusion
According to the principle of extrapolation, data from certain indication studies may be extrapolated to other indications that have already been approved for reference drugs. Eight adalimumab biosimilars showing PK, safety, efficacy and immunogenicity profiles comparable to those of adalimumab originators have been approved by the FDA, the EMA or health regulatory agencies worldwide. Therefore, biosimilars seem to be favorable alternatives to originator biologics, and on a wider scale, have the potential to lower the disease costs and increase the accessibility of biologic therapies. However, scientists have pointed out some concerns on issues with respect to extrapolation of indications and interchangeability. For the former issue, the EMA has defined it as a “well-established scientific principle”. Biosimilar manufacturers should provide bioequivalence data, including the mechanisms of action, PK, safety, efficacy, and immunogenicity, to support extrapolation. The FDA, through the BPCI Act, has created an abbreviated licensure pathway to provide the public with easier access to more economic biological products. The latter issue does not seem to be supported by adequate scientific evidence until now. Although there were several randomized controlled switching studies and some small-scale real-world transition studies of reference adalimumab and the respective biosimilars showing comparable safety and efficacy, and the consensus-based recommendations suggesting that a single switch from an originator to its biosimilar is safe and effective with the patient’s perspective considered, but the transposition of these results to real-life practice still seems debatable. It is well known that patients treated in clinical trials differ greatly from those in real-world practice. In a clinical trial, participants are strictly selected with limited concomitant diseases, as an analysis from the German biologics register RABBIT (Rheumatoid Arthritis: Observation of Biologic Therapy) showed that merely 21–33% of the registered patients would have been qualified for RCTs.49 In addition, data from post-marketing registries showed that patients switching from originators to their biosimilars have a slightly higher discontinuation rate than historical data show in the real-world setting.50,51 Thus, further data from controlled switching studies are needed to better address the efficacy and safety of the transition strategy. Although there are some remaining concerns, experts of the International Psoriasis Council advocate that dermatologists take an active role in the development of biosimilar prescribing policies to improve access to biosimilar drugs for psoriasis patients.1
A limitation of this review is that only a small number of clinical trials were conducted specifically to evaluate biosimilars in psoriasis populations and that only a few real-world observational studies on switching from reference adalimumab to the respective biosimilars have been published to date. More well-designed prospective studies and real-life observational cohorts in patients with psoriasis are needed to support the equivalence between biosimilars and their originators and the transition between the two in achieving desirable clinical results in this patient population. Clear guidance from medical regulatory authorities and data from post-marketing monitoring registries will also be helpful for the confidence of switching for both physicians and patients. Finally, the biosimilar drugs currently on the market are all TNF-α inhibitors. There are some ongoing clinical trials of ustekinumab biosimilars. After ustekinumab biosimilar, which is an IL-12/23p40 inhibitor, enters the market, doctors and patients will have more choices when a patient loses efficacy on a biologic agent and intends to switch to a biosimilar that does not target TNF-α. At the same time, monitoring of switching from one biosimilar to another (or from an RP to a biosimilar) targeting different inflammatory factors and post-marketing clinical trials are needed to ensure this practice.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2018YFC1705305); Clinical discipline innovation project (2019YXK028).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Cohen AD, Wu JJ, Puig L, et al. Biosimilars for psoriasis: worldwide overview of regulatory guidelines, uptake and implications for dermatology clinical practice. Br J Dermatol. 2017;177(6):1495–1502. doi:10.1111/bjd.15756
2. The European Medicines Agency, The European Commission. Biosimilars in the EU - Information guide for healthcare professionals. Prepared jointly by the European Medicines Agency and the European Commission. ©European Med Agency; 2019. Available from: https://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_en.pdf.
3. U.S. Food & Drug Administration. Biosimilar and Interchangeable Products. Available from: https://www.fda.gov/drugs/biosimilars/biosimilar-and-interchangeable-products#biosimilar.
4. Ruff L, Rezk M, Uhlig T, Gommers J. Budget impact analysis of an etanercept biosimilar for the treatment of rheumatoid arthritis In Europe. Value Heal. 2015;18(7):A639. doi:10.1016/j.jval.2015.09.2276
5. Gisondi P, Geat D, Pizzolato M, Girolomoni G. State of the art and pharmacological pipeline of biologics for chronic plaque psoriasis. Curr Opin Pharmacol. 2019;46:90–99. doi:10.1016/j.coph.2019.05.007
6. Menter A, Tyring SK, Gordon K, et al. Adalimumab therapy for moderate to severe psoriasis: a randomized, controlled phase III trial. J Am Acad Dermatol. 2008;58(1):106–115. doi:10.1016/j.jaad.2007.09.010
7. Saurat J-H, Stingl G, Dubertret L, et al. Efficacy and safety results from the randomized controlled comparative study of adalimumab vs. methotrexate vs. placebo in patients with psoriasis (CHAMPION). Br J Dermatol. 2008;158(3):558–566. doi:10.1111/j.1365-2133.2007.08315.x
8. Norman P. Humira: the impending patent battles over adalimumab biosimilars. Pharm Pat Anal. 2016;5(3):141–145. doi:10.4155/ppa-2016-0002
9. Puig L, López-Ferrer A. Biosimilars for the treatment of psoriasis. Expert Opin Biol Ther. 2019;19(10):993–1000. doi:10.1080/14712598.2019.1636963
10. Reynolds KA, Pithadia DJ, Lee EB, Liao W, Wu JJ. Safety and effectiveness of anti-tumor necrosis factor-alpha biosimilar agents in the treatment of psoriasis. Am J Clin Dermatol. 2020;0123456789. doi:10.1007/s40257-020-00507-1
11. Generics and Biosimilars Initiative (GaBI). Biosimilars approved in Europe. Available from: https://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-Europe.
12. Olteanu R, Zota A, Constantin M. Biosimilars: an update on clinical trials (review of published and ongoing studies). Acta Dermatovenerologica Croat. 2017;25(1):57–66.
13. Generics and Biosimilars Initiative (GaBI). Biosimilars approved in the US. Available from: https://www.gabionline.net/Biosimilars/General/Biosimilars-approved-in-the-US.
14. Kaur P, Chow V, Zhang N, Moxness M, Kaliyaperumal A, Markus R. A randomised, single-blind, single-dose, three-arm, parallel-group study in healthy subjects to demonstrate pharmacokinetic equivalence of ABP 501 and adalimumab. Ann Rheum Dis. 2017;76(3):526–533. doi:10.1136/annrheumdis-2015-208914
15. Papp K, Bachelez H, Costanzo A, et al. Clinical similarity of biosimilar ABP 501 to adalimumab in the treatment of patients with moderate to severe plaque psoriasis: a randomized, double-blind, multicenter, phase III study. J Am Acad Dermatol. 2017;76(6):1093–1102. doi:10.1016/j.jaad.2016.12.014
16. Cohen S, Genovese MC, Choy E, et al. Efficacy and safety of the biosimilar ABP 501 compared with adalimumab in patients with moderate to severe rheumatoid arthritis: a randomised, double-blind, phase III equivalence study. Ann Rheum Dis. 2017;76(10):1679–1687. doi:10.1136/annrheumdis-2016-210459
17. Cohen S, Pablos JL, Pavelka K, et al. An open-label extension study to demonstrate long-term safety and efficacy of ABP 501 in patients with rheumatoid arthritis. Arthritis Res Ther. 2019;21(1):1–10. doi:10.1186/s13075-019-1857-3
18. Wathion N Withdrawal of the Marketing Authorisation in the European Union; 2019. Available from: https://www.ema.europa.eu/en/documents/public-statement/public-statement-cyltezo-withdrawal-marketing-authorisation-european-union_en.pdf.
19. Wynne C, Altendorfer M, Sonderegger I, et al. Bioequivalence, safety and immunogenicity of BI 695501, an adalimumab biosimilar candidate, compared with the reference biologic in a randomized, double-blind, active comparator phase I clinical study (VOLTAIRE®-PK) in healthy subjects. Expert Opin Investig Drugs. 2016;25(12):1361–1370. doi:10.1080/13543784.2016.1255724
20. Cohen SB, Alonso-Ruiz A, Klimiuk PA, et al. Similar efficacy, safety and immunogenicity of adalimumab biosimilar BI 695501 and Humira reference product in patients with moderately to severely active rheumatoid arthritis: results from the phase III randomised VOLTAIRE-RA equivalence study. Ann Rheum Dis. 2018;77(6):914–921. doi:10.1136/annrheumdis-2017-212245
21. Cohen SB, Czeloth N, Lee E, Klimiuk PA, Peter N, Jayadeva G. Long-term safety, efficacy, and immunogenicity of adalimumab biosimilar BI 695501 and adalimumab reference product in patients with moderately-to-severely active rheumatoid arthritis: results from a Phase 3b extension study (VOLTAIRE-RAext). Expert Opin Biol Ther. 2019;19(10):1097–1105. doi:10.1080/14712598.2019.1645114
22. Efficacy, Safety and Immunogenicity of BI 695501 Versus Humira® in Patients With Moderate to Severe Chronic Plaque Psoriasis(identification no: NCT02850965); 2019. Available from: https://clinicaltrials.gov/ct2/show/NCT02850965.
23. The VOLTAIRE-X Trial Looks at the Effect of Switching Between Humira® and BI 695501 in Patients With Plaque Psoriasis(identification no: NCT03210259). Available from: https://clinicaltrials.gov/ct2/show/NCT03210259?term=BI695501&draw=2&rank=9.
24. Shin D, Lee Y, Kim H, Körnicke T, Fuhr R. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther. 2017;42(6):672–678. doi:10.1111/jcpt.12583
25. Weinblatt ME, Baranauskaite A, Niebrzydowski J, et al. Phase 3 randomized study of SB5, an Adalimumab biosimilar, versus reference adalimumab in patients with moderate to severe rheumatoid arthritis. Arthritis Rheumatol. 2017;38(1):42–49. doi:10.1111/ijlh.12426
26. Weinblatt ME, Baranauskaite A, Dokoupilova E, et al. Switching from reference Adalimumab to SB5 (adalimumab biosimilar) in patients with rheumatoid arthritis: fifty-two–week phase III randomized study results. Arthritis Rheumatol. 2018;70(6):832–840. doi:10.1002/art.40444
27. von Richter O, Lemke L, Haliduola H, et al. GP2017, an adalimumab biosimilar: pharmacokinetic similarity to its reference medicine and pharmacokinetics comparison of different administration methods. Expert Opin Biol Ther. 2019;19(10):1075–1083. doi:10.1080/14712598.2019.1571580
28. Blauvelt A, Lacour JP, Fowler JF, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179(3):623–631. doi:10.1111/bjd.16890
29. Clinical trial to compare treatment with GP2017 and Humira® in patients with rheumatoid arthritis (ADMYRA). (Identification no: NCT02744755). Available from: https://clinicaltrials.gov/ct2/show/NCT02744755?cond=gp2017&draw=2&rank=3.
30. Hercogová J, Papp KA, Chyrok V, Ullmann M, Vlachos P, Edwards CJ. AURIEL-PsO: a randomized, double-blind phase III equivalence trial to demonstrate the clinical similarity of the proposed biosimilar MSB11022 to reference adalimumab in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol. 2020;182(2):316–326. doi:10.1111/bjd.18220
31. Edwards CJ, Monnet J, Ullmann M, Vlachos P, Chyrok V, Ghori V. Safety of adalimumab biosimilar MSB11022 (acetate-buffered formulation) in patients with moderately-to-severely active rheumatoid arthritis. Clin Rheumatol. 2019;38(12):3381–3390. doi:10.1007/s10067-019-04679-y
32. Puri A, Niewiarowski A, Arai Y, et al. Pharmacokinetics, safety, tolerability and immunogenicity of FKB327, a new biosimilar medicine of adalimumab/Humira, in healthy subjects. Br J Clin Pharmacol. 2017;83(7):1405–1415. doi:10.1111/bcp.13245
33. Genovese MC, Glover J, Greenwald M, et al. FKB327, an adalimumab biosimilar, versus the reference product: results of a randomized, Phase III, double-blind study, and its open-label extension. Arthritis Res Ther. 2019;21(1):1–12. doi:10.1186/s13075-019-2046-0
34. Genovese MC, Kellner H, Arai Y, Muniz R, Alten R. Long-term safety, immunogenicity and efficacy comparing FKB327 with the adalimumab reference product in patients with active rheumatoid arthritis: data from randomised double-blind and open-label extension studies. RMD Open. 2020;6(1):e000987. doi:10.1136/rmdopen-2019-000987
35. Lee A, Shirley M. PF-06410293: an adalimumab biosimilar. BioDrugs. 2020;34(5):695–698. doi:10.1007/s40259-020-00445-8
36. Fleischmann RM, Alten R, Pileckyte M, et al. A comparative clinical study of PF-06410293, a candidate adalimumab biosimilar, and adalimumab reference product (Humira®) in the treatment of active rheumatoid arthritis. Arthritis Res Ther. 2018;20(1):1–12. doi:10.1186/s13075-018-1676-y
37. Yu K-S, Jang I-J, Lim H-S, et al. Pharmacokinetic equivalence of CT-P17 to high-concentration (100 mg/mL) reference adalimumab: a randomized phase I study in healthy subjects. Clin Transl Sci. 2021. doi:10.1111/cts.12967
38. Kay J, Jaworski J, Wojciechowski R, et al. Efficacy and safety of biosimilar CT-P17 versus reference adalimumab in subjects with rheumatoid arthritis: 24-week results from a randomized study. Arthritis Res Ther. 2021;23(1):1–12. doi:10.1186/s13075-020-02394-7
39. Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2017;33(12):2160–2172. doi:10.1007/s12325-016-0431-5
40. Teeple A, Ellis LA, Huff L, et al. Physician attitudes about non-medical switching to biosimilars: results from an online physician survey in the United States. Curr Med Res Opin. 2019;35(4):611–617. doi:10.1080/03007995.2019.1571296
41. Barbier L, Ebbers HC, Declerck P, Simoens S, Vulto AG, Huys I. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: a systematic review. Clin Pharmacol Ther. 2020;108:734–755. doi:10.1002/cpt.1836
42. Kay J, Schoels MM, Dörner T, et al. Consensus-based recommendations for the use of biosimilars to treat rheumatological diseases. Ann Rheum Dis. 2018;77(2):165–174. doi:10.1136/annrheumdis-2017-211937
43. Araújo FC, Sepriano A, Teixeira F, et al. The Portuguese Society of Rheumatology position paper on the use of biosimilars – 2017 update. Acta reumatologica portuguesa. 2017;42(3):219–228.
44. Bitish Society for Reumatology. January 2017 Position statement: biosimilar medicines. Available from: https://www.rheumatology.org.uk/Portals/0/Documents/Policy/Position%20statements/Biosimilars.pdf?ver=2019-02-27-170506-670.
45. U.S. Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Considerations in demonstrating interchangeability with a reference product-guidance for industry. Fda. 2019;(May):23. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-product-guidance-industry.
46. Di Cesare A, Tronconi G, Fastame TM, et al. SB5 adalimumab biosimilar in the treatment of psoriasis and psoriatic arthritis. Dermatol Ther. 2020;33(3):26–27. doi:10.1111/dth.13435
47. Bruni C, Bitti R, Nacci F, et al. Efficacy and safety of switching from reference adalimumab to SB5 in a real-life cohort of inflammatory rheumatic joint diseases. Clin Rheumatol. 2021;40(1):85–91. doi:10.1007/s10067-020-05199-w
48. Nielsen VW, Lund TT, Gniadecki R, et al. Effectiveness and safety of switching from originator to biosimilar adalimumab in patients with psoriasis. Dermatol Ther. 2020;33(6):1–2. doi:10.1111/dth.14258
49. Zink A, Strangfeld A, Schneider M, et al. Effectiveness of tumor necrosis factor inhibitors in rheumatoid arthritis in an observational cohort study: comparison of patients according to their eligibility for major randomized clinical trials. Arthritis Rheum. 2006;54(11):3399–3407. doi:10.1002/art.22193
50. Cantini F, Benucci M. Switching from the bio-originators to biosimilar: is it premature to recommend this procedure? Ann Rheum Dis. 2019;78(4):e23. doi:10.1136/annrheumdis-2017-212820
51. Fleischmann R. Therapy: biosimilars in rheumatology - why, how and when in 2017. Nat Rev Rheumatol. 2017;13(12):701–703. doi:10.1038/nrrheum.2017.179
52. Jamshidi A, Gharibdoost F, Vojdanian M, et al. A phase III, randomized, two-armed, double-blind, parallel, active controlled, and non-inferiority clinical trial to compare efficacy and safety of biosimilar adalimumab (CinnoRA®) to the reference product (Humira®) in patients with active rheumatoid arth. Arthritis Res Ther. 2017;19(1):1–9. doi:10.1186/s13075-017-1371-4
53. Jani RH, Gupta R, Bhatia G, et al. A prospective, randomized, double-blind, multicentre, parallel-group, active controlled study to compare efficacy and safety of biosimilar adalimumab (Exemptia; ZRC-3197) and adalimumab (Humira) in patients with rheumatoid arthritis. Int J Rheum Dis. 2016;19(11):1157–1168. doi:10.1111/1756-185X.12711
54. Su J, Li M, He L, et al. Comparison of the efficacy and safety of adalimumab (humira) and the adalimumab biosimilar candidate (HS016) in Chinese patients with active ankylosing spondylitis: a multicenter, randomized, double-blind, parallel, Phase III Clinical Trial. BioDrugs. 2020;34(3):381–393. doi:10.1007/s40259-020-00408-z
55. Phase 3 Study of M923 and Humira® in Subjects With Chronic Plaque-type Psoriasis. Available from: https://clinicaltrials.gov/ct2/show/results/NCT02581345?term=Adalimumab+Biosimilar&draw=4&rank=3&view=results.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.