")
Back to Archived Journals » Research and Reports in Biology » Volume 6
An update on the mechanisms and pathophysiological consequences of genomic instability with a focus on ionizing radiation
Authors Streffer C
Received 10 July 2015
Accepted for publication 8 September 2015
Published 1 December 2015 Volume 2015:6 Pages 225—233
DOI https://doi.org/10.2147/RRB.S72016
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Zvi Kelman
Christian Streffer
Institute for Medical Radiobiology, University Clinics Essen, Essen, Germany
Abstract: The genome of eukaryotic cells is generally instable. DNA damage occurs by endogenous processes and exogenous toxic agents. The efficient DNA repair pathways conserve the genetic information to a large extent throughout the life. However, exposure to genotoxic agents can increase the genomic instability. This phenomenon develops in a delayed manner after approximately 20 and more cell generations. It is comparatively thoroughly investigated after the exposure to ionizing radiation. The increase of genomic instability has been observed after exposures to ionizing radiation in vitro and in vivo as well as with many different types of radiation. The effect is induced over a wide dose range, and it has been found with cell death, chromosomal damage, cell transformations, mutations, double-strand breaks, malformations, and cancers. No specific chromosomes or genomic sites have been observed for such events. The increased genomic instability can be transmitted to the next generation. Possible mechanisms such as oxidative stress (mitochondria may be involved), reduced DNA repair, changes in telomeres, epigenetic effects are discussed. A second wave of oxidative stress has been observed after radiation exposures with considerably high doses as well as with cytotoxic agents at time periods when an increased genomic instability was seen. However, the increase of genomic instability also happens to much lower radiation doses. Hypoxia induces an increase of genomic instability. This effect is apparently connected with a reduction of DNA repair. Changes of telomeres appear as the most probable mechanisms for the increase of genomic instability. Syndromes have been described with a genetic predisposition for high radiosensitivity. These individuals show an increase of cancer, a deficient DNA repair, a disturbed regulation of the cell cycle, and an increased genomic instability. Several studies with cancer patients show an increased genomic instability in the blood lymphocytes before any treatment has been performed. It is generally accepted that the increased genomic instability is a promotor for carcinogenesis.
Keywords: cancer, DNA repair, genomic instability, hypoxia, ionizing radiation
Introduction
It was generally accepted after the publication of the double helical structure of DNA by Watson and Crick1 in 1953 that DNA is a relatively stable molecule. A structural change would unavoidably lead to a mutated cell or to cell death. Only during the following decades, it was realized that the large DNA molecules are rather unstable. However, it was soon observed that the breaks of the polynucleotide chains, loss of DNA bases and failures during the processes of DNA replication steadily occur to a high degree by intracellular, metabolic events, eg, oxidative processes in vivo, and the continuity of the genomic information is only established by very efficient DNA repair pathways.2 However, the DNA repair is frequently not complete and not always perfect; misrepair can occur. Therefore, cells with mutations and chromosome breaks including translocations appear, which are usually observed during the metaphase of mitotic cell division.3–5 These processes are seen as one of the dominating principles of evolution.6 These observations show that there is always some genomic instability developing in living cells. This article reviews and especially updates on which cellular processes and mechanisms the genomic instability is depending, which exogenous processes and mechanisms influence and especially increase genomic instability and what is the impact of genomic instability on certain pathophysiological events.
Increase of genomic instability by ionizing radiation
In 1922, Caspari described that the “Zustand” (condition) of the chromatin is decisive for the cellular radiosensitivity,7 and in 1943, Langendorff described the induction of chromosomal breaks, which were observed during mitotic cell division in plants and frogs after exposure to ionizing radiation.7,8 Interestingly, Langendorff also described in 1943 the “Heilung” (healing) of chromosomal damage, although the first molecular pathways of DNA repair were only discovered in the end of the 1950s and in the 1960s of the last century. Many publications9 followed where the dose-response of chromosomal aberrations after radiation exposures with different radiation qualities (different energies and different types of radiation) have been described. Therefore, the measurement of chromosomal aberrations has often been proposed as a technique for biodosimetry and recently strong efforts have been undertaken in this direction.9 It is generally accepted and strongly proposed that it is more appropriate to study the aberration rate in the first mitosis after radiation exposure, and it was assumed for a long time that cells have to be hit by an ionizing particle to express radiation-induced chromosomal aberrations (targeted effect).10 However, in 1989, it was reported that an increase of chromosomal aberrations occurred in cells, which were not exposed by radiation but their ancestors had been exposed many cell generations earlier, and this effect was described as an “increase of instability of the genome”.11
In these experiments, zygotes of mice and omnipotent stem cells were X-irradiated. A skin biopsy was taken from the newborn mice or from the mouse fetuses just before the birth, and the epithelial cells were cultured in vitro for 48 hours. After addition of colcemid, the metaphases, which develop during the mitotic cell division, were analyzed for chromosome aberrations. A significant increase of aberrations was observed in those epithelial cells that originated from the newborn mice with radiation exposure in the zygote stage in comparison with the epithelial cells from newborn mice, which had developed from zygotes without X-ray exposure. Thus, the affecting radiation exposure had occurred in many cell generations before the cytogenetic analysis. The increase of genomic instability expressed as chromosomal aberrations has developed during the course of the prenatal development. Therefore, it is expressed in many cell generations after the radiation exposure. In contrast to the former assumption that a radiation effect with changes of the chromatin structure does only occur in those cells, which have been directly hit by an ionizing particle, and these additional chromosomal changes have developed although the affected cell nuclei have not been exposed at all. These radiation effects are classified as “nontargeted effects”.10 These radiation effects are “delayed” in contrast to the direct, targeted effects, which are manifested shortly after radiation exposure.
Many experimental studies on genomic instability have been performed in the last century and in the following years. These data have extensively been reviewed by the “United Nations Scientific Committee on the Effects of Atomic Radiation” (UNSCEAR) in 2006.10 “Radiation-induced instability is observed in cells at delayed times after irradiation and manifests in the progeny of exposed cells of multiple generations after the initial insult (Figure 1). Instability is measured as chromosomal alterations, changes in ploidy, micronuclei formation, gene mutations and amplifications, mini- and microsatellite (short tandem repeat) instabilities, and/or decreased plating efficiency”.10
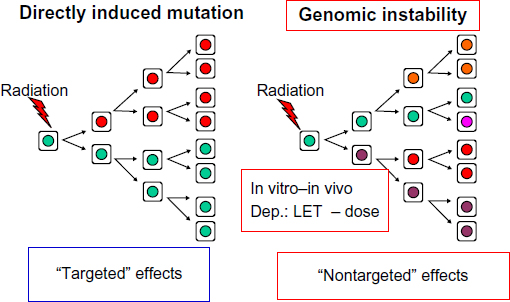 | Figure 1 Radiation-induced genomic instability as a new mechanism of mutagenesis. |
The increase of genomic instability has been observed in a wide range of cells and organisms, in vivo and in vitro with very different radiation qualities (different types and energies of ionizing radiation).12 After the above described experiments with radiation exposure to the zygotes of mice, Kadhim et al13 observed abnormalities of the karyotype in 40%–60% of hematopoietic stem cells of mice exposed to doses of alpha particles that would lead in the average to approximately one hit per cell. Sabatier et al14 found chromosomal instability in >80% of metaphase cells from human dermal fibroblasts irradiated with heavy ions (neon, argon, and lead). A wide range of high linear energy transfer (LET) radiations (386–13,600 keV/μ) was used in these experiments. This instability had developed after more than 20 passages of the cell culture. Similar to the studies with the aforementioned exposure of the zygotes to X-rays, Limoli et al15 observed that chromosomal instability was induced with X-rays (low LET radiation). In that way, approximately 3% of surviving hamster–human hybrid GM10115 clones showed an increase of genomic instability after exposure to 1 Gy X-rays.
Thus, the induction of the increased genomic instability by ionizing radiation is apparently a general phenomenon in living cells and tissues, which develops more than 20 cell generations after exposure to all qualities of ionizing radiation. Similar data have been reported with other genotoxic and cytotoxic agents. Very variable dose-response was found in different experiments.10 In some cases, the radiation effect significantly increased with the radiation dose; in other experiments, this did not happen. A significant linear dose-response was described after radiation exposure of mouse zygotes for the increase of genomic instability in skin epithelial cells from neonatal mice in the dose range of 0.5–2.0 Gy X-rays. This effect persisted over many further cell generations.16 Ullrich and Davis17 observed a significant dose response in the dose range of 0.1–1.0 Gy for delayed chromosomal aberrations in epithelial cells of the mammary gland after irradiation of BALB/c mice. The authors removed mammary gland tissue at different times after radiation exposure, cultured the tissue in vitro and then performed a cytogenetic analysis. The dose-response reached a plateau at higher radiation doses. It is interesting that the same specific differences could be observed for the mouse strain susceptibility with respect to the induction of mammary carcinomas as well as for the ranking for the increase of genomic instability.10
Very impressive experiments have been performed by Okada et al.18 This group studied the cell growth (increase of cell number) and the appearance of double-strand breaks (DSBs) in DNA (measured by immunofluorescence foci19 as a marker for DSB) after exposures to X-rays and to carbon ions with low radiation doses down to 1 mGy. Genomic instability significantly increased for both cell growth and foci for DSBs, after the 25th passage and for both radiation qualities. However, the effect was stronger after exposure to carbon ions than to X-rays when identical absorbed radiation doses (mGy) were compared. In case of the carbon ions such a radiation dose means, that one out of 18 cells is hit by an ionizing particle. These data show that low-absorbed radiation doses induce an increase of genomic instability in principle, but considering the local distribution of the radiation energy it means that the locally absorbed energy density is considerably higher with the carbon ions than with X-rays in the hit cell nucleus. Generally, it appears that a significant increase of genomic instability has only been observed after radiation doses higher than 100 mGy when low LET radiations (X-rays, photons, gamma- or beta-radiations) are used. However, under specific conditions, it is apparently possible that lower radiation doses are effective.18
With the studies of the cytogenetic effects after radiation exposures to the zygotes, it was also possible to investigate the quality of the damaged chromosomal structure. The analysis of the chromosome and the chromatid breaks showed after exposure to X-rays a higher number of chromosome breaks than of chromatid breaks in the metaphase of the first mitotic cell division (targeted, direct effect), while chromatid breaks were dominating in the metaphases of those mitotic cell divisions without radiation exposure.16,20 However, more chromatid breaks than chromosome breaks were also observed in the metaphases of the epithelial cells from the newborn mice independent whether the zygotes received an X-exposure (delayed, nontargeted effects) or not.11,16 Thus, these data show a difference in the quality of the chromosomal aberrations when the metaphases were analyzed directly or delayed (at the time of increased genomic instability) after the radiation exposure (Table 1). These findings are in agreement with observations of Little et al21 who studied the molecular structure of mutations in the Hprt locus of Chinese hamster ovary cells directly and delayed (delayed for 23 cell generations) after exposure to ionizing radiation (Table 2). It was concluded that as “thus the mutational spectrum for delayed mutations is very different from that of direct X-ray-induced mutations. It is, however, similar to the spectrum for spontaneously arising mutations.”21
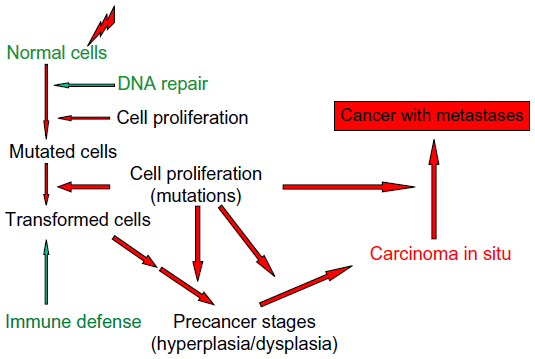 | Figure 2 Schematic diagram of the steps for the development of a malignant cancer after radiation exposure. |
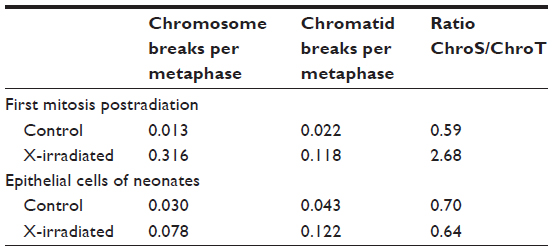 | Table 1 Chromosomal aberrations without X-ray exposure and after radiation exposure with 2 Gy X-rays of zygotes of HLG-mice |
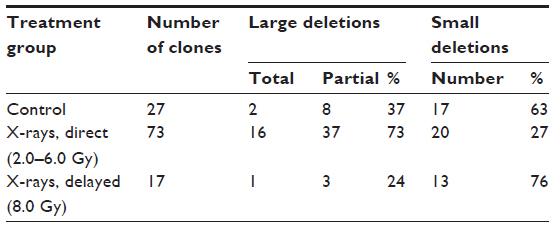 | Table 2 Molecular structure of direct and delayed Hprt mutations in Chinese ovary hamster cells after X-ray exposure (for direct effects: 4.0 Gy; for delayed effects: 8.0 Gy) |
Several studies have shown that the increased genomic instability can be transmitted to the next generation of mice or humans.10 Such effects have been observed with the induction of cancer, malformations, and other hereditary effects. Very interesting is the transmittance of mutations in mini-satellites of DNA and “expanded simple tandem repeats”. Dubrova et al22 observed the mutations of expanded simple tandem repeats in different mouse strains and determined a radiation doubling dose of approximately 0.5 Gy for such effects. Similar data were found with human studies after the Chernobyl accident.23
Some interesting data have been obtained after radiation exposures during pregnancy. Ohtaki et al24 observed no increase of chromosome aberrations in the hematopoietic cells of such individuals who were exposed to ionizing radiation during their prenatal development by the atomic bomb explosions in Hiroshima and Nagasaki, although their mothers showed an increase of aberrations in the lymphocytes. It was concluded that aberrant cells could be eliminated during the prenatal development. However, Nakano et al25 found that after prenatal exposure of rats, no increase of chromosomal aberrations was observed again in lymphatic (spleen) cells but an increase of chromosomal aberrations was found in epithelial cells of the mammary tissue in these prenatally exposed animals. Thus, there are differences between the tissues or organ systems during the prenatal development with respect to the development of an increased genomic instability. Uma Devi et al26 found an increased number of leukocytes with an increase of polyploid cells in 12-month old mice, which were irradiated in utero at the fetal stage. These animals developed leukemia later.
Cytogenetic studies with leukemia patients of the Japanese A-bomb survivors showed a higher rate of chromosomal aberrations after radiation doses >2 Gy than patients who were unexposed or exposed to lower radiation doses.27 Further studies of the strongly exposed A-bomb survivors with acute myeloid leukemia or myelodysplastic syndrome indicated chromosomal instability.28 Thus, it was concluded that the instability might influence the development of leukemia in humans exposed to ionizing radiation. Such an association will be discussed in detail later.
Genomic instability and genetic disposition
From clinical experience, it is very well-known that individual radiosensitivity can be very different. There are strong indications that the genetic predisposition is very important in this context. Thus, it has been observed by Scott29 that the number of chromosomal aberrations can differ tremendously in a range of 60–200 aberrations per 100 lymphocytes of breast cancer patients after X-ray exposure to 0.5 Gy. When studying the radiosensitivity of relatives from the patients with high numbers of chromosomal aberrations in their lymphocytes, it turned out that a large number of these relatives also developed many aberrations. A genetic predisposition for increased radiosensitivity is apparently involved in these processes.
During the treatment of cancer patients with cytostatic drugs or ionizing radiation, patients are observed in rare cases, who develop severe side effects although the same treatment schedules are well tolerated without these side effects in the vast majority of patients. Experimental studies with cells and tissues from the extremely sensitive patients have revealed that the genetic disposition is responsible for these severe side effects. In a number of cases, distinct syndromes could be classified and the genetic background with mutations in certain genes could be identified. Table 3 shows such syndromes are ataxia telangiectasia,10,27,30 Bloom’s syndrome,10 Fanconi anemia,10,28,30,31 Li–Fraumeni syndrome,10,29,30,32 neurofibromatosis,30–35 Nijmegen breakage syndrome,27,30,36 and retinoblastoma.30,37 Further syndromes caused by the genetic disposition have been described in the textbook of Weinberg.39
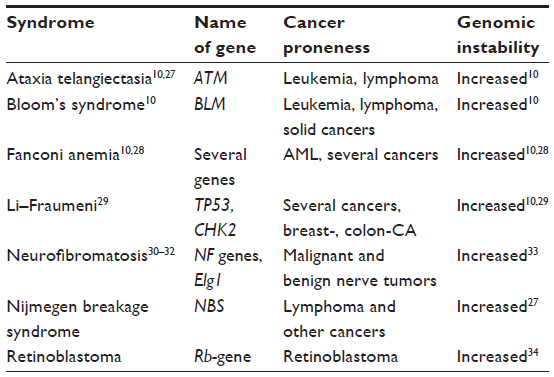 | Table 3 Human syndromes with genetic predisposition for increased sensitivity against cytostatic drugs and ionizing radiation, with cancer proneness and genomic instability |
In most cases, the involved genes have been described and quite often the locations of the mutations are known. Some of the involved genes are very large, and mutations can occur in different locations as well as different exons. Some of these genes with the corresponding gene products are important for key points in the regulation of the cell proliferation cycle.10,39 Thus, the gene products of the ATM gene, the TP53, or the Rb-gene are participating at a key point in the cell cycle where it is decided after a DNA damage whether the cell will go to the G1/S checkpoint, tries to repair the DNA damage and then starts DNA replication or whether the cell will undergo apoptosis and is eliminated. In patients with these syndromes, various DNA repair processes are also disturbed;38,39 therefore, certain DNA repair pathways are generally deficient. Most of these genetic syndromes are inherited in an autosomal recessive mode. The degree of sensitivity and genomic instability is always higher with the homozygote than with the heterozygote carriers.38 Several attempts have been undertaken to see whether specific chromosomes are more affected than others in cells with an increased genomic instability. Such specificity has not been observed. The distribution of chromosome breaks and possibly other lesions also seem to be comparatively randomly distributed over the whole genome.
Present knowledge about mechanisms for radiation-induced genomic instability
The mechanisms that are responsible for the development of genomic instability over many cell generations are not well understood until now. The following possibilities have been discussed:
- Oxidative stress – mitochondria involved
- Reduced DNA repair
- Changes in telomeres
- Epigenetic effects.
It is very well-known since a long time that ionizing radiation induces in water, especially in the presence of oxygen, and thus also in living cells highly reactive oxygen radicals (reactive oxygen species [ROS]), which lead to oxidative stress in living cells. This is not the only case in mitochondria through the endogenous metabolism but also through ionizing radiation and can lead to a perturbation of mitochondrial metabolism. Such metabolic changes have usually been studied shortly (hours and days) after radiation exposure.40 However, Limoli et al41 found that clones of a human–hamster hybrid cell line showed increased levels of ROS at later time periods after genotoxic exposures when an increased genomic instability was induced in these cells by genotoxic and cytotoxic agents. However, the radiation doses with 7.5–15 Gy were considerably high. This means a comparatively low survival of cells. An appreciable selection of cells had taken place. The authors suggested that the perturbation and the genomic instability may be linked to the state of chronic oxidative stress derived in part from dysfunctional mitochondria. These similar data have been recently reviewed by Azzam et al.42 It was concluded that the radiation-induced changes of oxidative metabolism persist for a long time, which leads finally to a damage of repair enzymes; consequently genomic instability is caused with in many cell generations after the exposure.
However, some arguments should be considered as follows:
- The dose ranges for the delayed metabolic perturbations of mitochondrial metabolism and the induction of genomic instability are different. The effects on oxidative stress have been observed only after radiation doses of several Gy and until now not with lower doses (<1 Gy) after which the increase of genomic instability has also been observed.
- Further, it should be considered that genomic instability is also induced in lymphocytes. These cells have very few mitochondria per cell and therefore the energy metabolism is quite different in these cells.40
- Furthermore, it should be considered that genomic instability is also induced by hypoxia. The situation with hypoxia is certainly very complex: chronic hypoxia does not induce oxidative stress. However, hypoxia-reoxygenation cycles, which occur in connection with the dynamics of changes in micro-vessel perfusion, can lead to oxidative stress in the hypoxic tissue regions and cells. Reoxygenation is necessary to induce oxidative stress.
Huang et al43 discussed that hypoxia-inducible factor-α (HIF-α) promotes angiogenesis after phosphorylation of HIF-α takes place to form the hypoxia-responsive element and the induction of vascular endothelial growth factor, whereas genomic instability is promoted when HIF-α is not phosphorylated, which then leads to a down-regulation of the DNA repair genes NBS1 and MSH2. It has further been shown that p53 also has a significant role in this mechanism. Deletion or knockdown of TP53 reduces the basal expression of MSH2 and MSH6.44 In this context, the supply and demand of the intracellular ATP for the energy metabolism, cell proliferation, and DNA repair is of high importance.44,45 Hypoxia does apparently not produce DSB in the polynucleotide chains that are necessary for chromosomal aberrations. The ROS induction occurs during/after the reoxygenation phase. The oxygen radicals produce mainly base damage in the DNA and possible DSBs occur only in later phases.46 However, the complex DSBs within clusters of ionization events, which are very important for the development of chromosomal breaks, are produced certainly to a smaller extent by ROS than by ionizing particles. Therefore, it must be assumed that decisive differences exist between the reaction of ROS and ionizing radiation especially in the low-dose range.
Hypoxia without reoxygenation does apparently not cause DNA damage via ROS production, but chronic, long-lasting hypoxia leads to downregulation of DNA repair enzymes.47,48 Besides the aforementioned downregulation of NBS1 and MSH2, in case HIF-α is not phosphorylated, it was also observed that the homologous repair-associated genes BRCA1 and RAD51 were downregulated by hypoxia. The decreased homology-dependent repair supports the induction of genomic instability.47 Undoubtedly ionizing radiation causes less DSBs under chronic hypoxic conditions than exposures in the presence of oxygen but the DSBs persist apparently longer.47 Therefore, it appears quite plausible that the changes of DNA repair processes are an important factor for the development of genomic instability in hypoxic conditions.
With respect to the reduction of DNA repair, it has already been mentioned that in humans with the defined syndromes and high radiosensitivity, DNA repair is very often reduced. Certain repair pathways are inactivated as discussed earlier, and this is apparently associated with an increased genomic instability. In these individuals, such processes have developed on the basis of the individual genetic predisposition with mutations in defined genes, eg, ATM and TP53 (Table 3).
There are certainly a number of strong arguments that telomeres and their possible changes are very much contributing to the development of genomic instability. Telomeres are well-known structures today, which stabilize the chromosomes in eukaryotic cells. They are mainly located at the ends of the chromosomes. The essential parts are repeats of 5′-TTAGGG-3′ nucleotide sequences.49 A protein complex consisting of six subunits is associated with these nucleotide sequences. This protein complex is named shelterin.50 The complex shelterin is protecting the telomeres at the end of the chromosomes and also recognizes possible damage in telomeres and DNA. Apparently, a very fine tuning and interplay between these protein complexes and DNA repair processes take place.50 The whole system of telomeres and the shelterin complex are very much involved in maintaining the genomic stability. In proliferating cells, the telomeres can be shortened when the cells go through the proliferation cycle. In many cells and tissues, especially in stem cells and regenerating tissues including cancers, the enzyme telomerase is extending the telomeres. The shelterin complex participates in the regulation of these processes.50 Telomere length decreases with age and has relations to many diseases.51 It has been reported that telomeres are shortened in irradiated cells, and this effect occurs in a delayed mode.52,53 An inverse relation between the induction of micronuclei after incubation of lymphocytes from cord blood with mitomycin-C and the length of telomeres has been observed in a significant manner.51 Again the increase of micronuclei can be taken as a sign of increased genomic instability. On the basis of such data, it has been postulated that the induction of an increase of genomic instability is caused by telomere dysfunction.52
In conclusion, there is apparently not the only one mechanism for the development of genomic instability. A deficiency in certain DNA repair pathways is very important. This may be caused by inheritance or by environmental factors, eg, hypoxia, which have an influence on the expression of such genes, which are involved in DNA repair. Energy metabolism plays a significant role. The ATP supply is very important in this context.38 Hypoxia may be a driver for the induction of genomic instability but this effect can also occur without hypoxia. Dysregulation of the cell cycle can be inherited or it can be induced by genotoxic as well as cytotoxic agents, which then can contribute to the increase of genomic instability. After radiation exposures with higher doses, oxidative stress will be significant. It should be always considered that the increase of genomic instability develops during the progress of a number of 20 cell cycles or even more and here apparently changes in the length as well as the structure of telomeres occur associated with genomic instability. The mechanism, which leads to the changes of telomeres remains open.
Genomic instability and pathophysiological consequences (health effects)
In principle, genomic instability has an influence on all health problems where a genetic impact is involved, as the probability for mutations is increasing with the instability.21,38 Most studies and data of such connections are available for cancer diseases and their development. Several references in this direction have already been made, and in the following, it will be continued to focus the discussion on the relation between genomic instability and cancer including the development of these diseases. Many experimental studies and clinical experiences clearly indicate that several mutations are usually necessary for the development from normal tissues/cells to a cancer with metastases (Figure 2).38,53 The following steps can be defined for such a development in a comparatively general way.
Exposures of cells/tissues to cytotoxic and genotoxic agents such as ionizing radiation cause DNA damage in the cell nucleus. DNA repair processes try to repair this damage, which may not be complete or even misrepair may occur. Proliferation of cells with incomplete DNA repair or misrepair leads to mutated cells and finally to malignantly transformed cells. The organism is equipped besides the protective instrument, DNA repair, with a further defense mechanism, the immune system, which can eliminate malignant cells by apoptosis or phagocytosis.54 Then follow a number of further mutations, which are different for specific cancers and where oncogenes, eg, ras- and myc-family as well as tumor-suppressor genes, eg, TP53 and other more specific genes for individual cancers are involved.55 The probability of such mutations increases with cells that have an increased genomic instability. Under these circumstances, it is quite obvious that an increased genomic instability will promote carcinogenesis, and this has been proposed quite frequently.38,39,46,47,51,53,55,56
Such an association has been already discussed in context with the genetically dependent syndromes such as ataxia telangiectasia and others. In all these symptoms, there was found an increase of cancer, a reduced DNA repair, changes in the regulation of the cell cycle, and an increased genomic instability.
Studies with uranium miners who received high exposures to the respiratory tract mainly by radon and its radioactive daughter nuclides have shown that genomic instability is increased in the lymphocytes of these miners measured by chromosomal aberrations or by the micronucleus-centromere method.38,45,57 The increased genomic instability was observed in the lymphocytes of miners with a bronchial carcinoma as well as to a lesser degree in miners without a cancer. An increase of genomic instability was observed decades after the loco-regional exposure to the radioactive nuclides and before any treatment of the cancer. Thus, the increased genomic instability had developed just as a consequence of the radiation exposure. Interestingly, an increased genomic instability was also found in the lymphocytes of patients with head and neck cancer again before any treatment of the cancer.38,45 These data clearly show that cancer is not a local disease but other cell systems are also affected to a certain degree. Furthermore, it may be possible that cancer develops preferably in individuals with a relatively high genomic instability. Thus, individuals with an increased genomic instability have apparently a higher risk to develop a cancer.
An interesting study was published with Hodgkin lymphoma (HL) patients.58 The telomere length (kb) and the chromosomal aberrations (CAs per cell) were measured in the lymphocytes of 30 healthy persons (controls) (group 1), of 73 patients with HL after radiotherapy without a secondary cancer (group 2), and of 28 patients with HL after radiotherapy with a secondary cancer (group 3). The telomere length decreases in the sequence group 1 (11.7 kb) > group 2 (8.3 kb) > group 3 (6.6 kb), whereas the number of chromosomal aberrations per cell increases in the same sequence (group 1: 0.003 CAs per cell; group 2: 0.026 CAs per cell and group 3: 0.164 CAs per cell). These are clear indications for an increase of genomic instability. During the course of the study, secondary cancers appeared in two patients of group 2. With these two patients, it was observed that the telomere length was in the lower range of group 2 (telomere length in these two patients was: 6 kb and 7.5 kb respectively). These observations go in line with the assumption that the telomere length is strongly associated with genomic instability, and this result is a predictor for the appearance of a secondary cancer.
Thus, it is generally accepted that genomic instability is a strong driver or promotor for the development of cancer after transformation of normal cells to malignant cells by endogenous or exogenous processes.38,39,53,55,56 Increased genomic instability is apparently an indicator for an enhanced probability to develop cancer. The German cell biologist Theodor Boveri suggested approximately 100 years ago that the loss of “key attributes” (today known as tumor suppressor genes) is a driving event in the development of cancer and genetic disposition could play a role in cancer susceptibility. As well, he predicted that chromosomal (genomic) instability is a cornerstone of cancer development.59
Acknowledgment
This work is dedicated in deep and sincere remembrance to Dr William Morgan, an eminent scientist and very good friend who died on November 12, 2015, much too early.
Disclosure
The author reports no conflicts of interest in this work.
References
Watson JD, Crick FHC. A structure for deoxyribose nucleic acid. Nature 1953;171:737–738. | |
Alberts B, Johnson A, Lewis J, et al. Molecular Biology of the Cell. New York: Garland Science; 2014. | |
Sumner AT. Chromosomes: Organization and Function. Oxford: Blackwell Science; 2003. | |
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. Washington, DC: ASM Press; 2006. | |
Martin CL, Warburton D. Detection of chromosomal aberrations in clinical practice: from karyotype to genome sequence. Annu Rev Genomics Hum Genet. 2015;16:309–326. | |
Krylov V, Tlapakova T. Xenopus cytogenetics and chromosomal evolution. Cytogenet Genome Res. 2015;145(3–4):192–200. | |
Streffer C, Herrmann T. Ein Jahrhundert Entwicklung in der Strahlenbiologie. [A century of development in radiation biology]. Strahlenther Onkol. 2012;188:231–244. German. | |
Langendorff H. Die Bedeutung von Zellreaktionen für die Analyse der biologischen Strahlenwirkung. [The significance of cell reactions for the analysis of biological radiation effects]. Strahlentherapie. 1943;73:181–195. German. | |
Kulka U, Ainsbury L, Atkinson M, et al. Realising the European network of biodosimetry: RENEB-status quo. Radiat Prot Dosimetry. 2015;164(1–2):42–45. | |
United Nations Scientific Committee on the Effects of Atomic Radiation (UNSCEAR). Non-targeted and Delayed Effects of Exposure to Ionizing Radiation. New York: United Nations; 2006. | |
Pampfer S, Streffer C. Increased chromosome aberration levels in cells from mouse fetuses after zygote X-irradiation. Int J Radiat Biol. 1989;55:85–92. | |
Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat Res. 2003;159:567–580. | |
Kadhim MA, MacDonald DA, Goodhead DT, Lorimore SA, Marsden SJ, Wright EG. Transmission of chromosomal instability after plutonium alpha-particle irradiation. Nature 1992;355:738–740. | |
Sabatier L, Dutrillaux B, Martin MB. Chromosomal instability. Nature 1992;357:548. | |
Limoli CL, Corcoran JJ, Milligan JR, Morgan WF. Critical target and dose and dose-rate responses for the induction of chromosomal instability by ionizing radiation. Radiat Res. 1999;151:677–685. | |
Streffer C. Bystander effects, adaptive response and genomic instability induced by prenatal irradiation. Mutat Res. 2004; 568(1):79–87. | |
Ullrich RL, Davis CM. Radiation induced cytogenetic instability in vivo. Radiat. Res. 1999;152:170–173. | |
Okada M, Okabe A, Uchihori Y, et al. Single extreme low dose/low dose rate irradiation causes alteration in lifespan and genome instability in primary human cells. Br J Cancer. 2007;96:1707–1710. | |
Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A. 2003;100:5057–5062. | |
Weissenborn U, Streffer C. Analysis of structural and numerical chromosomal anomalies at the first, second, and third mitosis after irradiation of one-cell mouse embryos with X-rays or neutrons. Int J Radiat Biol. 1988;54:381–394. | |
Little JB, Nagasawa H, Pfenning T, Vetrovs H. Radiation-Induced Genomic Instability: Delayed Mutagenic and Cytogenetic Effects of X Rays and Alpha Particles. Radiat Res. 1997;148:299–307. | |
Dubrova YE, Jeffreys AJ, Malashenko AM. Mouse minisatellite mutations induced by ionizing radiation. Nat Genet. 1993;5:92–94. | |
Dubrova YE, Nesterov VN, Krouchinsky NG, et al. Human minisatellite mutation rate after the Chernobyl accident. Nature 1996;380:683–686. | |
Ohtaki K, Kodama Y, Nakano M, et al. Human fetuses do not register chromosome damage inflicted by radiation exposure in lymphoid precursor cells except for a small but significant effect at low doses. Radiat Res. 2004;161:373–379. | |
Nakano M, Nishimura M, Hamasaki K, et al. Fetal irradiation of rats induces persistent translocations in mammary epithelial cells similar to the level after adult irradiation, but not in hematolymphoid cells. Radiat Res. 2014;181:172–176. | |
Uma Devi P, Hossain M, Satyamitra M. Low dose fetal irradiation, chromosomal instability and carcinogenesis in mouse. Int Congr Ser. 2003;1236:123–126. | |
Tanaka K, Kamada N. Leukemogenesis and chromosome aberrations: de novo leukemia in humans – with special reference to atomic bomb survivors. Nihon Ketsueki Gakkai Zasshi. 1985;48:1830–1842. | |
Nakanishi M, Tanaka K, Shintani T et al. Chromosomal instability in acute myelocytic leukemia and myelodysplastic syndrome patients among atomic bomb survivors. J Radiat Res. 1999;40:159–167. | |
Scott D. Chromosomal radiosensitivity, cancer predisposition and response to radiotherapy. Strahlenther Onkol. 2000;176:229–234. | |
Pollard JM, Gatti RA. Clinical radiation sensitivity with DNA repair disorders: an overview. Int J Radiat Oncol Biol Phys. 2009;74:1323–1331. | |
Takata M, Ishiai M, Kitao H. The Fanconi anemia pathway: insides from somatic cell genetics using DT 40 cell line. Mutat Res. 2009;668:92–102. | |
Tabori U, Nanda S, Druker H. Younger age of cancer initiation is associated with shorter telomere length in Li–Fraumeni syndrome. Cancer Res. 2007;67:1415–1418. | |
Kobayashi C, Oda Y, Takahira T. Chromosomal aberrations and microsatellite instability of malignant peripheral nerve sheath tumors: a study of ten tumors from nine patients. Cancer Genet Cytogenet. 2006;165(2):98–105. | |
Spyra M, Otto B, Schön G, Kehrer-Sawatzki H, Mautner VF. Determination of the mutant allele frequency in patients with neurofibromatosis type 2 and somatic mosaicism by means of deep sequencing. Genes Chromosomes Cancer. Epub 2015 May 29. doi: 10.1002/gcc.22259. | |
Thomas LE, Winston J, Rad E, et al. Evaluation of copy number variation and gene expression in neurofibromatosis type-1-associated malignant peripheral nerve sheath tumours. Hum Genomics. 2015;9:3. doi: 10.1186/s40246-015-0025-3. | |
Gazy I, Liefshitz B, Parnas O, Kupiec M. Elg1, a central player in genome stability. Mutat Res Rev Mutat Res. 2015; 763:267–279. doi: 10.1016/j.mrrev.2014.11.007. | |
Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17:1363–1372. | |
Streffer C. Strong association between cancer and genomic instability. Radiat Environ Biophys. 2010;49:125–131. | |
Weinberg AR. The Biology of Cancer. 2nd ed. New York: Garland Science; 2013. | |
Streffer C. Strahlen-Biochemie. Heidelberger Taschenbücher. [Radiation-Biochemistry]. Berlin: Springer-Verlag; 1969. German. | |
Limoli CL, Giedzinski E, Morgan WF, Swarts SG, Jones GD, Hyun W. Persistent oxidative stress in chromosomally unstable cells. Cancer Res. 2003;63:3107–3111. | |
Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60. | |
Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability, a calculated mechanism underlying tumor progression. J Mol Med. 2007;85:139–148. | |
To KKW, Koshiji M, Hammer ST, Huang LE. Genetic instability: the dark side of the hypoxic response. Cell Cycle. 2005;4:881–882. | |
Streffer C. Radiological protection: challenges and fascination of biological research. Strahlenschutzpraxis. 2009;2:2–12. | |
Bindra RS, Glazer PM. Genetic instability and the tumor microenvironment: towards the concept of microenvironment-induced mutagenesis. Mutat Res. 2005;569:75–85. | |
Glazer PM, Hegan DC, Lu Y, Czochor J. Scanlon SE. Hypoxia and DNA repair. Yale J Biol Med. 2013;86:443–451. | |
Scanlon SE, Glazer PM. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst). 2015;32:180–189. doi: 10.1016/j.dnarep.2015.04.030. | |
Blackburn EH, Gall, JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J Mol Biol. 1978;120:33–53. | |
de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. | |
Moreno-Palomo J, Creus A, Marcos R, Hernández A. Genomic instability in newborn with short telomeres. PLoS One. 2014;9(3):e91753. | |
Sabatier L, Lebeau J, Dutrillaux B. Chromosomal instability and alterations of telomeric repeats in irradiated human fibroblasts. Int J Radiat Biol. 1994;66:611–613. | |
Shim G, Ricoul M, Hempel WM, Azzam WI, Sabatier L. Crosstalk between telomere maintenance and radiation effects: a key player in the process of radiation-induced carcinogenesis. Mutat Res. 2014;760:1–17. | |
Multhoff G, Pockley AG, Streffer C, Gaipl US. Dual role of heat shock proteins (HSPs) in anti-tumor immunity. Curr Mol Med. 2012;12:1174–1182. | |
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou Sh, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. | |
Little JB, Lauriston S. Taylor lecture: nontargeted effects of radiation: implications for low-dose exposures. Health Phys. 2006;91:416–426. | |
Kryscio A, Müller WU, Wojcik A, Kotschy A, Grobelny S, Streffer C. A cytogenetic analysis of the long-term effect of uranium mining on peripheral lymphocytes using the micronucleus-centromere assay. Int J Radiat Biol. 2001;77:1087–1093. | |
M’kacher R, Bennaceur-Griscelli A, Girinski TH, et al. Telomere shortening and associated chromosomal instability in peripheral blood lymphocytes of patients with Hodgkin lymphoma prior to any treatment are predictive of second cancers. Int J Radiat Oncol Biol Phys. 2007;68:465–471. | |
Hansford S, Huntsman DG. Boveri at 100: Theodor Boveri and genetic predisposition to cancer. J Pathol. 2014;234:142–145. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.