")
Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
An update on gene therapy for the treatment of lipoprotein lipase deficiency
Received 25 October 2013
Accepted for publication 10 December 2013
Published 15 May 2014 Volume 2014:4 Pages 47—54
DOI https://doi.org/10.2147/ODRR.S39979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Andrew E Libby, Hong Wang
Division of Endocrinology, Metabolism, and Diabetes, School of Medicine, University of Colorado at Denver, Aurora, CO, USA
Abstract: Lipoprotein lipase (LPL) is responsible for clearance of triglyceride-rich lipoproteins from the blood. Deficiency or defects in this enzyme result in profound hypertriglyceridemia and susceptibility to chronic, life-threatening pancreatitis. Management of LPL deficiency has traditionally been restricted to palliative care and strategies to reduce the risk of pancreatitis, including severe dietary restrictions of fat. Recently, the European Commission approved the first gene therapy treatment in the West to treat this rare disease. Alipogene tiparvovec (Glybera®) was granted marketing authorization in November 2012 to treat LPL deficiency in a subset of patients that are at increased risk for pancreatitis. Designed as a one-time treatment, the drug uses adeno-associated virus (AAV1) delivery of transgenic LPL to muscle in patients lacking functional enzyme. Although statistically significant reduction of serum triglycerides was initially observed in trial subjects, this effect was found to be transient, with triglyceride levels eventually rebounding to basal levels by 26 weeks in all participants. Nevertheless, despite the return of triglycerides to pretreatment levels, alipogene tiparvovec was found to have a long-term impact on postprandial chylomicron metabolism by lowering the fraction of triglyceride found in this subset of lipoproteins. Furthermore, the drug led to a clinically significant reduction in the incidence of pancreatitis in LPL-deficient patients. The regulatory approval of alipogene tiparvovec was a historic process and serves as an example of the challenges that future orphan drugs will face.
Keywords: lipoprotein lipase deficiency, gene therapy, AAV, chylomicron, pancreatitis
Introduction
Found on the luminal surface of capillaries, lipoprotein lipase (LPL) is the rate-limiting enzyme for clearance of circulating chylomicrons and very-low-density lipoprotein (VLDL) from the plasma. LPL is primarily expressed in cardiac, adipose, muscle, and brain tissues, and is important for lipid uptake for storage, energy production, signaling, learning and memory, and regulation of energy balance.1–4 Many naturally-occurring polymorphisms have been identified in the LPL gene, although very few of them lead to truly deficient or catalytically inactive enzyme.5 Human LPL deficiency (LPLD) is a rare, autosomal-recessive disease that affects approximately one in 1 million people. Patients deficient in LPL exhibit pronounced hypertriglyceridemia, with high plasma triglycerides (TGs) leading to hepatosplenomegaly, xanthomata, and lipemia retinalis.6 However, the largest complication for LPLD patients is typically pancreatitis, with this condition often presenting early in childhood and recurring throughout adulthood.7 LPLD is unresponsive to standard therapeutics such as fibrates, and the only course of treatment is a diet severely restricted in fat (<15% of daily caloric intake). Even with dietary compliance, though, TGs often remain elevated above 10 mmol/L and can reach higher than 20 mmol/L. Due to the fact that this disease cannot be treated with traditional methods, it is clear that novel approaches to gene therapy were needed for this disease.8
In November 2012, a major milestone was achieved when the European Commission approved the first gene therapy treatment.8 Developed to treat LPLD in a subset of patients with chronic pancreatitis, alipogene tiparvovec (Glybera®; uniQure Biopharma BV, Amsterdam, the Netherlands) works by using an adeno-associated virus (AAV) to deliver the LPL gene to patients without a functional version of this enzyme. Glybera’s journey from bench to bedside was a long one, with many challenges and hurdles to overcome. Slated for rollout in 2014, Glybera tested the limits and boundaries of the regulatory approval process and demonstrated many of the unique difficulties that future gene therapy treatments must necessarily deal with. The goal of this review is to provide a general overview of the development of Glybera from its proof-of-concept and clinical trials through its approval last year by the European Medicines Agency (EMA). Extensive reviews of each phase of Glybera’s development have been provided previously.9–13
Early proof of principle
In 1997, Excoffon et al showed that hypertriglyceridemia and impaired fat tolerance could be corrected in mice heterozygous for LPLD by liver-targeted AAV gene transfer of human LPL (hLPL).14 By day 7 post-injection, plasma TGs were significantly reduced relative to control mice, and LPLD mice exhibited improved oral and intravenous fat-load tolerance. Remarkably, analysis of plasma lipoproteins in mice that underwent AAV-mediated gene transfer showed that VLDL-derived TGs were significantly reduced. However, this correction was temporary, and plasma TG returned to endogenous levels by day 42. Furthermore, this model of gene deficiency correction was not able to be tested in mice truly homozygous for LPLD since mice totally deficient in LPL do not survive after birth. Nevertheless, the study suggested that AAVs could be employed as efficient gene delivery vehicles to correct genetic deficiencies such as LPLD.
Shortly thereafter, Liu et al tested this liver-targeted AAV-mediated LPL delivery approach in an animal model that more closely mimics the human disorder.15 This study was conducted in a naturally-occurring feline phenotype that is homozygous for LPLD (LPL -/-). Unlike mice with complete LPLD, cats with this disorder survive into adulthood and exhibit chronic pancreatitis, more closely resembling the condition observed in man. The gene delivery approach was successful with a 10-fold reduction in plasma TGs and enhanced clearance of intravenously introduced fat loads. Like the previous studies in mice, though, this reduction in plasma TG was ultimately temporary, with the effects lasting 2 weeks. Importantly, this group was able to identify several problems that would be critical for subsequent development of AAV-mediated therapies. They were able to detect AAV-neutralizing antibodies by 7 days, and antibodies against hLPL were identified by 14 days. These observations provided a possible explanation for the temporary effect of the therapy.
Preclinical development
To address the need for a mouse model of LPLD that more closely resembles the condition in humans, Ross et al rescued LPL (-/-) mice at birth by somatic gene transfer of LPL via adenovirus.16 This was accomplished via intramuscular injection of an adenoviral vector carrying the S447X variant of LPL. The S447X mutation truncates full-length LPL by one residue, replacing the codon for a terminal serine with a stop codon. This mutation – carried by 20% of the human population – has been shown to be a gain-of-function mutation that confers several benefits, including decreased plasma TG, increased high-density lipoprotein cholesterol (HDL-C), and a reduction in coronary artery disease risk.17 By contrast, when the group attempted to use wild-type LPL to rescue mice at birth, it was unsuccessful. Though they survived into adulthood, mice rescued at birth with the adenoviral-LPLS447X construct ultimately developed an LPL-deficient phenotype, displaying markedly increased plasma TG (>200-fold), low HDL-C, etc. Importantly, since this adenoviral-mediated rescue was initiated right after birth, it was found that these animals did not display the immune response to the viral vector or transgenic LPL, unlike mice from earlier studies. It was therefore concluded that this animal would be a good model for testing gene therapy approaches to treat LPLD.
Ross et al then showed that long-term correction of LPLD could be achieved in the rescued LPL knockout mice using AAV1-mediated gene transfer.18 The transgene is delivered by a non-replicating and non-integrating vector made up of AAV1 capsid proteins and AAV2-derived inverted terminal repeats. Expression of the transgene is controlled by a cytomegalovirus promoter and a woodchuck hepatitis post-transcriptional regulatory element.12 Unlike previous studies, it was decided that muscle is a better target than the liver for LPL gene delivery due to the fact that it is a natural site of LPL production and activity. Providing further support for muscle as the ideal tissue for LPL introduction was work showing that even intramuscular injection of naked plasmid DNA containing LPL on a muscle-specific promoter could lead to significant decreases in plasma TG.19 Intramuscular administration of AAV-LPLS447X resulted in detectable LPL activity in post-heparin plasma in these animals (up to 33% of normal levels). Plasma TG levels were lowered dramatically to near-normal levels, and HDL-C was found to be significantly increased. Of great significance is the fact that the effect on plasma TG and lipid clearance was long-lasting; clearance after intravenous lipid challenge was near-normal even at 8 months post-injection, and plasma TG levels were near-normal for at least 1 year. This is in stark contrast to earlier experiments in which AAV-mediated gene delivery to the liver induced effects for only 42 days. No antibodies to hLPL were observed in treated mice (likely due to tolerance introduced shortly after birth), although antibodies targeting AAV were observed. Although LPL expression slowly declined 12 weeks post-injection, the reduction in plasma TGs persisted for over a year, indicating that even partial replacement of activity (<10% of normal) can have dramatic lipid-lowering effects.
Subsequent work focused on characterizing the safety and biodistribution of AAV1-LPLS447X in wild-type mice.20 It was found that muscle is well-suited to dose-dependent LPL expression and that skeletal muscle is tolerant to high viral loads.18 Additionally, acute inflammatory elements are only transiently elevated post-injection in mice. Although injected mice displayed elevated spleen hyperplasia, this effect was reversible and dose-dependent. Biodistribution analysis showed that long-term persistence of AAV-derived hLPL DNA was limited to the intramuscular injection site and draining lymph nodes, while short-term vector leakage from the site of injection into the circulation was efficiently cleared via hepatic routes.
Preclinical work also included efficacy testing of AAV-LPLS447X on the LPLD feline model.21 Like previous studies in mice, the authors observed dose-dependent expression of LPL in injected muscle. The study observed an initial reduction of plasma TG by 99% to normal levels, but this effect was transient and lasted only 3 weeks. Antibodies to both hLPL and AAV were detected, and the authors predicted that this immune response was likely limiting the duration of action of the injected vector. This study was the first to include oral dosing of cyclophosphamide for a subset of injected subjects in order to inhibit immune responses. Although cyclophosphamide led to a greater reduction in plasma TGs, it did not affect the duration of efficacy. In addition to antibody production, it was speculated that cellular immune responses may have contributed to the transient nature of this treatment, as local accumulation of mononuclear cells were observed near the injection sites. Nevertheless, it was predicted that immune responses would be limited in human subjects due to the fact that most LPLD patients have some level of circulating inactive LPL that would minimize the risk of antibody production.
Clinical development of Glybera
Phase I/II (CT-AMT-010-01)
Amsterdam Molecular Therapeutics (AMT; uniQure Biopharma BV, Amsterdam, the Netherlands) undertook clinical trial studies of AAV1-LPLS447X in mid-2005. The first clinical study was an open-label trial which consisted of a dose-response study, CT-AMT-010-01. The goal of this trial was to establish efficacy, monitor toxicity, and examine the biodistribution of AAV1-LPLS447X in humans.11 Eight LPLD subjects were selected for participation in this phase. All subjects had confirmed mutations in the LPL gene, significantly reduced LPL activity (>20% of normal), detectable LPL mass in circulating post-heparin plasma (≥5% of normal), and fasting plasma TGs of >10 mmol/L.13 AMT-010-IM was administered in four subjects at 1 × 1011 gc/kg, while the remaining subjects were given a higher dose of 3 × 1011 gc/kg. The vector was administered as multiple injections into the quadriceps muscle. The endpoint for this study was defined as a reduction in fasting plasma TG to ≤10 mmol/L or a fasting plasma TG reduction of ≥40%. There were no serious adverse events in any of the subjects monitored. Twelve weeks post-injection, one subject in the low-dose cohort and two subjects in the high-dose cohort showed significantly reduced fasting plasma TGs past the defined endpoint. Although enzymatic activity and protein levels were found in muscle biopsies taken from around the injection sites 26–36 weeks after injection, the reduction in plasma TG proved to be transient and was absent at long-term follow-up. Because antibodies were detected to AAV1 coat proteins, it was speculated that the transient nature of the TG reduction could be due to a mounted immune response.22 No antibodies were detected against hLPLS447X, however.
Phase II/III (CT-AMT-011-01)
After the first trial phase, it was speculated that efficacy could likely be improved by increasing the injected dose to 1 × 1012 gc/kg. AMT chose a baculovirus-based manufacturing process and gave the drug used for all subsequent trials the name alipogene tiparvovec (Glybera). A second dose-escalation trial was initiated, CT-AMT-011-01, in which participants were selected based on criteria from the first trial as well as having a history of pancreatitis. The goal of this phase of study was to determine whether a higher dose of alipogene tiparvovec would be more efficacious and to examine whether the drug could reduce incidence of recurrent pancreatitis. Fourteen participants were selected for the trial and divided into two cohorts. The first group (six subjects) received a dose of 3 × 1011 gc/kg, while the second group received 1 × 1012 gc/kg virus. Additionally, it was decided that immunosuppressants would be included in this phase of study in order to minimize potential immunogenicity of the viral capsid proteins and/or transgenic LPL. Accordingly, a combination of 3 mg/kg cyclosporine and 2 g/day mycophenolate mofetil were chosen to combat immune responses. Four of six subjects in the low-dose cohort and all eight subjects in the high-dose group were coadministered immunosuppressants along with alipogene tiparvovec. Like the first study, subjects were examined for fasting TG levels 12 weeks post-injection.
Seven of the participants (50%) in CT-AMT-011-01 achieved either median fasting plasma TG levels ≤10 mmol/L or a 40% reduction in median fasting plasma TG between 3 and 12 weeks. Like the first study, though, this effect was only transient, and fasting plasma TG levels reverted to baseline levels by week 26. Although no antibodies to hLPLS447X were detected, it was speculated that immunosuppressants were likely ineffective at increasing the duration of the TG reduction response. Interestingly, although TG levels reverted back to normal by 26 weeks, analysis of the fasting plasma lipoprotein fractions revealed an overall reduction in fasting chylomicron TG content with a concomitant increase in VLDL up to 52 weeks. This suggested that, although total serum TG levels ultimately increased post-injection, the lipoprotein composition had been altered.10
Phase II/III (CT-AMT-011-02)
Upon review of the data generated by the initial clinical studies of Glybera, it was debated whether reduction of total fasting TG levels were an appropriate endpoint for evaluating efficacy of the drug. Thus far, TG levels had eventually returned to baseline in all enrolled subjects regardless of dosage or coadministration of immunosuppresants. Despite this fact, there did appear to be emerging evidence that alipogene tiparvovec had a long-lasting effect on plasma lipoprotein composition. Follow-up of subjects from the first studies showed that, at 52 weeks post-injection, participants had significantly reduced chylomicron TG content and increased VLDL. Importantly, it is thought that large, TG-rich chylomicrons might be the principal trigger for pancreatitis presentation in LPLD patients.23 Large, buoyant chylomicrons may become trapped in small pancreatic microcapillaries and subjected to the action of pancreatic lipase.10 Free fatty acids that are subsequently released may trigger an inflammatory response in the pancreatic acinar cells and capillary beds.24 It was decided that long-lasting beneficial effects on chylomicron metabolism may be a better primary endpoint for evaluating the efficacy of Glybera, with reduction in total fasting TG levels a secondary measure of efficacy. Additionally, data from long-term follow-up of the first two clinical studies indicated that incidence of pancreatitis may have been reduced in participants that received Glybera, although the numbers were too small to reach statistical significance.13
A third trial was designed, CT-AMT-011-02, that primarily tested whether Glybera had long-lasting effects on postprandial chylomicron metabolism.23 A dose of 1 × 1012 gc/kg was chosen for coadministration with cyclosporine, mycophenolate mofetil, and a pre-dose bolus injection of methylprednisolone. Five participants who met previous eligibility criteria and who had histories of chronic pancreatitis were chosen for this phase of study. Two weeks before injection and 14 weeks afterward, subjects were given a low fat meal containing a 3H palmitate tracer, and blood samples were drawn each hour in order to examine the tracer and TG content of lipoprotein fractions. Data from LPLD patients was compared to six healthy controls.
At 14 weeks, all five patients who received alipogene tiparvovec exhibited near-normal chylomicron clearance rates and chylomicron TG levels comparable to control subjects. Although total plasma TG was already returning to baseline by the 14-week time-point, postprandial chylomicron metabolism showed significant improvement compared to pre-injection. Of the original five subjects enrolled in the study, three of them repeated tracer experiments at 52 weeks post-injection. Enhanced chylomicron clearance was still observed almost 1 year after alipogene tiparvovec administration, although the increase in clearance was dampened compared to the 14-week time point. The data from CT-AMT-011-02 indicated that patients receiving alipogene tiparvovec experience long-term benefits in chylomicron metabolism even after total serum TG levels rebound to baseline. Additionally, chylomicron clearance was deemed to be a valid efficacy endpoint for alipogene tiparvovec. This would prove to be controversial during subsequent regulatory approval for the drug.
CT-AMT-011-03
The final phase of clinical studies for alipogene tiparvovec was designed specifically to examine the effect of the drug on pancreatitis. Given that the drug was shown to improve long-term chylomicron clearance and metabolism, AMT moved to examine whether pancreatitis incidence was reduced in LPLD patients. Although not statistically significant, data from previous trials had suggested that patients who received one-time injections of alipogene tiparvovec experienced reduced incidence of pancreatitis. Additionally, although not admissible in the regulatory approval process, self-reporting from trial patients themselves during the first three trials showed reduced abdominal discomfort and pain.
The fourth study, CT-AMT-011-03, consisted of long-term follow-up and retroanalysis of 17 of the 22 patients enrolled in the first three studies in order to examine the effect of Glybera on recurrence of pancreatitis in LPLD individuals. This phase of study examined incidence of pancreatitis over a period of 3 years before and after treatment. This analysis proved controversial, as these observations were made in a limited number of patients and relied on a relatively short period of time before and after treatment. Analysis of the 2012 EMA Assessment of Glybera by Bryant et al concluded that eight of the 17 analyzed subjects were pancreatitis-free after injection of Glybera, but had also been pancreatitis-free for an equal period of time before treatment.13,25 Equally important, three of the 17 subjects accounted for eleven of 41 (27%) pretreatment pancreatitis incidents. Likewise, the majority of pancreatitis presentations posttreatment involved only three individuals. A summary of clinical trial data for alipogene tiparvovec is provided in Table 1.
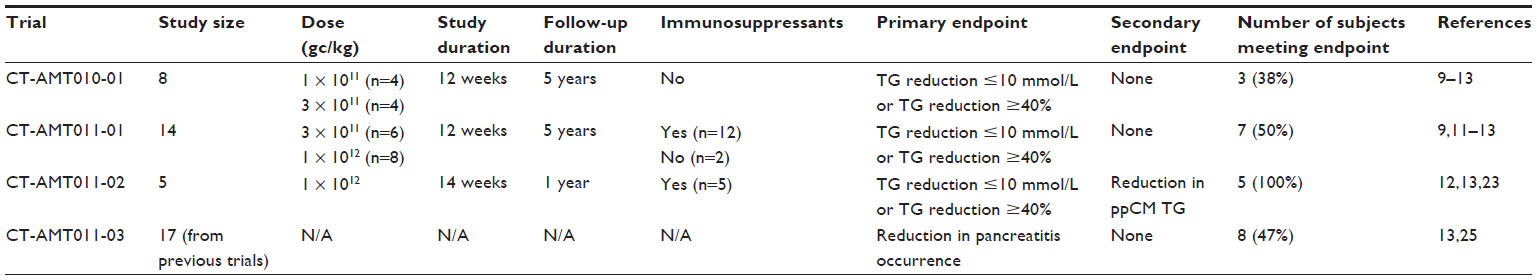 | Table 1 Summary of clinical trial data for alipogene tiparvovec |
Regulatory approval of Glybera
AMT submitted an application for marketing approval of Glybera to the EMA in January 2010.26 During the 3 years that Glybera spent in review, important obstacles arose that nearly derailed the approval of the drug.27 Importantly, challenges that any future gene therapy agents – and orphan drugs, in general – ultimately face during regulation were thrust into the public spotlight. As this was the first gene therapy agent approved in the West, the limits and boundaries of the European regulatory approval process were tested, and scrutiny of this approval process experienced unprecedented levels.
For novel biological therapeutics, marketing approval is a multilayered process in the European Union. Drugs like alipogene tiparvovec are first reviewed by the EMA’s Committee for Advanced Therapeutics (CAT), an entity created to review gene and cell therapy products. The CAT then passes on a recommendation to the main regulatory arm of the EMA, the Committee on Human Medicinal Products (CHMP) for further review, and it is the CHMP that makes the recommendation for market authorization to the European Commission for final acceptance or rejection. Like other novel biological agents, initial concern was placed on the safety and manufacturing consistency of the drug.13 However, preclinical toxicology as well as data from clinical trials showed that it did not exhibit acute toxicity or biodistribution problems, and these initial concerns were put aside. Nevertheless, both the CAT and CHMP initially rejected AMT’s application for alipogene tiparvovec. AMT then appealed and sought reexamination of the drug by the EMA. After additional review, the CAT reversed its original negative opinion and recommended to the CHMP that alipogene tiparvovec be approved. To the surprise of many, however, the CHMP once again rejected approval in October 2011. The CHMP held the opinion that alipogene tiparvovec had not shown efficacy in being able to lower blood TG and that there were too few patients from whom long-term data could be gleaned.13
Indeed, LPLD is an ultrarare disease, affecting only one in 1 million people, and clinical trials included a total of only 27 participants. One of the main reservations for the CHMP was that there was insufficient data available to recommend approval. Yet, this is a dilemma that any orphan drug faces. Since very small populations exist to test any drug developed to treat rare diseases, the commonly-held convention is that the agent must show clear efficacy for meeting the established endpoints during human trials. In the case of alipogene, it had failed to show long-term lowering of blood TGs. All patients eventually rebounded to baseline serum TG levels. Additionally, postprandial chylomicron metabolism was a previously invalidated endpoint with no precedence. After its application was rejected twice, AMT was faced with financial hardships that prompted it to sell its assets to a newly-formed private company, UniQure.
In a rare move, the European Commission, the entity which usually ratifies the CHMP recommendation, asked the CHMP to reexamine alipogene tiparvovec yet again in January 2012. This time, the committee was asked to review the drug for use in a subset of LPLD patients who had experienced chronic and recurring pancreatitis. This action was rather unexpected and controversial. Data from AMT’s fourth trial, CT-AMT-011-03, had shown a clinically significant reduction in incidence of pancreatitis of 17 patients who were monitored 3 years posttreatment. Furthermore, data from CT-AMT-011-02 had shown that alipogene tiparvovec can alter long-term lipoprotein characteristics by reducing TG content in the chylomicron fraction. This was important because it is thought that the large, newly-formed, TG-rich chylomicrons get trapped in pancreatic microcapillaries and restrict blood flow, thereby inducing subsequent pancreatitis. As postprandial chylomicron metabolism was a previously invalidated endpoint, however, this third round of review by the CHMP again resulted in a negative opinion for approval, although the number of committee members in favor of approval was higher than in previous reviews.25 Reevaluation of the use of alipogene tiparvovec for pancreatitis-prone patients had limited the available clinical data even further, and concerns were raised over whether postprandial chylomicron metabolism was an adequate measure of efficacy, as this data was gleaned from only five patients. Recognizing the inherently limited patient data available for such a rare disease, UniQure was encouraged to apply a fourth time for reexamination of alipogene tiparvovec. This time, the CHMP recommended approval by a narrow majority, and the decision was ratified by the European Commission in November 2012 with market rollout of Glybera slated for 2014.
Conclusion
LPLD is a serious genetic disorder characterized by profound hypertriglyceridemia that can cause severe, recurring pancreatitis – even when patients follow severely fat-restricted diets. There are several reasons why LPLD is a good target for novel gene replacement therapies. First, there have traditionally been no effective treatments for the disorder, thereby necessitating the development of novel approaches to treat the disease. Additionally, animal models of LPLD are available which make development of novel therapeutics easier to test and study in vivo. Although the adenoviral-rescued mouse model does not reflect the human disease concerning pancreatitis, the LPLD feline model provides an excellent simulation for the disorder in man. Importantly, the main hallmark of LPLD – elevated TGs – provides a straightforward metric that can be monitored to assess the efficacy of novel therapeutics.
Although the approval of alipogene tiparvovec as the first gene therapy treatment in the West was an important moment in history, many questions still remain. Most important among them is why the serum TG-lowering effect is only temporary. Because long-term correction of hypertriglyceridemia was not observed, the drug was only approved for a small subset of LPLD patients with histories of severe chronic pancreatitis, and many experts still question whether alipogene tiparvovec is effective as a one-time treatment. Nevertheless, the fact that fasting TG levels rebound by 26 weeks after treatment certainly warrants further study. It is unlikely that the temporary effect is due to a mounted immune response, as improvement in duration was not improved with immunosuppressants. Nevertheless, recent advances in immunological monitoring at the cellular level may shed light as to why many AAV-mediated therapies may show only short-term efficacy.28 The temporary effects of alipogene tiparvovec suggest that the biology of long-term transgene introduction and expression may be more complex than originally thought, and this area of study may be important for other AAV-based therapeutics in development. Ultimate long-term correction of hypertriglyceridemia in LPLD patients may likely come from next-generation gene therapy approaches such as the introduction of induced pluripotent stem cells and LPL gene editing in muscle satellite stem cells. Last, the mechanism by which alipogene tiparvovec affects long-term postprandial chylomicron metabolism even after serum TG levels have rebounded merits further study.
Whether or not alipogene tiparvovec is truly effective at reducing incidence of pancreatitis – the indication for which the drug was approved – is another area of concern. Due to the inherently small size of the study populations, it is difficult to determine with confidence whether or not the drug reduces recurrence of this symptom. Although eight of 17 analyzed subjects in CT-AMT-011-03 were pancreatitis-free after injection of the drug, they had also been pancreatitis-free for an equal period of time before treatment.13,25 Equally important, three of the 17 subjects accounted for eleven of the 41 pretreatment pancreatitis incidents. The question of whether alipogene tiparvovec is an effective prophylactic for pancreatitis should be addressed eventually as new patients receive treatment and are monitored over longer periods of time.
Questions also remain about how the regulatory approval process for alipogene tiparvovec will impact future development of novel gene therapies. The drug provides an excellent example of a fundamental challenge that new gene therapies to treat orphan diseases will face in the coming years: small study populations. Demonstrating efficacy with clear statistical significance can be difficult for drugs developed to treat diseases that affect such small percentages of the general population. In this case, this difficulty was acknowledged by the EMA and was a major factor in the decision to approve the drug. Equally important is the rocky path to approval that alipogene tiparvovec faced. Multiple rounds of applications for approval proved very costly and eventually led to financial hardships from which AMT could not recover. Luckily, private investors (UniQure) were able to step in and provide a route by which Glybera could continue advancing through the regulatory process. Whether other companies will hesitate to submit novel gene therapies for fear of similar regulatory distress is unknown. Last, development costs for gene therapy treatments like Glybera are especially high. Glybera is expected to cost $1.6 million per treatment, making this novel therapeutic the most expensive drug in the world when it is rolled out in 2014.29 Reducing development costs will likely prove to be a major challenge for companies advancing future gene therapy treatments for orphan diseases. Alipogene tiparvovec has served as a valuable example for the way novel gene therapies will be developed and approved in the coming years, and this work will be extremely valuable to future investigators pursuing new avenues in the treatment of LPLD.
Disclosure
The authors report no conflicts of interest in this work. No sources of funding were used to assist in the preparation of this manuscript.
References
Wang H, Astarita G, Taussig MD, et al. Deficiency of lipoprotein lipase in neurons modifies the regulation of energy balance and leads to obesity. Cell Metab. 2011;13(1):105–113. | |
Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297(2):E271–E288. | |
Wang H, Eckel RH. What are lipoproteins doing in the brain? Trends Endocrinol Metab. Epub November 2, 2013. | |
Wang H, Eckel RH. Lipoprotein lipase in the brain and nervous system. Annu Rev Nutr. 2012;32:147–160. | |
Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43(12):1997–2006. | |
Gotoda T, Yamada N, Kawamura M, et al. Heterogeneous mutations in the human lipoprotein lipase gene in patients with familial lipoprotein lipase deficiency. J Clin Invest. 1991;88(6):1856–1864. | |
Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet. 2008;371(9607):143–152. | |
Kastelein JJ, Ross CJ, Hayden MR. From mutation identification to therapy: discovery and origins of the first approved gene therapy in the Western world. Hum Gene Ther. 2013;24(5):472–478. | |
Burnett JR, Hooper AJ. Alipogene tiparvovec, an adeno-associated virus encoding the Ser(447)X variant of the human lipoprotein lipase gene for the treatment of patients with lipoprotein lipase deficiency. Curr Opin Mol Ther. 2009;11(6):681–691. | |
Gaudet D, Méthot J, Déry S, et al. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther. 2013;20(4):361–369. | |
Gaudet D, de Wal J, Tremblay K, et al. Review of the clinical development of alipogene tiparvovec gene therapy for lipoprotein lipase deficiency. Atheroscler Suppl. 2010;11(1):55–60. | |
Bruno M. Gene therapy coming of age – prevention of acute pancreatitis in lipoprotein lipase deficiency through alipogene tiparvovec. Eur Gastroenterol Hepatol Rev. 2010;6(1):48–53. | |
Bryant LM, Christopher DM, Giles AR, et al. Lessons learned from the clinical development and market authorization of Glybera. Hum Gene Ther Clin Dev. 2013;24(2):55–64. | |
Excoffon KJ, Liu G, Miao L, et al. Correction of hypertriglyceridemia and impaired fat tolerance in lipoprotein lipase-deficient mice by adenovirus-mediated expression of human lipoprotein lipase. Arterioscler Thromb Vasc Biol. 1997;17(11):2532–2539. | |
Liu G, Ashbourne Excoffon KJ, Wilson JE, et al. Phenotypic correction of feline lipoprotein lipase deficiency by adenoviral gene transfer. Hum Gene Ther. 2000;11(1):21–32. | |
Ross CJ, Liu G, Kuivenhoven JA, et al. Complete rescue of lipoprotein lipase-deficient mice by somatic gene transfer of the naturally occurring LPLS447X beneficial mutation. Arterioscler Thromb Vasc Biol. 2005;25(10):2143–2150. | |
Rip J, Nierman MC, Ross CJ, et al. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol. 2006;26(6):1236–1245. | |
Ross CJ, Twisk J, Meulenberg JM, et al. Long-term correction of murine lipoprotein lipase deficiency with AAV1-mediated gene transfer of the naturally occurring LPL(S447X) beneficial mutation. Hum Gene Ther. 2004;15(9):906–919. | |
Schlaepfer IR, Eckel RH. Plasma triglyceride reduction in mice after direct injections of muscle-specific lipoprotein lipase DNA. Diabetes. 1999;48(1):223–227. | |
Rip J, Nierman MC, Sierts JA, et al. Gene therapy for lipoprotein lipase deficiency: working toward clinical application. Hum Gene Ther. 2005;16(11):1276–1286. | |
Ross CJ, Twisk J, Bakker AC, et al. Correction of feline lipoprotein lipase deficiency with adeno-associated virus serotype 1-mediated gene transfer of the lipoprotein lipase S447X beneficial mutation. Hum Gene Ther. 2006;17(5):487–499. | |
Mingozzi F, Meulenberg JJ, Hui DJ, et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood. 2009;114(10):2077–2086. | |
Carpentier AC, Frisch F, Labbé SM, et al. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab. 2012;97(5):1635–1644. | |
Gan SI, Edwards AL, Symonds CJ, Beck PL. Hypertriglyceridemia-induced pancreatitis: A case-based review. World J Gastroenterol. 2006;12(44):7197–7202. | |
European Medicines Agency. Assessment Report: Glybera. London: European Medicines Agency; 2012. | |
Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20(10):1831–1832. | |
Melchiorri D, Pani L, Gasparini P, et al. Regulatory evaluation of Glybera in Europe – two committees, one mission. Nat Rev Drug Discov. 2013;12(9):719. | |
Britten CM, Walter S, Janetzki S. Immunological monitoring to rationally guide AAV gene therapy. Front Immunol. 2013;4:273. | |
Whalen J. Gene-therapy approval marks major milestone. Wall Street Journal. November 2, 2012. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.