")
Back to Journals » Journal of Hepatocellular Carcinoma » Volume 8
An Overview of the Genomic Characterization of Hepatocellular Carcinoma
Received 14 May 2021
Accepted for publication 18 August 2021
Published 7 September 2021 Volume 2021:8 Pages 1077—1088
DOI https://doi.org/10.2147/JHC.S270533
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Ahmed Kaseb
Sun Young Yim,1 Ju-Seog Lee2
1Department of Internal Medicine, Korea University College of Medicine, Seoul, Korea; 2Department of Systems Biology, The University of Texas M. D. Anderson Cancer Center, Houston, TX, 77030, USA
Correspondence: Ju-Seog Lee
Department of Systems Biology, The University of Texas M. D. Anderson Cancer Center, 6565 MD Anderson Blvd., Houston, Tx, 77030, USA
Tel +1 713 834 6154
Fax +1 713 563 4235
Email [email protected]
Abstract: Tumor classifications based on alterations in the genome, epigenome, or proteome have revealed distinct tumor subgroups that are associated with clinical outcomes. Several landmark studies have demonstrated that such classifications can significantly improve patient outcomes by enabling tailoring of therapy to specific alterations in cancer cells. Since cancer cells accumulate numerous alterations in many cancer-related genes, it is a daunting task to find and confirm important cancer-promoting alterations as therapeutic targets or biomarkers that can predict clinical outcomes such as survival and response to treatments. To aid further advances, we provide here an overview of the current understanding of molecular and genomic subtypes of hepatocellular carcinoma (HCC). System-level integration of data from multiple studies and development of new technical platforms for analyzing patient samples hold great promise for the discovery of new targets for treatment and correlated biomarkers, leading to personalized medicine for treatment of HCC patients.
Keywords: gene expression profile, RNA-seq, proteomics, mutations, genomic subtypes
Introduction
The development of hepatocellular carcinoma (HCC) is a highly complex process involving the accumulation of epigenetic and genetic alterations in numerous genomic loci, leading to dramatic changes in signaling pathways. Adding further complexity, genetic aberrations of HCC show considerable heterogeneity derived from differences in ethnicity, etiology, and environmental exposures.1–4 The complex linkages between various risk factors and hepatocarcinogenesis were not well understood until the recent developments in genomic technologies that have enabled us to gain a systemic overview of genomic profiles, providing insight into the pathogenesis of HCC. Substantial efforts have been carried out to uncover molecular subtypes of HCC with differences in clinical outcomes.
HCC arises in the background of a fibrotic or cirrhotic liver following infection with hepatitis C or B viruses; it less commonly occurs with other conditions such as alcohol abuse, hemochromatosis, and non-alcoholic steatohepatitis.5,6 These conditions trigger a series of genetic and epigenetic alterations that lead to transformation of normal cells into pre-malignant cells.7 Rapid progress in the use of genomic and proteomic data for understanding HCC development at the molecular level has brought the promise of new clinical utility for treating HCC patients. In analyzing genomic and proteomic data, there are two approaches for discovering subtypes correlated with clinical characteristics.8 In a supervised approach, the data are arranged to uncover a set of variables, such as genetic alterations or expression of genes, that are significantly associated with clinical outcomes such as response to treatment, overall survival, and recurrence. An unsupervised approach seeks to discover new subtypes of HCC patients that have not previously been recognized. The ultimate goal of both of these approaches is to uncover the underlying biology of disease that is associated with clinical outcomes and identify reliable markers for classifying HCC patients for better management in the clinic.
While a wide range of options, including several multi-targeted tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs), can be offered for the treatment of HCC, not all patients benefit from these treatments, and response rates to these systemic treatments are typically lower than those observed in other cancer types.6,9,10 However, recent success in a clinical trial of a combination of a targeted drug (bevacizumab) and an ICI (atezolizumab) encourages us to continue seeking better combination treatments in future studies.6,11–13 Although the effects of this new combination treatment were very promising, many patients still did not respond,12 further emphasizing that the identification and validation of biomarkers or genetically homogenous subtypes that can help to identify better responders to treatment remain of critical importance.
Mutational Landscape of HCC
There have been many genomic studies for cataloguing driver genes activated in HCC (Table 1).14–23 The TERT gene encodes an enzymatic subunit of telomerase that is required for preserving telomere length and plays important roles in cancer.24,25 TERT expression is largely suppressed in most somatic cells, except for stem cells and regenerating hepatocytes.26,27 However, cancer cells express TERT stably, which is reactivated and essential for limitless growth of cancer cells.28–30 TERT is regulated transcriptionally, and the promoter region has several binding sites for transcription factors such as MYC and Sp1.31–35 Unlike other oncogenes, in which most mutations are in coding regions, the most frequent mutations in TERT were found in the promoter sequences,36–38 providing novel insight into the means of tumor-specific activation of TERT. In fact, more than 50% of HCC tumors have mutations in the TERT promoter, making them one of the most frequently mutated sites in HCC.21,22,39 Mutated sequences become a potential binding site for ETS/TCF transcription factors which can increase promoter activity and expression of TERT.
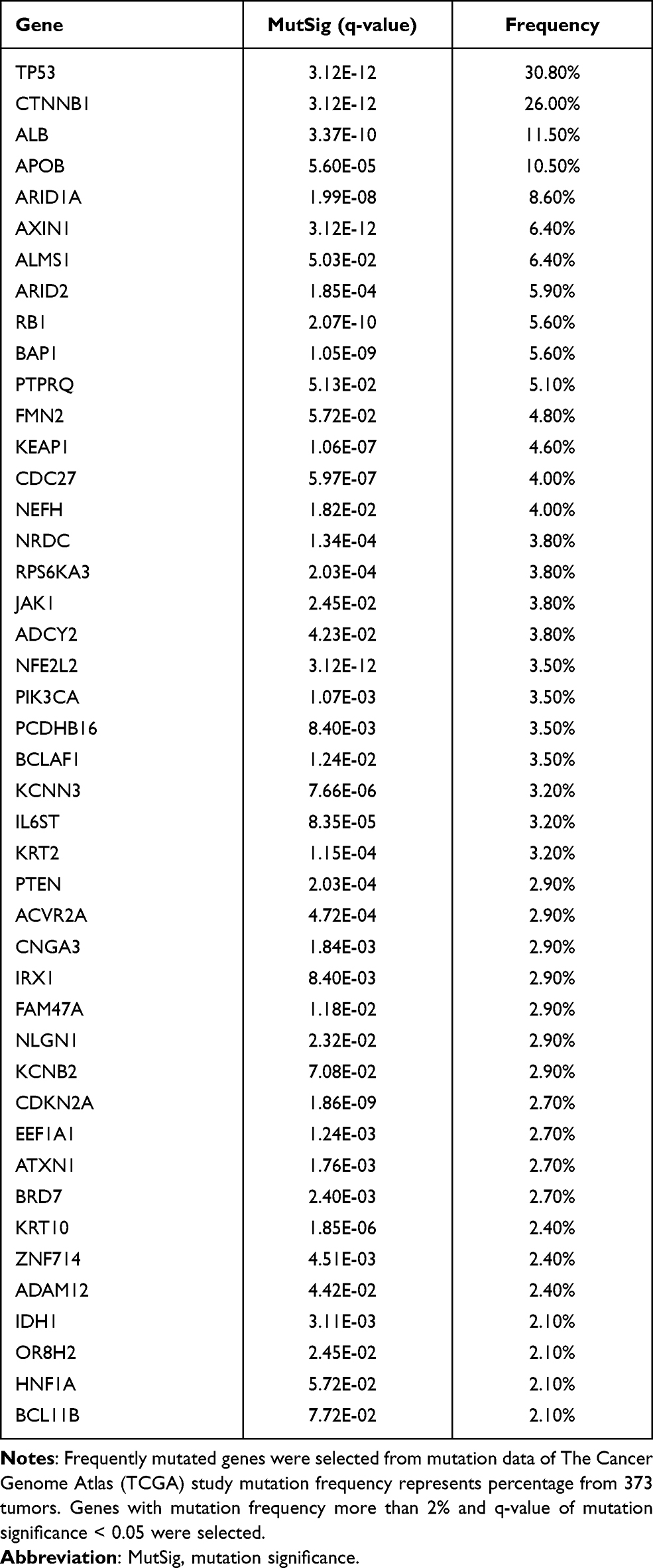 |
Table 1 Most Frequently Mutated Genes in HCC (from TCGA Study) |
Mutations in TP53 occur in more than 30% of HCC tumors.16,21–23,40,41 TP53 is best known as a tumor suppressor gene that, to preserve the genome integrity, can initiate apoptosis, cell-cycle arrest, senescence, and ferroptosis in response to cellular stresses such as genomic damage, activation of oncogenes, and hypoxia.42–45 TP53 can function both as an activator and a repressor of gene expression, controlling expression of a large number of genes involved in ferroptosis, apoptosis, cell cycle arrest, and senescence. However, recent studies showed that TP53 also regulates expression of genes playing key roles in stem cell maintenance, oxidative stress, autophagy, and cellular metabolism.46 The vast majority of TP53 mutations are located in its DNA-binding region, causing a weak interaction of TP53 with the promoters of target genes. However, some mutations are gain-of-function alterations that give rise to new molecular activity that are not seen with wild-type TP53, such as metastasis, angiogenesis, and resistance to standard therapies.46,47 As would be expected, mutations in TP53 are significantly correlated with poor survival of HCC patients.41
CTNNB1 (coding β-catenin) is a subunit of the cellular cadherin complex acting as a downstream effector of the WNT pathway.48,49 In the absence of WNT signaling, the intracellular level of β-catenin is low due to phosphorylation-dependent ubiquitination/degradation that is regulated by the APC-AXIN-CK1-GSK3 complex. When WNT signaling is active, stabilized β-catenin is translocated to the nucleus and forms a complex with TCF/LEF transcription factors to turn on the expression of genes regulating cell proliferation. Aberrant activation of β-catenin due to mutations has been observed in more than 20% of HCC tumors.21–23,50 Most mutations are located in or near phosphorylation sites,21–23,49–51 enabling β-catenin to escape phosphorylation-dependent degradation. Thus, mutated β-catenin is constitutively activated. These observations indicate that tumors with mutated β-catenin may represent a clinically different subtype of HCC. In addition to β-catenin, AXIN1 is another frequently mutated gene in the WNT pathway in HCC (occurring in 5–10% of tumors).16,20–23,52 Intriguingly, CTNNB1 mutations are most significantly associated with TERT mutations, but are almost mutually exclusive from mutations in TP53,16,21,39,53 suggesting a potential working interface among these three genes in HCC development.
Mutations in ARID1A and ARID2 are also frequently observed in HCC (~20% of tumors).14,20–22 Both genes are members of the ARID family.54 By interacting with several other proteins, they make DNA accessible to cellular complexes regulating DNA replication, DNA repair, and transcription. Mutations in ARID1A are also frequently observed in many other cancer types, such as gastric, esophageal, breast, gynecologic, prostate, bladder, and pancreatic cancers.35,54–58 The vast majority of mutations in ARID1A appear to be loss-of-function mutations, strongly indicating that ARID1A is a tumor suppressor. Intriguingly, Arid1a-knockout mice showed a surprising and complex phenotype: unexpectedly, HCC development was significantly reduced in these mice,59 indicating that Arid1a may play roles in transformation of hepatocytes. In contrast to the earlier stage of HCC development, in the later stage Arid1a knockout enhanced progression and metastasis of HCC tumors, also suggesting that the roles of Arid1a in HCC development are complicated and context dependent.59 Tumor-suppressive activity of ARID1A in the liver was further supported by recent analysis of genomic data from tumors. Inactivation of ARID1A was significantly correlated with shorter overall survival of patients and consistently related to poor-prognostic genomic subtypes such as high-recurrence subtypes, hepatic stem cell, highly proliferative, and inactivation of the Hippo pathway.60 Intriguingly, The Cancer Genome Atlas (TCGA) project showed that ARID1A and ARID2 mutations were mutually exclusive from TP53 mutations, suggesting that these genes may interact.22 However, more studies are needed to understand how TP53 interacts with ARID1A or whether TP53 can modulate ARID1A or ARID2 activity.
Reactive oxygen species (ROS) are typically generated by cellular metabolism and can cause mutations leading to the initiation of tumorigenesis by interacting with genomic DNA.61 NRF2 (NFE2L2), a member of the leucine zipper transcription factor family, is a major defense gene against cellular oxidative stress.62–64 In normal conditions, cellular NRF2 is maintained at a low level by KEAP1, which negatively regulates NRF2 by targeting it for E3 ligase–mediated ubiquitination and proteasomal degradation. Under oxidative conditions from metabolic stress, NRF2 is stabilized by inactivation of KEAP1 and translocated to the nucleus, in which it binds to the promoters of the downstream genes and increases their expression. Because NRF2 can prevent damage of genomic DNA and mutations, it was previously considered a tumor suppressor. However, recent studies showed that NRF2 can protect not only untransformed cells but also transformed cancer cells, resulting in protection of cancer cells from oxidative stress–mediated cell death.65–67 While KEAP1 and NRF2 are not highly mutated genes in HCC (3–5% of tumors),17,20–23 NRF2 is significantly amplified and overexpressed in HCC.68 Importantly, mutations in NRF2 are located in the KEAP1-binding sites, which are necessary for degradation of NRF2, showing that mutations make NRF2 constitutively active in HCC. In good agreement with this, several KEAP1 mutations cause loss of its interaction with NRF2, making NRF2 constitutively active. During tumor progression, high metabolic activity in cancer cells substantially increases the burden of oxidative stress that causes apoptotic cell death or growth arrest. Thus, activation of NRF2 would deliver great benefits to cancer cells under oxidative stress. Several studies showed that NRF2 activation is significantly associated with shorter survival as a result of high cellular level of cytoprotective proteins in cancer cells,66,69 further supporting the notion that NRF2 is an important gene for protecting cancer cells. Once cells are transformed, cancer cells steal the NRF2 system to decrease cellular stress and have an advantage in growth. In addition, NRF2 can translocate to the nucleus to form dimers with sMAF proteins and bind antioxidant response elements for the transcription of its target genes.70 Interestingly, a recent drug screening study showed that NRF2 activity is potentially regulated by the oncosuppressor GNMT.71 Taken together, these observations show that NRF2 is a main regulator of the antioxidative and detoxification responses and propels the progression of cancer, formation of metastases, and resistance to therapy.72
In addition to mutations in cancer-related genes, HCC tumors harbor mutations in serum proteins such as albumin (ALB) and apolipoprotein B (APOB), which are not frequently mutated in other cancer types. Because many mutations appear to confer loss of function,22,73 they may play roles in the development of HCC. However, the functional consequences of these mutations have not yet been examined. Interestingly, mutations in APOB were also discovered in familial hypobetalipoproteinemia (FHBL) with reduce level of low-density lipoprotein cholesterol in plasma.74 High incidence of hepatic steatosis, liver cirrhosis, and hepatocarcinoma have been observed in individuals with FHBL attributed to mutations in APOB.75 In a recent study from our lab, we carried out an analysis with systematic integration of genomic data from mouse models and human HCC tumors and found that APOB ablation in HCC is associated with poor survival of HCC patients.76 Interestingly, silencing of APOB expression with siRNAs significantly increased cell growth in multiple HCC cells, indicating that APOB may have weak tumor-suppressive activity. However, the underlying biology of APOB-mediated growth suppression is currently unknown.
Genomic Subtypes of HCC with Prognostic Association
Recurrence of HCC is a very serious problem after curative removal of the primary HCC and occurs in around half of cases within 3 years after the resection.77 Many studies have used supervised or unsupervised approaches to genomic data to identify genes or subgroups associated with recurrence or overall survival.
Iizuka et al analyzed gene profiling data from a small number of HCC tumors as a training set; they identified 12 recurrence-associated genes in a supervised learning manner and generated a prediction model.78 When the prediction model was tested in an independent cohort, it identified 93% of patients with early recurrence. This was the first study that demonstrated the prognostic association of genomic markers in HCC. Similarly, Kurokawa et al identified genes associated with recurrence by analyzing gene expression in HCC tumors from 100 patients.79 Their trained model with 20 genes also successfully identified patients with early recurrence in an independent test cohort.
By applying a supervised machine-learning algorithm, Ye et al identified 153 genes significantly associated with intrahepatic metastasis of HCC.80 They showed that primary tumors with metastatic nodules had gene expression patterns similar to those of their surrounding metastatic tumors, suggesting that activation of genes favoring invasion began in the primary HCC tumors. In a functional validation experiment, an antibody specific to osteopontin, a leading gene product in the signature, successfully inhibited invasion of HCC cells and suppressed metastasis of HCC tumors in a mouse model. This genomic metastasis predictor was further validated in an independent cohort.81 In a Cox analysis that included clinical features, the metastasis signature was a significant prognostic indicator, particularly for early recurrence after treatment, but not for late recurrence, which is mainly attributed to the field effect in remnant livers.
Another study applied a similar approach to selected genes associated with early recurrence after curative treatment.82 Around 600 genes were selected from hepatitis B–associated HCC tumors, and their prognostic significance was tested in a validation cohort of HCC patients. Network mapping analysis with selected genes discovered that transcription factor SP1 is a potential upstream regulator of genes correlated with high risk of early recurrence.
By using an unbiased approach for genomic data from primary tumors, investigators at the National Cancer Institute (NCI) identified two subtypes that are distinctive in terms of overall survival of the patients.83 Their statistical analysis yielded 947 genes correlated with overall survival. As gene set enrichment analysis of selected genes revealed that proliferation of cancer cells is most characteristic of a poor-prognostic subtype, it was named the NCI Proliferation (NCIP) signature. The poor-prognostic subtype showed elevated expression of genes related to histone modification and ubiquitination. The robustness of the signature in identifying patients with poor prognosis was further validated in multiple studies.84–86
As part of a follow-up study of the NCIP signature, Kim et al identified a small number of genes that were significantly correlated with patients’ prognosis by using two previously discovered prognostic signatures.85 This meta-analysis selected 65 genes that were conserved in both signatures.82,83 The authors developed recurrence-risk scoring with a dynamic range from 0 to 100 for better translation of the signature to the clinic. In validation cohorts, the score was a highly significant indicator of both overall survival and recurrence-free survival. Gene network analysis with integration of mutation data revealed that mutations in β-catenin were significantly associated with low risk scores and activations of the AKT and IGF1R pathways were significantly associated with high risk scores.
Vascular invasion is significantly correlated with prognosis, as reflected in high recurrence after treatment.87 A large-scale analysis with >1000 cases clearly demonstrated that hepatic vascular invasion should be a critical criterion in selecting patients for curative treatment.88 Minguez et al identified 35 genes whose expression is significantly correlated with hepatic vascular invasion and developed a model that can predict HCC patients with vascular invasion.89 One of the best correlated genes is CD24, which is expressed in endothelial cells.90 Interestingly, metabolic genes were negatively associated with vascular invasion.
Systematic analysis of HCC genomic data identified five genes (RAMP3, TAF9, KRT19, HN1, and RAN) with strong prognostic association and developed predictive scoring.91 Because these five genes well reflect dysregulation of key signaling pathways in HCC, the authors proposed that the new score can be used for improving selection of patients for liver transplantation by extending the Milan criteria for better-prognostic tumors even if the size is larger than 5 cm.92
Genomic Subtypes of HCC Associated with Hepatic Stem Cell Features
The HCC subtype with hepatic stem cell features was discovered by co-analysis of genomic data from primary HCC and fetal liver stem cells.86 Intriguingly, this subtype is a subset of the previously discovered high-proliferation subtypes in the NCIP classification. Informatics analysis of the signature predicted that JUNB, FOS, and FOSL2 are highly active in the hepatic stem cell subtype. High similarity of the signature to stem cells indicates that this subtype might be derived from stem or progenitor cells in the liver. In agreement with this notion, expression of marker genes for hepatic progenitor cells, such as VIM, KRT19, and KRT7, is highly elevated in this subtype.93
EpCAM is expressed predominantly in hepatic stem or progenitor cells.94,95 In an effort to discover a subgroup of HCC tumors with hepatic stem cell features, investigators selected EpCAM-positive HCC and identified 70 genes whose expression correlated with EpCAM.96 With use of the EpCAM signature reflecting different hepatic cell lineages, authors suggested classifying HCC tumors into four subtypes: EpCAM+/AFP+ (hepatic stem cell–like), EpCAM+/AFP– (bile duct epithelium–like), EpCAM–/AFP+ (hepatocytic progenitor–like), and EpCAM–/AFP– (mature hepatocyte–like). Expression of marker genes for hepatic stem cells, such as KIT and KRT19, is elevated in hepatic stem cell–like HCC. In contrast, expression of marker genes for mature hepatocytes, such as CYP3A4, is higher in mature hepatocyte–like HCC. Not surprisingly, EpCAM+ tumors are highly invasive, and EpCAM increased motility and invasion of HCC cells in functional assays.97
By analyzing genomic data from HCC and cholangiocarcinoma tumors together, Woo et al discovered that a fraction of HCC tumors shared gene expression patterns specific to cholangiocarcinoma and called the new subtype cholangiocarcinoma-like HCC (CLHCC).98 The most distinct features of these tumors are elevated expression of hepatic progenitor cell markers such as PROM1, EpCAM, and KRT19. Not surprisingly, validation of the CLHCC subtype in several independent cohorts showed that the subtype is significantly associated with poor survival of patients. Comparison of the signature with other studies showed that it is highly similar to multiple embryonic stem cell signatures and the previously discovered hepatic stem cell signature.86,99
Another group of investigators used a similar approach for finding genes correlated with hepatic progenitor or stem cells and developed a stem cell predictive signature.100 The signature was then tested in independent cohorts by using real-time RT-PCR with RNA from tumor tissues first and later by immunostaining paraffin-embedded tissues.
Genomic Subtypes of HCC Associated with Underlying Liver Biology
Hoshida et al found in a systemic review of multiple genomic studies that three subtypes of HCC tumors–namely, S1, S2, and S3—could be identified. They showed substantial association with conventional clinical parameters such as blood AFP level, cellular differentiation status, and number of tumors.101 Subtypes S1 and S2 are associated with poor prognosis, and subtype S3 is associated with better prognosis, as reflected in smaller and more differentiated tumors and preserved liver function. Gene set analysis showed that the TGF-β pathway is activated in subtype S1, while the WNT/β-catenin pathway is activated in subtype S3. Most interestingly, subtype S2 is associated with characteristics of hepatic stem cells as reflected by high expression of hepatic stem cell markers such as AFP, EPCAM, and GPC3. Moreover, subtype S2 is strongly associated with a vascular invasion signature and hepatoblastoma-like signature.89,96,97
Six genomic subtypes of HCC with distinct characteristics of genetic alterations–namely, G1 to G6–were uncovered by an unsupervised approach.102 Subtypes G1, G2, and G3 were associated with low chromosomal stability compared to the rest of the subtypes. CTNNB1 is the most frequently mutated gene in subtypes G5 and G6, while TP53 is the most frequently mutated gene in subtypes G2 and G3. PIK3CA is the most frequently mutated gene in subtype G2. Promoter regions of CDKN2A and CDH1 are most frequently methylated in subtypes G3, G5, and G6.
Co-analysis of gene expression data with genomic copy number alteration from the same tumors revealed five subtypes: namely, poly 7, proliferation, INF, CTNNB1, and unannotated subtypes.103 Subtype Poly 7 showed unique features: chromosome 7 polysomy and the concomitant high expression of genes on that chromosome. Interestingly, these tumors are further characterized by lack of chromosome 8q amplification, which is one of the most frequent gains of chromosomes in HCC.104,105 A subtype with activated β-catenin was characterized by activation mutations in CTNNB1. IGF-IR and RPS6 phosphorylation is enriched in the subtype with proliferation features. Interestingly, tumors in the INF-related subtype are smaller than those of other subtypes.
Analysis with pooled genomic data from HCC tumors revealed four molecular subtypes that are well correlated with liver zonation: namely, the perivenous (PV) subtype, periportal (PP) subtype, stem cell (STEM) subtype, and extracellular matrix (ECM) subtype.106 Subtype PV is associated with frequent mutations in CTNNB1 and shows elevated expression of genes typical for hepatic zone 3 such as HAL, VNN1, and GLUL. Similarly, tumors in subtype PP express genes typical for hepatic zone 1, such as GLS2 and ARG1. The STEM subtype is highly similar to the previously discovered hepatic stem cell subtype.86 The ECM subtype is characterized by high expression of genes involved in extracellular matrix remodeling and epithelial-mesenchymal transition.
Recent analysis of TCGA HCC genome data uncovered interesting subtypes that were not previously recognized. They discovered that tumors with IDH1 and IDH2 mutations have a highly distinct gene signature.22 Intriguingly, a substantial fraction of tumors lacking IDH mutations have a gene signature similar to those with IDH mutations. When directly compared with previously identified genomic subtypes, the IDH-like subtype showed the highest resemblance to the poor-prognostic hepatic stem cell subtype.86 Likewise, these tumors exhibited resemblance to the S2 subtype of Hoshida’s classification101 and the CLHCC subtype,98 and had very high scores using the 65-gene-based recurrence risk score classifier.85 Unlike previous studies, they used the iCluster strategy, which uses most of the collected genomic data, including mRNA expression, mutations, miRNA expression, copy number alteration, and DNA methylation data.107 This new approach uncovered three subtypes: namely, iC1, iC2, and iC3.22 Subtype iC1 is associated with female sex, Asian ethnicity, and younger age. These tumors also show poor-prognostic characteristics, such as macrovascular invasion and higher tumor grade. Moreover, subtype iC1 is associated with low frequency of CTNNB1 mutations, CDKN2A silencing, and mutations in the TERT promoter. Unlike iC1, subtypes iC2 and iC3 are high in CTNNB1 mutations, CDKN2A silencing, HNF1A mutation, and TERT promoter mutations. Subtype iC2 is correlated with clinical variables such as less microvascular invasion and low-grade tumors. Subtype iC3 is associated with higher mutations of TP53, low chromosomal stability, and under-methylation. In comparison with Hoshida’s classification, iC1 is highly similar to subtype S2, whereas iC3 is highly similar to subtype S3.
Proteomic Subtype
Although genomic approaches uncovered many HCC subtypes with significant association with clinical features and unique underlying biology at the molecular level, these studies are limited by lack of insight on protein activity that is the functional end product of all genetic elements coupled with phenotypes. Moreover, post-translational modifications of gene products such as ubiquitination, phosphorylation, and methylation that are critical in regulating cellular activity cannot be faithfully represented solely by profiling the genome.
By applying an unsupervised approach to HCC proteomic data generated with use of mass spectrometry, a recent study uncovered three proteomic subtypes: namely, subtypes S-I, S-II, and S-III.23 Subtype S-I is characterized by high expression of metabolism-related proteins typical for retaining liver function. Not surprisingly, among the three proteomic subtypes, S-I has the best prognosis. As seen in the genomic subtype with good prognosis, tumors with subtype S-I showed very high genome stability. Subtype S-III is characterized by increased expression of proteins regulating cell proliferation, such as MKI-67, PCNA, TOP2A, and PARP1, and is associated with poor prognostic clinical features such as larger tumor size, tumor thrombus, and advanced tumor stage. In agreement with the expression patterns of proliferation proteins, patients with subtype S-III showed the poorest survival after surgery. Unlike S-I and S-III, subtype S-II lacks a specific proteomic signature, as reflected in having an intermediate expression pattern of metabolic and proliferative proteins. However, S-II is characterized by downregulation of immunity-related proteins such as CD4, ITGB3, and CD8A.
Not surprisingly, some of the proteomic subtypes were subsets of previously recognized genomic subtypes (Figure 1). Subtype S-III is highly similar to high-proliferation subtype of the NCIP classification, while S-I and S-II are subsets of low-proliferation subtype. Likewise, subtype S-III is similar to EpCAM-high HCC tumors, while S-I and S-II are subsets of EpCAM-low tumors. Interestingly, subtype S-III is split into S1 and S2 of Hoshida’s classification, indicating that genomic data can help to identify subsets of proteomic subtypes. More interestingly, no proteomic subtype showed similarity to the TCGA iC3 subtype, indicating that the biological features in subtype iC3 may not be well reflected in proteomic data. Together, these relationships suggest that no single-platform data set can fully represent clinical and biological features of HCC tumors. As data from both platforms are complementary to each other, we should analyze them together to achieve a full spectrum of tumor features.
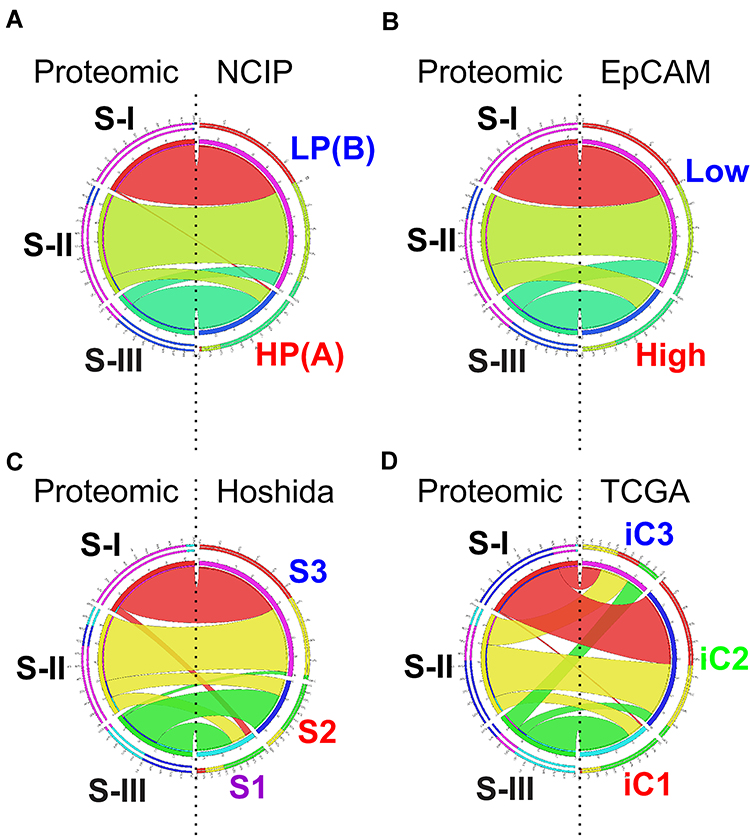 |
Figure 1 Circos plots of the relationship of proteomic subtypes with genomic subtypes in Zhongshan HCC cohort. (A) Comparison of proteomic subtypes with NCIP subtype. (B) Comparison of proteomic subtypes with EpCAM subtype. (C) Comparison of proteomic subtypes with Hoshida subtype. (D) Comparison of proteomic subtypes with TCGA subtype. Circular ideogram of pairwise comparison of two classifications. Proteomic subtypes are displayed in left and matched subtypes from previous study are displayed in right. Ribbons indicate matched samples between two classifications. Graphic is built using Circos (http://circos.ca/). Abbreviations: NCIP, National Cancer Institute Proliferation; HP (also known as (A)), high proliferation; LP (also known as (B)), low proliferation; EpCAM, Epithelial Cell Adhesion Molecule subtype; Hoshida, Hoshida classification; TCGA, The Cancer Genome Atlas. |
Summary and Discussion
Genomic, epigenomic, and proteomic analyses of HCC tumors have discovered clinically distinct molecular subtypes, characteristic molecular changes correlated with subtypes, and prediction models that can identify patients in each subtype. These new analyses have begun to advance our understanding of oncogenesis as well as clinical decision-making for treatment of patients.
Although some of these findings are very promising, there are considerable barriers in moving discovered subtypes to clinical use. Even though these genomic or proteomic subtypes are significantly associated with clinical outcomes, their clinical usefulness is hindered by lack of concordance, which is possibly because of difference in classification algorithms, etiology of patients, collection and storage of samples, and technological platforms used. However, several subtypes have been rediscovered multiple times. For example, the hepatic stem cell subtype is a subset of the high-proliferation NCIP subtype and similar to EpCAM+ tumors, IDH-like HCC, and the CLHCC subtype.22 The S3 subtype in Hoshida’s classification is highly similar to the CTNNB1 subtype from Barcelona’s classification and the PV subtype.103 Despite substantial differences among molecular classifications, similarities among subsets of tumors strongly indicate that it is possible to uncover good consensus from independently defined classification of HCC tumors.
A major limitation of these subtypes is lack of clinically useful knowledge that is necessary for treating HCC patients. For example, although sorafenib has been used for treatment of HCC patients for a long time,108,109 no markers and subtypes have yet been found to be useful in predicting response to sorafenib treatment. Similarly, many other treatments for HCC also lack predictive markers associated with molecular subtypes. The development of antibodies blocking ICIs such as CTLA-4 or PD-1/PD-L1 represents a major breakthrough for treatment of cancer patients.110 While the response rates for ICIs or their combination with targeted drugs for HCC are significantly better than response rates for standard systemic treatment,11–13 not all patients benefit from these treatments, and those with greatest potential benefit from such treatments are currently unknown. Therefore, there is an urgent need for identifying biomarkers or molecular subtypes that can help to predict response or resistance to ICI therapy or related immunotherapies.
There are several issues that need to be addressed in the near future to fill our knowledge gap in optimizing currently available treatments for HCC patients. First, molecular subtypes that are homogeneous in underlying biology as well as having significant association with clinical outcomes need to be clearly defined. Although many different genomic or molecular subtypes have been discovered from previous studies, they lack consensus, and robustness of subtypes has yet to be properly tested and validated. Integration and co-analysis of discovered subtypes together might be a first step for finding subtypes with good consensus that can be later validated in pooled large data sets from previous studies. Second, biomarkers tightly associated with subtypes or durable response to standard treatments should be validated in prospective cohorts. As HCC is a heterogeneous disease, it is unlikely that a single biomarker will be good enough to identify patients with durable response. Therefore, non-redundant multiple biomarkers such as a mutation profile from cell-free DNA, serum markers, or metabolites need to be analyzed together and validated in prospective cohorts of patients. Finally, finding the best preclinical models that mimic the underlying biology of particular subtypes would be necessary for rational preclinical studies evaluating new treatments in the future.
Funding
This study was supported by National Cancer Institute grant R01-CA237327, P50-CA217674 (HCC SPORE), P30-CA016672 (Cancer Center Support Grant), the Duncan Cancer Prevention Research Seed Funding Program at MD Anderson Cancer Center, the MD Anderson Sister Institute Network Fund, and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07040523).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Lee JS, Thorgeirsson SS. Genetic profiling of human hepatocellular carcinoma. Semin Liver Dis. 2005;25:125–132. doi:10.1055/s-2005-871192
2. Lee JS. The mutational landscape of hepatocellular carcinoma. Clin Mol Hepatol. 2015;21:220–229. doi:10.3350/cmh.2015.21.3.220
3. Yim SY, Lee JS. Genomic perspective on mouse liver cancer models. Cancers. 2019;11:1648. doi:10.3390/cancers11111648
4. Caruso S, O’Brien DR, Cleary SP, et al. Genetics of HCC: novel approaches to explore molecular diversity. Hepatology. 2020;2:458.
5. Yang JD, Hainaut P, Gores GJ, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
6. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7:6.
7. Ding X, He M, Chan AWH, et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology. 2019;157:1630–1645.e1636. doi:10.1053/j.gastro.2019.09.005
8. Libbrecht MW, Noble WS. Machine learning applications in genetics and genomics. Nat Rev Genet. 2015;16(6):321–332. doi:10.1038/nrg3920
9. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–1462. doi:10.1056/NEJMra1713263
10. Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: looking outside the box. J Hepatol. 2020;72:342–352. doi:10.1016/j.jhep.2019.09.010
11. Rizzo A, Ricci AD, Brandi G. Atezolizumab in advanced hepatocellular carcinoma: good things come to those who wait. Immunotherapy. 2021;13:637–644. doi:10.2217/imt-2021-0026
12. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382:1894–1905. doi:10.1056/NEJMoa1915745
13. Rizzo A, Ricci AD, Brandi G. Immune-based combinations for advanced hepatocellular carcinoma: shaping the direction of first-line therapy. Future Oncol. 2021;17:755–757. doi:10.2217/fon-2020-0986
14. Li M, Zhao H, Zhang X, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–829. doi:10.1038/ng.903
15. Totoki Y, Tatsuno K, Yamamoto S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011;43:464–469. doi:10.1038/ng.804
16. Ahn SM, Jang SJ, Shim JH, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972–1982. doi:10.1002/hep.27198
17. Cleary SP, Jeck WR, Zhao X, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–1702. doi:10.1002/hep.26540
18. Jhunjhunwala S, Jiang Z, Stawiski EW, et al. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol. 2014;15:436–0436.
19. Kan Z, Zheng H, Liu X, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi:10.1101/gr.154492.113
20. Schulze K, Imbeaud S, Letouze E, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi:10.1038/ng.3252
21. Totoki Y, Tatsuno K, Covington KR, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi:10.1038/ng.3126
22. Ally A, Balasundaram M, Carlsen R; The.Cancer.Genome.Atlas.Research.Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–1341. doi:10.1016/j.cell.2017.05.046
23. Gao Q, Zhu H, Dong L, et al. Integrated proteogenomic characterization of HBV-Related hepatocellular carcinoma. Cell. 2019;179:561–577.e522. doi:10.1016/j.cell.2019.08.052
24. Bryan TM, Sperger JM, Chapman KB, et al. Telomerase reverse transcriptase genes identified in Tetrahymena thermophila and Oxytricha trifallax. Proc Natl Acad Sci U S A. 1998;95:8479–8484. doi:10.1073/pnas.95.15.8479
25. Smogorzewska A, de Lange T. Regulation of telomerase by telomeric proteins. Annu Rev Biochem. 2004;73:177–208. doi:10.1146/annurev.biochem.73.071403.160049
26. Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–622. doi:10.1038/nrg1656
27. Lin S, Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018;556:244–248. doi:10.1038/s41586-018-0004-7
28. Harley CB. Telomerase and cancer therapeutics. Nat Rev Cancer. 2008;8:167–179. doi:10.1038/nrc2275
29. Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi:10.1126/science.7605428
30. Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33:787–791. doi:10.1016/S0959-8049(97)00062-2
31. Cong YS, Wen J, Bacchetti S. The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet. 1999;8:137–142. doi:10.1093/hmg/8.1.137
32. Horikawa I, Cable PL, Afshari C, et al. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999;59:826–830.
33. Takakura M, Kyo S, Kanaya T, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res. 1999;59:551–557.
34. Kyo S, Takakura M, Taira T, et al. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT). Nucleic Acids Res. 2000;28:669–677. doi:10.1093/nar/28.3.669
35. Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi:10.1158/2159-8290.CD-12-0361
36. Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi:10.1126/science.1230062
37. Huang FW, Hodis E, Xu MJ, et al. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. doi:10.1126/science.1229259
38. Killela PJ, Reitman ZJ, Jiao Y, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110:6021–6026. doi:10.1073/pnas.1303607110
39. Nault JC, Mallet M, Pilati C, et al. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi:10.1038/ncomms3218
40. Meng X, Franklin DA, Dong J, et al. MDM2-p53 pathway in hepatocellular carcinoma. Cancer Res. 2014;74:7161–7167. doi:10.1158/0008-5472.CAN-14-1446
41. Woo HG, Wang XW, Budhu A, et al. Association of TP53 mutations with stem cell-like gene expression and survival of patients with hepatocellular carcinoma. Gastroenterology. 2011;140:1063–1070. doi:10.1053/j.gastro.2010.11.034
42. Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–1286. doi:10.1101/gad.190678.112
43. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014;14:359–370. doi:10.1038/nrc3711
44. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–317. doi:10.1016/j.ccr.2014.01.021
45. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi:10.1038/nature14344
46. Hager KM, Gu W. Understanding the non-canonical pathways involved in p53-mediated tumor suppression. Carcinogenesis. 2014;35:740–746. doi:10.1093/carcin/bgt487
47. Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov. 2014;4:405–414.
48. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192–1205. doi:10.1016/j.cell.2012.05.012
49. Monga SP. Beta-catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology. 2015;148:1294–1310. doi:10.1053/j.gastro.2015.02.056
50. Guichard C, Amaddeo G, Imbeaud S, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi:10.1038/ng.2256
51. Takigawa Y, Brown AM. Wnt signaling in liver cancer. Curr Drug Targets. 2008;9:1013–1024. doi:10.2174/138945008786786127
52. Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000;24:245–250. doi:10.1038/73448
53. Pilati C, Letouze E, Nault JC, et al. Genomic profiling of hepatocellular adenomas reveals recurrent FRK-activating mutations and the mechanisms of malignant transformation. Cancer Cell. 2014;25:428–441. doi:10.1016/j.ccr.2014.03.005
54. Lin C, Song W, Bi X, et al. Recent advances in the ARID family: focusing on roles in human cancer. Onco Targets Ther. 2014;7:315–324.
55. Guan B, Mao TL, Panuganti PK, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011;35:625–632. doi:10.1097/PAS.0b013e318212782a
56. Jones S, Wang TL, Shih I, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi:10.1126/science.1196333
57. Wiegand KC, Lee AF, Al-Agha OM, et al. Loss of BAF250a (ARID1A) is frequent in high-grade endometrial carcinomas. J Pathol. 2011;224:328–333. doi:10.1002/path.2911
58. Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi:10.1056/NEJMoa1008433
59. Sun X, Wang SC, Wei Y, et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell. 2017;32:574–589.e576. doi:10.1016/j.ccell.2017.10.007
60. Yim SY, Kang SH, Shin JH, et al. Low ARID1A expression is associated with poor prognosis in hepatocellular carcinoma. Cells. 2020;1:9.
61. Cadenas E. Biochemistry of oxygen toxicity. Annu Rev Biochem. 1989;58:
62. Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi:10.1146/annurev.pharmtox.46.120604.141046
63. Moi P, Chan K, Asunis I, et al. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930. doi:10.1073/pnas.91.21.9926
64. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564–571. doi:10.1038/nrc3278
65. Padmanabhan B, Tong KI, Ohta T, et al. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol Cell. 2006;21:689–700. doi:10.1016/j.molcel.2006.01.013
66. Shibata T, Kokubu A, Gotoh M, et al. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology. 2008;135:1358–1368, 1368. doi:10.1053/j.gastro.2008.06.082
67. Wang XJ, Sun Z, Villeneuve NF, et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235–1243. doi:10.1093/carcin/bgn095
68. Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43.
69. Solis LM, Behrens C, Dong W, et al. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res. 2010;16:3743–3753. doi:10.1158/1078-0432.CCR-09-3352
70. Carvajal-Yepes M, Himmelsbach K, Schaedler S, et al. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J Biol Chem. 2011;286:8941–8951. doi:10.1074/jbc.M110.186684
71. Wu HC, Cheng MJ, Yen CH, et al. Chemical constituents with GNMT-promoter-enhancing and NRF2-reduction activities from Taiwan Agarwood Excoecaria formosana. Molecules. 2020;2:25.
72. Yen CH, Hsiao HH. NRF2 is one of the players involved in bone marrow mediated drug resistance in multiple myeloma. Int J Mol Sci. 2018;4:19.
73. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi:10.1158/2159-8290.CD-12-0095
74. Welty FK. Hypobetalipoproteinemia and abetalipoproteinemia. Curr Opin Lipidol. 2014;25:161–168. doi:10.1097/MOL.0000000000000072
75. Buryska S, Ahn JC, Allen A, et al. Familial hypobetalipoproteinemia: an underrecognized cause of lean NASH. Hepatology. 2021. doi:10.1002/hep.31988
76. Lee G, Jeong YS, Kim DW, et al. Clinical significance of APOB inactivation in hepatocellular carcinoma. Exp Mol Med. 2018;50:1–12. doi:10.1038/s12276-018-0187-x
77. Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379(9822):1245–1255. doi:10.1016/S0140-6736(11)61347-0
78. Iizuka N, Oka M, Yamada-Okabe H. Oligonucleotide microarray for prediction of early intrahepatic recurrence of. Lancet. 2003;361:923–929. doi:10.1016/S0140-6736(03)12775-4
79. Kurokawa Y, Matoba R, Takemasa I. Molecular-based prediction of early recurrence in hepatocellular carcinoma. J Hepatol. 2004;41(2):284–291. doi:10.1016/j.jhep.2004.04.031
80. Ye QH, Qin L-X, Forgues M. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using. Nat Med. 2003;9:416–423. doi:10.1038/nm843
81. Roessler S, Jia H-L, Budhu A. A unique metastasis gene signature enables prediction of tumor relapse in. Cancer Res. 2010;70:10202–10212. doi:10.1158/0008-5472.CAN-10-2607
82. Woo HG, Park ES, Cheon JH. Gene expression-based recurrence prediction of hepatitis B virus-related human. Clin Cancer Res. 2008;14:2056–2064. doi:10.1158/1078-0432.CCR-07-1473
83. Lee JS, Chu IS, Heo J, et al. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatology. 2004;40:667–676. doi:10.1002/hep.20375
84. Kim JH, Sohn BH, Lee HS, et al. Genomic predictors for recurrence patterns of hepatocellular carcinoma: model derivation and validation. PLoS Med. 2014;11:e1001770. doi:10.1371/journal.pmed.1001770
85. Kim SM, Leem SH, Chu IS, et al. Sixty-five gene-based risk score classifier predicts overall survival in hepatocellular carcinoma. Hepatology. 2012;55:1443–1452. doi:10.1002/hep.24813
86. Lee JS, Heo J, Libbrecht L, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi:10.1038/nm1377
87. Llovet JM, Di Bisceglie AM, Bruix J. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(10):698–711. doi:10.1093/jnci/djn134
88. Mazzaferro V, Llovet JM, Miceli R. Predicting survival after liver transplantation in patients with hepatocellular. Lancet Oncol. 2009;10:35–43. doi:10.1016/S1470-2045(08)70284-5
89. Minguez B, Hoshida Y, Villanueva A. Gene-expression signature of vascular invasion in hepatocellular carcinoma. J Hepatol. 2011;55(6):1325–1331. doi:10.1016/j.jhep.2011.02.034
90. Sagiv E, Memeo L, Karin A. CD24 is a new oncogene, early at the multistep process of colorectal cancer. Gastroenterology. 2006;131:630–639. doi:10.1053/j.gastro.2006.04.028
91. Nault JC, De Reyniès A, Villanueva A. A hepatocellular carcinoma 5-gene score associated with survival of patients. Gastroenterology. 2013;145:176–187. doi:10.1053/j.gastro.2013.03.051
92. Clavien PA, Lesurtel M, Bossuyt PM, Gores GJ, Langer B, Perrier A. Recommendations for liver transplantation for hepatocellular carcinoma: an. Lancet Oncol. 2012;13:e11–22LID. doi:10.1016/S1470-2045(11)70175-9
93. Roskams TA. Progenitor cells in diseased human liver. Semin Liver Dis. 2003;23:385–396.
94. Schmelzer E, Wauthier E, Reid LM. The phenotypes of pluripotent human hepatic progenitors. Stem Cells. 2006;24(8):1852–1858. doi:10.1634/stemcells.2006-0036
95. Schmelzer E, Zhang L, Bruce A. Human hepatic stem cells from fetal and postnatal donors. J Exp Med. 2007;204(8):1973–1987. doi:10.1084/jem.20061603
96. Yamashita T, Forgues M, Wang W. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of. Cancer Res. 2008;68:1451–1461. doi:10.1158/0008-5472.CAN-07-6013
97. Yamashita T, Ji J, Budhu A, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012–1024. doi:10.1053/j.gastro.2008.12.004
98. Woo HG, Lee J-H, Yoon J-H. Identification of a cholangiocarcinoma-like gene expression trait in. Cancer Res. 2010;70:3034–3041. doi:10.1158/0008-5472.CAN-09-2823
99. Ben-Porath I, Thomson MW, Carey VJ. An embryonic stem cell-like gene expression signature in poorly differentiated. Nat Genet. 2008;40:499–507. doi:10.1038/ng.127
100. Yang XR, Xu Y, Yu B. High expression levels of putative hepatic stem/progenitor cell biomarkers. Gut. 2010;59:953–962. doi:10.1136/gut.2008.176271
101. Hoshida Y, Nijman SM, Kobayashi M, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009;69(18):7385–7392. doi:10.1158/0008-5472.CAN-09-1089
102. Boyault S, Rickman DS, de Reyniès A. Transcriptome classification of HCC is related to gene alterations and to new. Hepatology. 2007;45:42–52. doi:10.1002/hep.21467
103. Chiang DY, Villanueva A, Hoshida Y. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi:10.1158/0008-5472.CAN-08-0742
104. Patil MA, Gütgemann I, Zhang J. Array-based comparative genomic hybridization reveals recurrent chromosomal. Carcinogenesis. 2005;26:2050–2057. doi:10.1093/carcin/bgi178
105. Okamoto H. PTK2 and EIF3S3 genes may be amplification targets at 8q23-q24 and are associated. Hepatology. 2003;38:1242–1249. doi:10.1053/jhep.2003.50457
106. Desert R, Rohart F, Canal F, et al. Human hepatocellular carcinomas with a periportal phenotype have the lowest. Hepatology. 2017;66:1502–1518. doi:10.1002/hep.29254
107. Shen R, Olshen AB, Ladanyi M. Integrative clustering of multiple genomic data types using a joint latent. Bioinformatics. 2009;25:2906–2912. doi:10.1093/bioinformatics/btp543
108. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi:10.1056/NEJMoa0708857
109. Cheng AL, Kang Y-K, Chen Z. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with. Lancet Oncol. 2009;10:25–34. doi:10.1016/S1470-2045(08)70285-7
110. Hoos A. Development of immuno-oncology drugs - from CTLA4 to PD1 to the next generations. Nat Rev Drug Discov. 2016;15:235–247. doi:10.1038/nrd.2015.35
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.