")
Back to Journals » Cancer Management and Research » Volume 11
An integrated prediction model of recurrence in endometrial endometrioid cancers
Authors Miller MD , Salinas EA, Newtson AM, Sharma D, Keeney ME, Warrier A, Smith BJ , Bender DP, Goodheart MJ, Thiel KW, Devor EJ, Leslie KK , Gonzalez-Bosquet J
Received 23 January 2019
Accepted for publication 22 March 2019
Published 6 June 2019 Volume 2019:11 Pages 5301—5315
DOI https://doi.org/10.2147/CMAR.S202628
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Beicheng Sun
Marina D Miller,1 Erin A Salinas,1 Andreea M Newtson,1 Deepti Sharma,1 Matthew E Keeney,2 Akshaya Warrier,1 Brian J Smith,3 David P Bender,1,4 Michael J Goodheart,1,4 Kristina W Thiel,1 Eric J Devor,1,4 Kimberly K Leslie,1,4 Jesus Gonzalez-Bosquet1,4
1Department of Obstetrics and Gynecology, University of Iowa Carver College of Medicine, Iowa City, IA, USA; 2Department of Pathology, University of Iowa Carver College of Medicine, Iowa City, IA, USA; 3Department of Biostatistics, University of Iowa College of Public Health, Iowa City, IA, USA; 4Holden Comprehensive Cancer Center, University of Iowa Carver College of Medicine, Iowa City, IA, USA
Objectives: Endometrial cancer incidence and mortality are rising in the US. Disease recurrence has been shown to have a significant impact on mortality. However, to date, there are no accurate and validated prediction models that would discriminate which individual patients are likely to recur. Reliably predicting recurrence would be of benefit for treatment decisions following surgery. We present an integrated model constructed with comprehensive clinical, pathological and molecular features designed to discriminate risk of recurrence for patients with endometrioid endometrial adenocarcinoma.
Subjects and methods: A cohort of endometrioid endometrial cancer patients treated at our institution was assembled. Clinical characteristics were extracted from patient charts. Primary tumors from these patients were obtained and total tissue RNA extracted for RNA sequencing. A prediction model was designed containing both clinical characteristics and molecular profiling of the tumors. The same analysis was carried out with data derived from The Cancer Genome Atlas for replication and external validation.
Results: Prediction models derived from our institutional data predicted recurrence with high accuracy as evidenced by areas under the curve approaching 1. Similar trends were observed in the analysis of TCGA data. Further, a scoring system for risk of recurrence was devised that showed specificities as high as 81% and negative predictive value as high as 90%. Lastly, we identify specific molecular characteristics of patient tumors that may contribute to the process of disease recurrence.
Conclusion: By constructing a comprehensive model, we are able to reliably predict recurrence in endometrioid endometrial cancer. We devised a clinically useful scoring system and thresholds to discriminate risk of recurrence. Finally, the data presented here open a window to understanding the mechanisms of recurrence in endometrial cancer.
Keywords: endometrial cancer, recurrence risk, prediction model
Introduction
Endometrial cancer is the most common gynecologic malignancy in developed countries. In 2017, it was estimated to affect 61,380 women in the United States, accounting for 10,920 deaths.1 Up to 75% of endometrial cancers are of endometrioid histology, or endometrial endometrioid cancer (EEC). This histology is considered more indolent than papillary serous and clear cell endometrial cancers. Thus, 70% of women with EEC are diagnosed at an early stage, when surgery alone is typically curative and prognosis is excellent with a 5-year survival of 81.3%.1 Despite these favorable aspects of the disease, endometrial cancer mortality has risen in the United States by 1.4% per year between 2005 and 20141,2 and is projected to increase another 55% by 2030 due to the obesity epidemic.3 One of the contributors to mortality in EEC is recurrence. Indeed, recurrence occurs in approximately in 10–15% of patients.4 The prognosis for recurrent EEC is poor and treatment is only undertaken with curative intent when disease is isolated to the vagina.4 Thus, identifying patients who might benefit from additional surveillance and treatment to prevent recurrence would be of great value.
Prior studies have suggested that certain clinical, immunologic and even radiologic features of endometrial tumors are associated with disease recurrence. Some of these clinical characteristics have been used in risk stratification for recurrence and spread of disease.5–8 However, there are no validated and accurate prediction models that asses individual probability for recurrence in any given patient with EEC. Prior attempts at predicting local and distant recurrences utilizing data from PORTEC trials yielded accuracies between 59% and 73%.9 Other models, using a combination of clinical and pathological variables, were able to predict recurrence with an area under the curve (AUC) around 80%.7 Thus, we hypothesize that a comprehensive method to assess a patient’s risk of recurrence that includes clinical, pathological as well as molecular characteristics of the tumors themselves would improve accuracy in prediction and validation. Thus, we have constructed an integrated and comprehensive recurrence risk prediction model composed of clinical, pathological and molecular features. Further, we have validated this model using data from the Cancer Genome Atlas (TCGA). Finally, we created a scoring system with these clinical-pathological-molecular risk factors to translate our models into a clinically useful risk assessment tool.
Subjects and methods
Definition of recurrence
For the purpose of this study, we defined disease recurrence as EEC diagnosed in any location after completing treatment and a subsequent period with no evidence of disease. Only those patients with recurrence within two years after completion of treatment were considered to have disease recurrence. This decision was based on two factors: 1) Ideally, to construct a classifier or prediction model, the outcome of interest should be dichotomous, non-time-dependent; 2) 90% of patients from University of Iowa Hospitals and Clinics (UIHC) experienced a recurrence with 2 years of initial treatment (8 out of 9); the other patient had a recurrence after almost 4 years; additionally, 91% (or 49 out of 54) of TCGA patients recurred before 2 years; the rest recurred either after 3 years (1 patient) or 4 years (the remaining 3). Thus, for statistical purposes and to detect the characteristics most typically associated with the majority of recurrent patients while excluding outliers, we set two years as the cutoff.
Endometrial cancer cohorts
University of Iowa cohort
A cohort of 125 patients diagnosed with EEC at UIHC was assembled under approval by the Institutional Review Board (IRB# 201607815). An outline of the study population is shown in Figure 1. Patient charts were reviewed and clinical variables extracted. Pre-operative characteristics included body mass index (BMI), age at diagnosis, pre-operative pathology diagnosis, pre-operative hemoglobin, serum creatinine, albumin, chest x-ray, electrocardiogram, comorbidities (coronary artery disease, diabetes mellitus, congestive heart disease, history of cardiovascular accident, tobacco use) and Charlson morbidity index. Intraoperative characteristics included type of surgery (laparoscopic, robotic, laparotomy, vaginal), operative time and estimated blood loss. Post-operative characteristics extracted included final pathology diagnosis, disease stage, estrogen and progesterone receptor status, surgical complications, adjuvant therapy (including radiation therapy), recurrence and death.
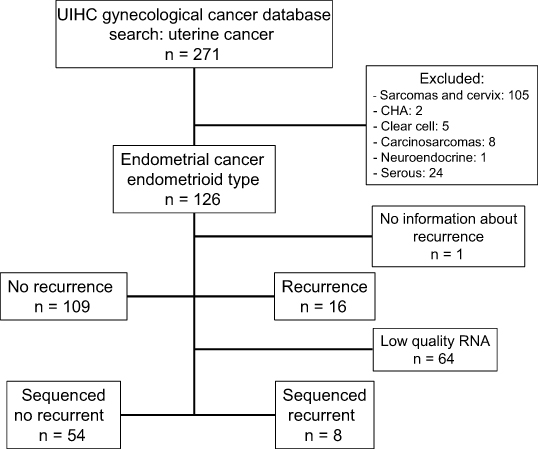 | Figure 1 Flow chart of patients included in the UIHC endometrial cancer study cohort. There were 126 patients with endometrial cancer, endometrioid type. 62 had sufficient quantity and quality of purified RNA for RNA sequencing. Abbreviation: CHA, complex endometrial hyperplasia with atypia; UIHC, University of Iowa Hospitals and Clinics. |
We considered EEC patients with high risk for poor outcomes those with presenting factors that have previously been associated with such outcomes, such as spread of disease, involvement of lymph nodes, recurrence of disease and poor survival. These factors associated with poor outcomes are largely based on the results and criteria from the Gynecologic Oncology Group (GOG/NRG) study GOG 33, the GOG 99 clinical trial and subsequently modified in the PORTEC trials.10–12 Thus, high-risk patients were classified as those presenting with Stages II, III and IV as defined by 2009 FIGO classification13 and patients with initial Stage I and high-intermediate risk features by GOG 99 criteria. Low-risk patients were the remaining Stage I patients, either with no myometrial invasion and no risk factors, or low-intermediate risk features.14
TCGA cohort
Data from TCGA dataset for endometrial cancer were downloaded from the National Cancer Institute (NCI) database in accordance with TCGA Human Subject Protection and Data Access Policies, adopted by the NCI and the National Human Genome Research Institute (NHGRI). Data were downloaded with the NCI database of genotypes and phenotypes approval (dbGaP#16003). Patients with non-endometrioid histology were excluded. Clinical and molecular data (gene and miRNA expression, gene copy number and mutation analysis) were obtained from 394 patients diagnosed with EEC, of which 49 experienced recurrence of disease as defined above (Table S1).
RNA purification and sequencing
Primary tumor tissue samples were available for 125 patients identified in the UIHC EEC cohort. All tissues were collected under written informed consent in accordance with the Declaration of Helsinki (IRB #200910784 and IRB#200209010). Total tissue RNA was extracted from these available primary tumors in the UIHC cohort using the mirVana mRNA isolation kit (AM1560, Thermo Fisher Scientific, Waltham, MA) following manufacturer’s recommendations. RNA concentration and purity were assessed using a NanoDrop Lite spectrophotometer (ND-LITE-PR, Thermo Fisher Scientific, Waltham, MA) and a Model 2100 Bioanalyzer (G2939BA, Agilent, Santa Clara, CA). RNAs of sufficient mass and RNA Integrity (RIN>7.0) were obtained from 62 of our 125 specimens, and were submitted for RNA sequencing (RNAseq).15 The primary reason for failure to meet the RNA QC threshold was delay in preservation of tissue between the operating room and the laboratory.
RNA sequencing was performed in the Genomics Division of the University of Iowa Institute for Human Genetics (IIHG). Total cellular RNA (500 ng) was fragmented, converted to cDNA and ligated to bar-coded sequencing adaptors from the Illumina TruSeq stranded total RNA library preparation kit (20020596, Illumina, Inc., San Diego, CA). Molar concentrations of the indexed libraries were measured on the Model 2100 Agilent Bioanalyzer and combined equally into pools for sequencing. Concentrations of the pools were measured using the Illumina Library Quantification Kit (KR0405, KAPA Biosystems, Wilmington, MA). Sequencing was carried out on the Illumina HiSeq 4000 platform using a 150 bp paired-end SBS chemistry.
File pre-processing of diverse biological data
Reads generated from all sequencing studies were analyzed with a local deployment of the Galaxy tool shed for RNAseq in the High-Performance Computing Environment at the University of Iowa.16 Additional tools from BRB-SeqTools were also used for pre-processing and data analysis.17 Briefly, sequence reads were mapped and aligned to the human reference genome (GRCh38) using paired-end enabled algorithms such as TopHat2.18 BAM files were produced after alignment. We used the Cufflinks isoform assembly and quantitation algorithm to estimate the relative abundance of transcripts and featureCount to measure gene expression from BAM files.19 After the gene counts were generated, we used DESeq2 package to import, normalize and prepare the data for analysis.20 We independently used gene expression (which is referred to as mRNA) and miRNA expression (miRNA) for the recurrence association analysis.
BAM files for each sample were also used for mutation discovery and calling against the human reference genome utilizing SAMtools and BCFtools.21 Results were annotated with ANNOVAR and formatted to display number of mutations per gene and sample.21 We included only non-synonymous somatic mutations: frame shift insertions and deletions, in-frame insertions or deletions, missense, nonsense and nonstop mutations, silence, splice site and translation start site mutations. Copy number variation (CNV) was determined with SAMtools and CopywriteR, using BAM files as input. CopywriteR extracts copy number information from targeted sequencing by utilizing off-target reads, and can be used without reference and applied to sequencing data obtained from various techniques.22
Variable selection for prediction modeling
In the prediction model, we only included those variables that could be assessed at baseline, right after completion of initial treatment, including adjuvant therapy. Variable selection for all dimensions of clinical, pathological and biological data (gene and miRNA expression, gene copy number and mutation analysis) were performed using k-fold cross-validation with the “caret” R package.23 Cross-validation was used to determine the subset of variables considered in the multivariable lasso regression analysis and to create the best prediction models. Only predictors that were informative in each fold of the cross-validation process were selected and considered for the multivariable lasso prediction model. Using regression models without cross-validation in the variable selection process may over-estimate model performances, thus rendering these models difficult to be validated externally.24 The initial univariate filtering resulted in 162 gene expressions, 47 miRNAs, 963 somatic mutations and 472 gene CNVs. These biological variables were then statistically associated (cross-validation) with recurrence of disease.
Prediction model construction
Selected clinical and molecular variables from univariate filtering, as detailed previously, were analyzed by individual categories and in combinations to determine their prediction potential for disease recurrence. The lasso method (least absolute shrinkage and selection operator), as implemented in the glmnet R package,25 was used to develop multivariate regression models to predict recurrent versus non-recurrent patients. We selected lasso because it is a multivariate regression method that allows simultaneous selection and estimation of the effects of variables while accounting and adjusting for confounding factors. We evaluated the performance of our model using the Receiver Operating Characteristic area under the curve (AUC) and its 95% confidence interval (CI). AUC was estimated with k-fold cross-validation of the combined univariate filtering and lasso modeling to avoid over-fitting of the model (internal validation).26 Bias-corrected and accelerated bootstrap CIs were computed for resulting AUCs and other diagnostic parameters (specificity, sensitivity, negative and positive predictive value and accuracy). AUC may be interpreted as the probability that recurrent patients have a higher predicted probability of recurrence of disease than non-recurrent patients. A value of 0.5 indicates a lack of model predictive performance, and 1.0 indicates perfect predictive performance. Analyses were performed with the R statistical software27 and the caret23 add-on package.
Cancer Genome Atlas replication and validation
RNAseq reads from endometrial adenocarcinomas were downloaded from the TCGA and analyzed with the same bioinformatic tools and software as above. Only CNV data input and pre-processing differ from UIHC analysis because TCGA had available genotype files that made for easier computation of CNVs. Samples from Agilent Human Genome CGH Microarray 244A (Agilent Technologies, Santa Clara, CA) were processed and Circular Binary Segmentation was used to identify regions with altered copy number in each chromosome.28 The copy number at a genomic location was computed based on the segmentation mean log ratio data. We found regions with frequent CNV among all samples by performing genomic identification of significant targets in cancer analysis.29
For external replication and validation of UIHC prediction models of recurrence, we used only those variables resulting from the UIHC lasso analysis that contributed to the performance of the model and selected them from the TCGA sample set. Then, the same lasso analysis was carried out with these variables only to assess the performance of the models in terms of AUC and 95% CIs. As we did with UIHC datasets, we replicated lasso prediction analyses with individual and combinations of data categories. Comparison between performance of the UIHC cohort prediction models and TCGA cohort clinical and molecular models was assessed with AUCs and their respective 95% CIs.
For validation of the best UIHC prediction models for recurrence in TCGA dataset, we took the best UIHC models that replicated well in TCGA and applied them to the TCGA dataset to obtain a predicted probability for each patient.23 Then, we used the R package pROC to determine thresholds for the UIHC model applied to the TCGA data.30 Thresholds are values for the score of the model above which patients were classified as recurrent and below which as non-recurrent. Thresholds were treated as a tuning parameter for which values were sought to produce a final classification model. Threshold values that yielded specificities around 80% were ranked from highest to lowest sensitivity and negative predictive value. Among the ranked results, the top-ranked set of tuning parameters was used to fit a final score of the model to the entire set of patients and define the classification rule.
Survival analysis
Survival analyses for Table 1 and Figure S1 were carried out using Cox proportional hazard regression with statistical significance set at a p-value of 0.05. Patients with no evidence of disease at their last visit were treated as censored observations in the analysis.
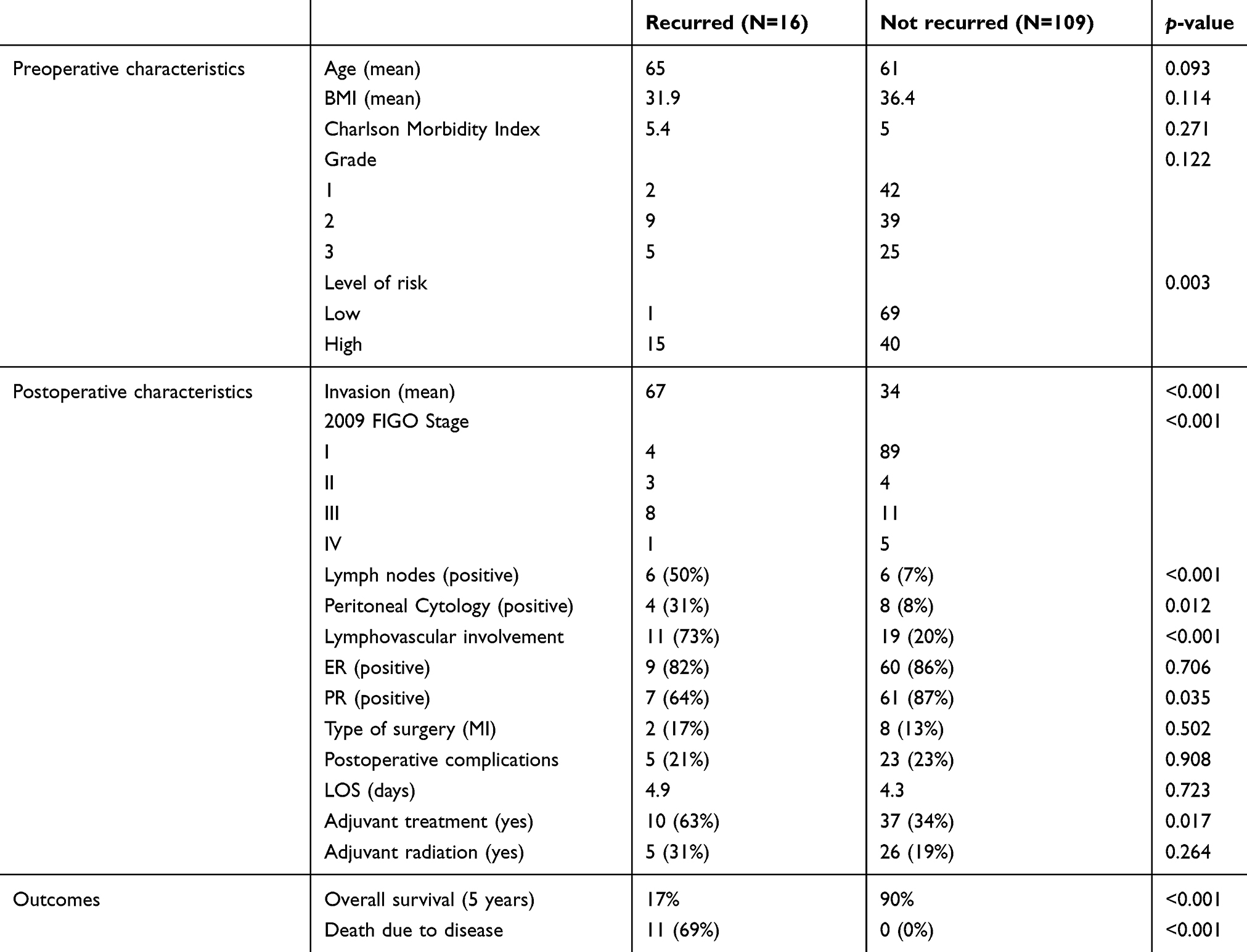 | Table 1 Clinical and pathological characteristics of the UIHC cohort of patients included in this study |
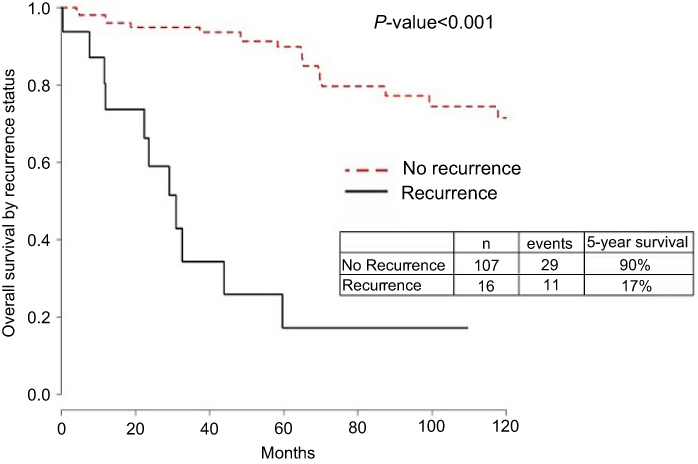 | Figure S1 Overall survival by recurrence status. |
Comparisons between Kaplan–Meier survival curves were performed with the log-rank test. Two-sided p-values are reported in the tables.
Results
Clinical variables associated with recurrence in the UIHC cohort
One hundred and twenty-five patients diagnosed and treated for EEC at UIHC met the criteria for inclusion in the present study (Table 1). Of these patients, 109 (87.2%) were non-recurrent whereas 16 (12.8%) had disease recurrence with an average of 75.6 months of follow-up. Overall survival (OS) was significantly impacted by recurrence, with an OS of 17% for patients who recurred, compared with 90% in patients without recurrent disease (Figure S1). Clinical variables significantly associated with recurrence in the univariate cox analysis included high-risk status, myometrial invasion, stage as determined by 2009 FIGO, positive lymph node, positive peritoneal cytology, lymphovascular space invasion, positive progesterone receptor status and adjuvant treatment (Table 1). The number of patients treated with radiation after surgery was not different in recurrent and non-recurrent patients.
Selection of molecular variables predictive of recurrence in the UIHC cohort
From the initial 125 patients, a total of 62 primary patient tumor tissues produced RNA of sufficient quality to be submitted for RNA sequencing. This sub-cohort included tissue from 8 patients whose disease recurred and 54 patients who did not experience recurrence within the follow-up period. RNAseq analysis produced mRNA expression data for 26,336 genes and 1,916 miRNAs, along with identification of 12,340 somatic mutations and 26,720 segments with CNV. Only predictors that were informative in the cross-validation process were selected. This panel included 162 expressed genes, 47 expressed miRNAs, 963 gene somatic mutations and 472 gene CNVs that were associated with recurrence (Figure 2). These variables were selected to be included in the prediction analysis.
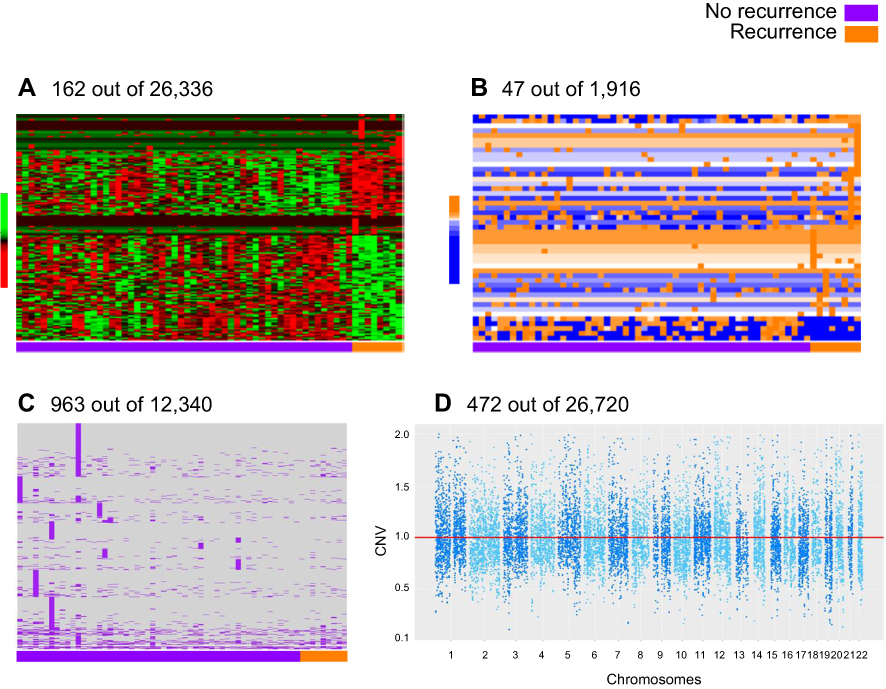 | Figure 2 Representation of differential gene expression (A), miRNA expression (B), somatic mutation (C), and copy number variation (D) by recurrence in the UIHC patient cohort (n=62 patients total; n=8 recurrence and n=54 no recurrence). Abbreviation: CNV, copy number variation; UIHC, University of Iowa Hospitals and Clinics. |
Categories of data predictive of recurrence within the UIHC prediction models
A recurrence prediction model utilizing only UIHC clinical data predicted recurrence with an AUC of 0.90 (95% CI: 0.87, 0.92, Table 2). Similarly, models predicting recurrence based upon single molecular data categories were modestly successful, with miRNA being the most predictive (AUC=0.81; 95% CI: 0.77, 0.86) and mutation being the least predictive (AUC=0.60; 95% CI: 0.49, 0.70, Table 2). Increasing predicting power was achieved by constructing models with multiple data categories consisting of the clinical variable plus one or more of the molecular data categories. The highest performing models contained clinical data augmented with one molecular category or with two molecular categories (Table 2). The addition of molecular tumor characteristics to the available clinical data contributes as much as 10% to recurrence risk prediction performance.
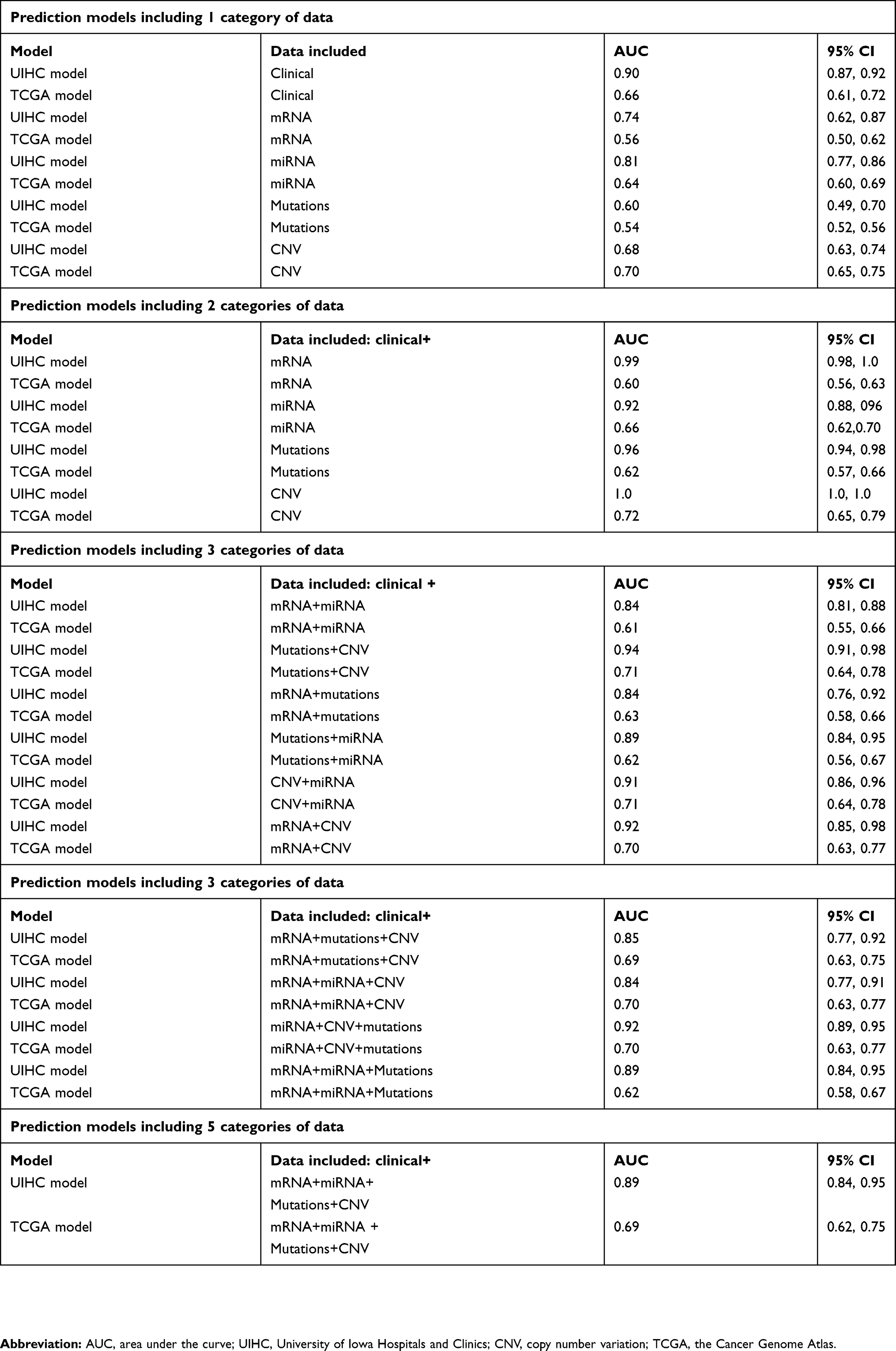 | Table 2 Prediction models for recurrence incorporating clinical, pathological and molecular data, as well as external replication of prediction models in TCGA |
Replicating the UIHC prediction model with TCGA data
As the results from our prediction models were derived from data from a single institution, we replicated the UIHC-based model in an independent dataset. The best available independent endometrial endometrioid adenocarcinoma cohort is TCGA, as the available information includes similar clinical and molecular data to the UIHC cohort. None of the models performed as well with the TCGA cohort as with the UIHC cohort (Table 2). Overall, performance of the UIHC models using TCGA data as measured by AUC is reduced by approximately 20%.
Validating the UIHC prediction model with TCGA data
For external validation of the recurrence prediction model in TCGA, we chose the best performing models: 1) with two categories of data, including clinical and miRNA, and clinical and CNV; 2) with three categories of data including clinical, miRNA and CNV. For each model, we created a score with the value of the included variables and their relative weight on the model. Table 3 shows each variable included in the validation model. We then found a threshold value for the score above which patients were classified as recurrent and below which as non-recurrent for each of the final three models. Prediction model validation and their performance in TCGA data are summarized in Table 4.
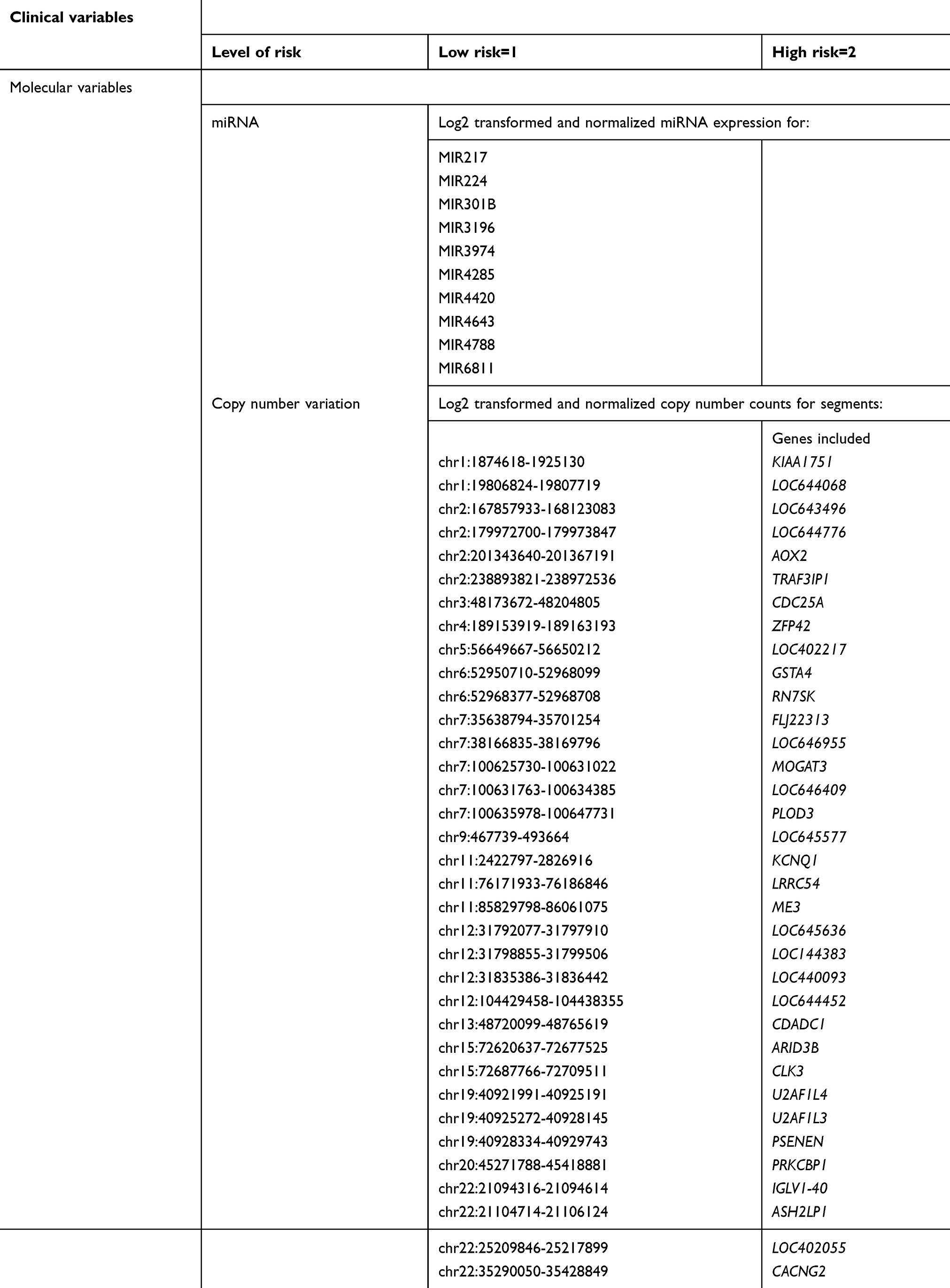 | Table 3 Values for each individual variable used to construct the prediction model score |
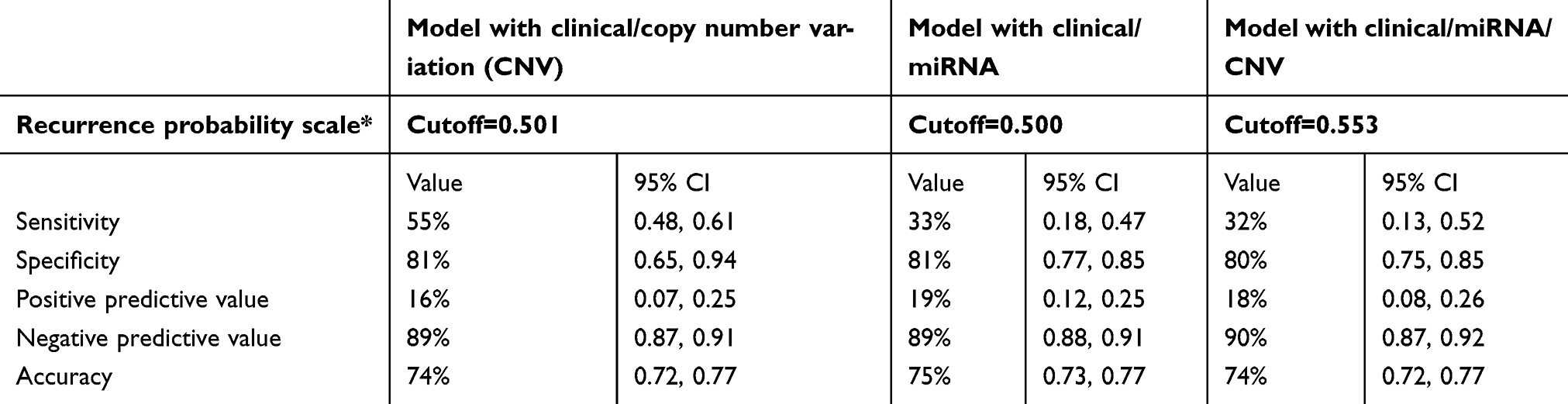 | Table 4 Validation of the prediction model of recurrence in TCGA |
Discussion
Prior studies have established clinical factors that are associated with an increased risk of endometrial cancer recurrence. Lee et al established that age at diagnosis, deep myometrial invasion, high FIGO grade, lymphovascular space invasion and cervical stromal invasion are clinical factors associated with disease recurrence in endometrial cancer.5 These clinical characteristics have helped guide adjuvant therapy. Other groups have found an association between histopathologic, patho-immunologic and radiologic features of endometrial tumors and recurrence.6–8 However, an association with recurrence does not necessarily translate to the ability to accurately and reliably predict recurrence. Statistical methods such as those employed in this study are necessary in order to construct and validate prediction models or classifiers.23 Senol et al showed that tumor diameter was a significant predictor of recurrence with an AUC of 0.77. Versluis et al showed that lymphovascular space invasion (LVSI) combined with FIGO stage, cytotoxic T-cells and memory T-cells were significant predictors of disease-free survival with Harrell’s C-indices of 0.71, 0.60 and 0.61, respectively. They also analyzed these factors in combination yielding the highest predictive power with the combination of FIGO stage, LVSI and memory T-cells, with a C-index of 0.83.7 Despite these promising results, a prediction model with accuracy in the lower 80s is unlikely to translate into a widely accepted clinical test. Furthermore, no clinical test, score for recurrence or threshold to classify patients has been defined and validated to implement a diagnostic tool that would determine whether or not an individual patient will recur.
Although the clinical characteristics shown here to be associated with disease recurrence have previously been described by other groups, our study is the first to combine such clinical characteristics with pathological and comprehensive molecular tumor data to construct a prediction model for endometrial cancer recurrence. Further, this prediction model can be applied in a diagnostic test for disease recurrence in an individual patient. Initial prediction models constructed utilizing data from our own institution predicted recurrence with high accuracy as noted by AUCs approaching 1. The high accuracy of these models provides the ability to translate models into a clinically useful tool for recurrence. Based on these UIHC prediction models that were validated with TCGA data, we created scoring systems with specific thresholds that would discriminate between recurrent and non-recurrent patients. These scoring systems and thresholds could be used clinically to predict a patient’s chance of recurrence with specificity over 80% and negative predictive value of 90%.
Replication of the same analysis in TCGA yielded less accurate results, yet with similar trends. For example, in both the UIHC and the TCGA datasets, the highest performing model included some of the same data categories: clinical information and gene CNV. The high predictive performance of these categories of data is intriguing and merits further investigation. A potential explanation as to why the TCGA model did not predict recurrence as efficiently as the UIHC model is that follow-up time was much longer in UIHC patients when compared to TCGA patients. Even though our group tried to account for that by defining recurrence at 2 years, in TCGA close to 80% of patients had less than 2 years of follow-up compared to 25.5% of UIHC patients. Thus, recurrence in the UIHC model is more accurately represented, whereas in the TCGA population it is likely underestimated. Additionally, the UIHC patient population may not be representative of the general or the TCGA population. The state of Iowa comprised primarily Caucasians (>90%) according to the United States Census Bureau.31 Tumors evaluated by TCGA were derived from various institutions across the United States and therefore this population is unlikely to be as homogeneous as the Iowa population. Finally, the prediction models derived from UIHC data included more comprehensive tumor molecular data as more genes, miRNAs and gene CNVs were found to be significantly associated with increased risk of recurrence.
gThe limitations of this study include its retrospective nature, as well as the lack of tumor tissue for all of the patients for whom we had clinical information. Given the interval between the time of tissue collection for the tumor bank (time of surgery) and RNA extraction and preparation, as well as the limited quantity of tissue available in EEC, we were able to acquire RNA of enough quantity and quality in only half of the patients with frozen tumor. We anticipate that if the tissue were collected prospectively with the intention of RNA extraction for sequencing, specialized and timely tissue handling would improve the RNA yield and quality. Therefore, prospective validation of this model would require instituting such specialized tissue handling techniques. Further, if validated prospectively, we propose that this model be utilized not only to provide patients with information on their disease’s prognosis, but also to guide further studies involving the molecular pathways associated with disease recurrence. Another limitation of the study is that the models were created and validated with few patients experiencing recurrence: 8 for UIHC and 49 in TCGA. However, only 10–15% of patients with EEC will be diagnosed with a disease recurrence, which is a limitation for all groups that study this outcome.4 To date, prediction models for recurrence have only used clinical and/or pathological features which resulted in performances in the lower 80s as measured by AUCs.6,7,9 Our study represents the only study describing prediction models for recurrence that includes a comprehensive analysis of biological features using RNA sequencing, in addition to clinical and pathological characteristics, and is externally validated using similar comprehensive features and methods.
The work presented here elucidates molecular characteristics of endometrial endometrioid tumors that could potentially inform the mechanisms by which these tumors develop recurrence. The specific loci contributing to the models are summarized in Table 3. Some of the miRNAs and regions with CNV found to be associated with disease recurrence have previously been reported in the literature as having cancer associations. Of the miRNAs that we found to be linked with recurrence risk, miR-217 and miR-224 have been shown to be involved in disease prognosis in other cancers. In the case of miR-217, it was found to target KRAS in pancreatic cancer and low levels of miR-217 in gastric cancers was associated with metastasis and poor prognosis.32,33 There is significant literature to support the involvement of miR-224 in cancer as well. It has been shown to be involved with cell cycle regulation, cell migration, and invasion in colorectal, lung and prostate cancers. In colorectal cancer, it promotes G1/S transition by repressing P21WAF1/CIP1 expression,34 whereas in lung cancer, it plays an oncogenic role by targeting CASP3 and 7, promoting cell proliferation and migration.35 Finally, in prostate cancer, it was shown to target TPD52. When silenced, TPD52 inhibits cancer cell migration and invasion.36
Finally, multiple genes were found to be affected by CNV as seen in Table 3, several of which have a well-established relationship with cancer. The few that stand out for their significant impact in tumorigenesis, disease recurrence and resistance to therapy include ZMYND8, KCNQ1, CDC25A, GSTA4 and ARID3B. The zinc-finger myeloid, Nervy and DEAF-1-type containing 8 (ZMYND8) is a key component of the transcription regulatory network known as a chromatin reader. Silencing ZMYND8 in neuroblastoma cells results in substantial anti-proliferative effects.37 This result was in support of the demonstration that ZMYND8 antagonized the expression of metastasis-linked genes, and its knockdown increased the invasiveness of prostate cancer cells.38 KCNQ1 is a potassium channel that can serve as a tumor suppressor. Loss of its expression has been associated with poor clinical outcomes in colorectal cancer, including an increased risk of disease recurrence and shows promise in this patient population as a biomarker for disease recurrence.39 CDC25A is a key regulator of cell cycle progression. It dephosphorylates and activates cyclin-CDK complexes and its overexpression has been shown to accelerate G1/S and G2/M transitions, thus promoting tumorigenesis.40 GSTA4 has been shown to be associated with resistance to therapy with cisplatin when its expression is increased in leukemia, mammary and ovarian cancer cell lines.41 ARID3B is involved in chromatin remodeling and regulation of gene expression. It is highly expressed in ovarian cancer and increases tumor growth.42 It was found to induce expression of genes that are associated with metastasis and cancer stem cells.
The available literature on these miRNAs and genes found to be affected by CNV in our study supports their role in disease aggressiveness. However, none of the available literature investigates their role in endometrial cancer specifically. Further study of these genes in endometrial cancer may be of great benefit to gaining better understanding of the molecular mechanisms guiding disease recurrence.
Conclusion
In conclusion, by constructing a model utilizing comprehensive clinical and molecular data, we were able to reliably predict recurrence in endometrioid endometrial cancer. We devised scoring systems as well as thresholds to discriminate between recurrent and non-recurrent patients. We assert that the data presented here potentially open a window to the mechanisms of recurrence in endometrial cancer. Moreover, not only do these models inform treatment and surveillance decisions, they point the way to future studies of the molecular processes driving recurrence, which has the potential to reveal new drug targets and treatment options for endometrial cancer patients.
Informed consent
Tumor samples were obtained under written informed consent in accordance with the Declaration of Helsinki after approval by the University of Iowa Institutional Review Board: IRB# 200910784 and 200209010.
Acknowledgments
The authors would like to thank the Genomic Division of the University of Iowa Institute of Human Genetics for their assistance with this project, specifically Mary Boes and Garry Hauser; and Dr. Donna Santillan, the director of the Department of Obstetrics & Gynecology Women’s Health Tissue Repository and Gynecologic Malignancy Repository. This work was supported in part by the NIH grant R01 CA99908 and R01 CA184101 to Kimberly K Leslie, and the basic research fund from the Department of Obstetrics & Gynecology at the University of Iowa.
Author contributions
All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Marina D Miller reports a patent Prediction model for recurrence pending; Dr Kristina W Thiel is a shareholder in Immortagen Inc.; Dr Jesus Gonzalez-Bosquet reports a patent P12659US00 pending. The authors report no other conflicts of interest in this work.
References
1. SEER Cancer Stat Facts: Endometrial Cancer. Bethesda, MD: National Cancer Institute. Available from:
2. Endometrial Cancer Treatment (PDQ®) – Health Professional Version. Bethesda, MD: National Cancer Institute. Available from:
3. Sheikh MA, Althouse AD, Freese KE, et al. USA endometrial cancer projections to 2030: should we be concerned? Future Oncol. 2014;10(6):2561–2568. doi:10.2217/fon.14.192
4. Del Carmen MG, Boruta DM, Schorge JO. Recurrent endometrial cancer. Clin Obstet Gynecol. 2011;54(2):266–277. doi:10.1097/GRF.0b013e318218c6d1
5. Lee KR, Vacek PM, Belinson JL. Traditional and nontraditional histopathologic predictors of recurrence in uterine endometrioid adenocarcinoma. Gynecol Oncol. 1994;54(1):10–18. doi:10.1006/gyno.1994.1158
6. Senol T, Polat M, Ozkaya E, Karateke A. Tumor diameter for prediction of recurrence, disease free and overall survival in endometrial cancer cases. Asian Pc J Cancer Prev. 2015;16(17):7463–7466. doi:10.7314/APJCP.2015.16.17.7463
7. Versluis MA, de Jong RA, Plat A, et al. Prediction model for regional or distant recurrence in endometrial cancer based on classical pathological and immunological parameters. Br J Cancer. 2015;113(5):786–793. doi:10.1038/bjc.2015.268
8. Kang SY, Cheon GJ, Lee M, et al. Prediction of recurrence by preoperative intratumoral FDG uptake heterogeneity in endometrioid endometrial cancer. Transl Oncol. 2017;10(2):178–183. doi:10.1016/j.tranon.2017.01.002
9. Creutzberg CL, van Stiphout RG, Nout RA, et al. Nomograms for prediction of outcome with or without adjuvant radiation therapy for patients with endometrial cancer: a pooled analysis of PORTEC-1 and PORTEC-2 trials. Int J Radiat Oncol Biol Phys. 2015;91(3):530–539. doi:10.1016/j.ijrobp.2014.11.022
10. Creasman WT, Morrow CP, Bundy BN, Homesley HD, Graham JE, Heller PB. Surgical pathologic spread patterns of endometrial cancer. A gynecologic oncology group study. Cancer. 1987;60(8):2035–2041.
11. Keys HM, Roberts JA, Brunetto VL, et al. A phase III trial of surgery with or without adjunctive external pelvic radiation therapy in intermediate risk endometrial adenocarcinoma: a gynecologic oncology group study. Gynecol Oncol. 2004;92(3):744–751. doi:10.1016/j.ygyno.2003.11.048
12. Creutzberg CL, van Putten WL, Koper PC, et al. Surgery and postoperative radiotherapy versus surgery alone for patients with stage-1 endometrial carcinoma: multicenter randomized trial. PORTEC Study Group. Post operative radiation therapy in endometrial carcinoma. Lancet. 2000;355(9213):1404–1411.3.
13. Pecorelli S. Revised FIGO staging for carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol Obstet. 2009;105(2):103–104.
14. Dai D, Thiel KW, Salinas EA, Goodheart MJ, Leslie KK, Gonzalez Bosquet J. Preoperative stratification of endometrioid endometrial cancer patients into risk levels using somatic mutations. Gynecol Oncol. 2016;142(1):150–157. doi:10.1016/j.ygyno.2016.05.012
15. Schroeder A, Mueller O, Stocker S, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol Biol. 2006;7:3. doi:10.1186/1471-2199-7-3
16. Afgan E, Baker D, van Den Beek M, et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44:W3–W10. doi:10.1093/nar/gkw343
17. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007;3:11–17. doi:10.1177/117693510700300022
18. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14(4):R36. doi:10.1186/gb-2013-14-4-r36
19. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi:10.1093/bioinformatics/btt656
20. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi:10.1186/gb-2010-11-11-r110
21. Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi:10.1093/bioinformatics/btp352
22. Kuilman T, Velds A, Kemper K, et al. CopywriteR: DNA copy number detection from off-target sequence data. Genome Biol. 2015;16:49. doi:10.1186/s13059-015-0667-4
23. Kuhn M. Building predictive models in r using the caret package. J Stat Softw. 2008;28:1–26. doi:10.18637/jss.v028.i07
24. Subramanian J, Simon R. Overfitting in prediction models – is it a problem only in high dimensions? Contemp Clin Trials. 2013;36:636–641. doi:10.1016/j.cct.2013.06.011
25. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22.
26. Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J Clin Oncol. 2005;23(29):7332–7341. doi:10.1200/JCO.2005.02.8712
27.
28. Olshen AB, Venkatraman ES, Lucito R, Wigler M. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5(4):557–572. doi:10.1093/biostatistics/kxh008
29. Beroukhim R, Getz G, Nghiemphu L, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104(50):20007–20012. doi:10.1073/pnas.0710052104
30. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi:10.1186/1471-2105-12-77
31.
32. Zhao WG, Yu SN, Lu ZH, Ma YH, Gu YM, Chen J. The miR-217 microRNA functions as a potential tumor suppressor in pancreatic ductal adenocarcinoma by targeting KRAS. Carcinogenesis. 2010;31(10):1726–1733. doi:10.1093/carcin/bgq160
33. Chen D-L, Zhang D-S, Lu Y-X, et al. microRNA-217 inhibits tumor progression and metastasis by downregulating EZH2 and predicts favorable prognosis in gastric cancer. Oncotarget. 2015;6(13):10868–10879. doi:10.18632/oncotarget.3451
34. Zhang X, Zhang X, Liu C, Jia N, Li X, Xiao J. miR-224 promotes colorectal cancer cells proliferation via downregulation of P21WAF1/CIP1. Mol Med Rep. 2014;9(3):941–946. doi:10.3892/mmr.2014.1900
35. Cui R, Kim T, Fassan M, et al. MicroRNA-224 is implicated in lung cancer pathogenesis through targeting caspase-3 and caspase-7. Oncotarget. 2015;6(26):21802–21815. doi:10.18632/oncotarget.5224
36. Goto Y, Nishikawa R, Kojima S, et al. Tumour-suppressive microRNA-224 inhibits cancer cell migration and invasion via targeting oncogenic TPD52 in prostate cancer. FEBS Lett. 2014;588(10):1973–1982. doi:10.1016/j.febslet.2014.04.020
37. Basu M, Khan MW, Chakrabarti P, Das C. Chromatin reader ZMYND8 is a key target of all trans retinoic acid-mediated inhibition of cancer cell proliferation. Biochim Biophys Acta. 2017;1860(4):450–459. doi:10.1016/j.bbagrm.2017.02.004
38. Li N, Li Y, Lv J, et al. ZMYND8 reads the dual histone mark H3K4me1-H3K14ac to antagonize the expression of metastasis-linked genes. Mol Cell. 2017;63(3):470–484. doi:10.1016/j.molcel.2016.06.035
39. Den Uil SH, Coupé VM, Linnekamp JF, et al. Loss of KCNQ1 expression in stage II and stage III colon cancer is a strong prognostic factor for disease recurrence. Br J Cancer. 2016;115(12):1565–1574. doi:10.1038/bjc.2016.376
40. Dozier C, Mazzolini L, Cénac C, et al. CyclinD-CDK4/6 complexes phosphorylate CDC25A and regulate its stability. Oncogene. 2017;36(26):3781–3788. doi:10.1038/onc.2016.506
41. Kalinina EV, Berozov TT, Shtil AA, et al. Expression of genes of glutathione transferase isoforms GSTP1-1, GSTA4-4, and GSTK1-1 in tumor cells during the formation of drug resistance to cisplatin. Bull Exp Biol Med. 2012;154(1):64–67.
42. Cowden Dahl KD, Dahl R, Kruichak JN, Hudson LG. The epidermal growth factor receptor responsive miR-125a represses mesenchymal morphology in ovarian cancer cells. Neoplasia. 2009;11(11):1208–1215.
Supplementary materials
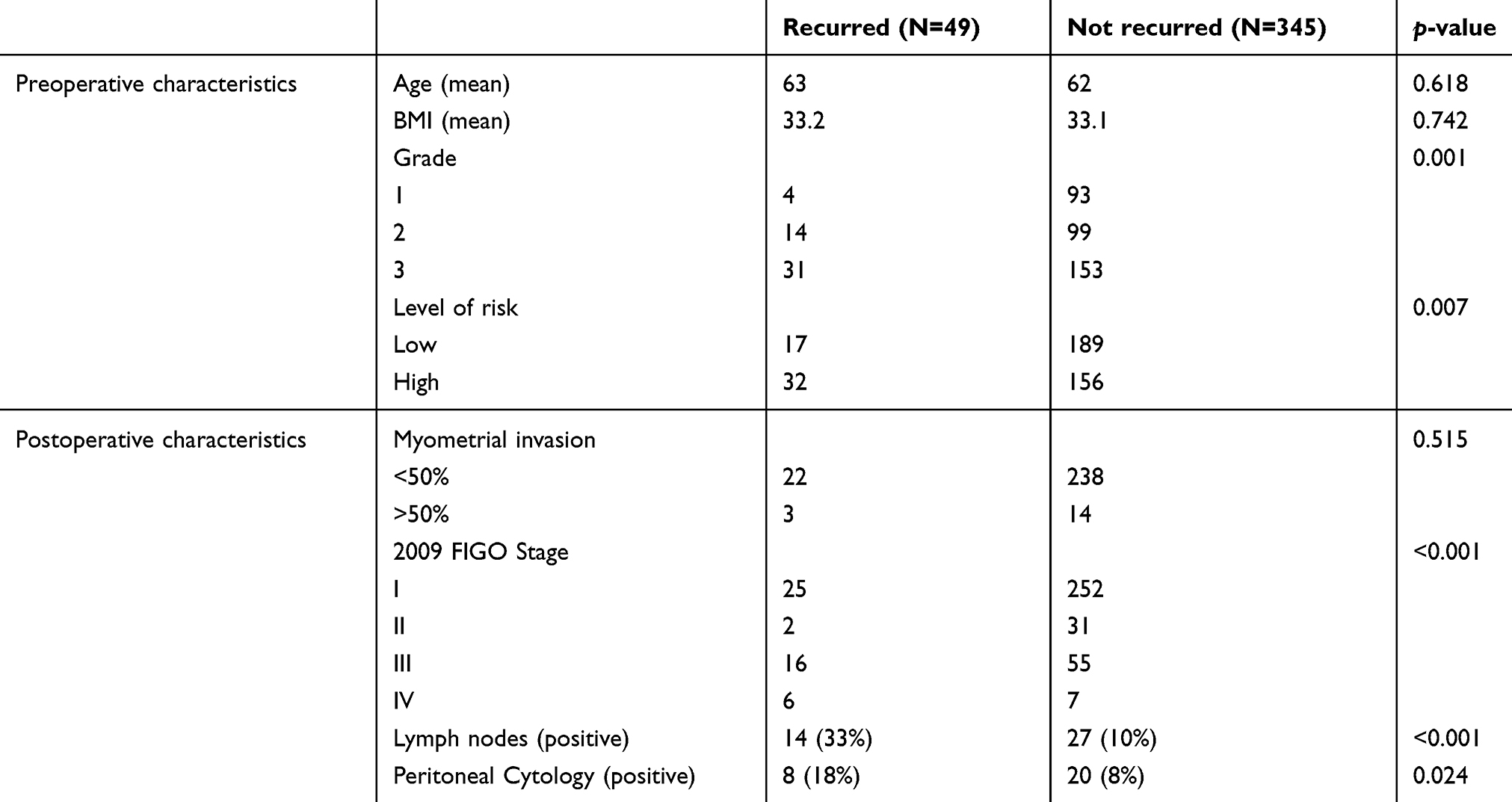 | Table S1 TCGA Patient clinical and pathological characteristics (N=394). Univariate analysis with Cox proportional Hazard ratio was used to assess differences between both groups |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.