")
Back to Journals » Cancer Management and Research » Volume 10
An expression signature model to predict lung adenocarcinoma-specific survival
Authors Shi X , Tan H, Le X, Xian H, Li X, Huang K, Luo VY, Liu Y, Wu Z , Mo HY, Chen AM , Liang Y, Zhang J
Received 11 December 2017
Accepted for publication 9 April 2018
Published 24 September 2018 Volume 2018:10 Pages 3717—3732
DOI https://doi.org/10.2147/CMAR.S159563
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lu-Zhe Sun
Xiaoshun Shi,1,2,* Haoming Tan,3 Xiaobing Le,4,5,* Haibing Xian,6,* Xiaoxiang Li,1 Kailing Huang,4,5 Viola Yingjun Luo,4,5 Yanhui Liu,4,5 Zhuolin Wu,7 Haiyun Mo,8 Allen M Chen,4,5,* Ying Liang,9 Jiexia Zhang1
1National Clinical Research Center for Respiratory Disease, State Key Laboratory of Respiratory Disease, Department of Medicine, Guangzhou Institute of Respiratory Disease, Guangzhou Institute of Respiratory Health, Guangzhou 510120, China; 2Department of Thoracic Surgery, Nanfang Hospital, Southern Medical University, Guangzhou 510515, China; 3Department of Thoracic Surgery, Shunde Lecong Affiliated Hospital of Guangzhou Medical University, Guangdong 528315, China; 4Mendel Genes Inc, Guangzhou 510515, China; 5Mendel Genes Inc, Manhattan Beach, CA 90266, USA; 6Department of Head and Neck/Thoracic Medical Oncology, The First People’s Hospital of Foshan, Guangdong 528000, China; 7Department of Biomedical Engineering, University of Minnesota, Twin Cities, MN, USA; 8Department of Public Health, Guangzhou Medical University, Guangzhou 510000, China; 9Department of Medical Oncology, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Guangzhou 510060, China
*These authors contributed equally to this work
Background: The current TNM staging system plays a central role in lung adenocarcinoma (LUAD) prognosis. However, it may not adequately stratify the risk of tumor recurrence. With the aid of gene expression profiling, we identified 31 lncRNAs whose expressions in tumor tissues could be used as a risk indicator for the guidance of lung cancer therapy. This exploratory analysis may shed new light on identification of potential prognostic factors.
Materials and methods: A survival prediction scoring model was developed from the data that are publicly available in The Cancer Genome Atlas (TCGA) LUAD RNA Sequencing dataset. Multivariate Cox regression analysis and Kaplan–Meier analysis were performed on a cohort of 254 stage I lung carcinoma patients with survival records.
Results: Our model indicates that the panels comprising 31 lncRNAs are highly associated with overall survival (OS): 18.9% (95% CI: 10.4%–34.5%) and 89.5% (95% CI: 80.7%–99.2%) for the high- and low-risk group, respectively. The specificity and sensitivity of the model are verified, which show that the area under receiver operating characteristic curve yields 0.881, meaning our model has good accuracy and it is feasible for further applications.
Conclusion: The 31-lncRNA model might be able to predict OS in patients with LUAD with high accuracy. Its further applications in biomolecular experiments using clinical samples with independent cohorts of patients are needed to verify the results.
Keywords: lung adenocarcinoma, lncRNA, signature, survival analysis, prognosis, RNA-seq
Background
Lung adenocarcinoma (LUAD) is the most commonly occurring histological type of lung cancer, which often has major driver oncogenes such as EGFR mutation1 and ALK fusion.2 Previous studies have shown that the genomic alterations in LUAD are different from those in other lung cancer subtypes.3,4 The current prediction systems for non-small-cell lung cancer (NSCLC) such as tumor, node, and metastases (TNM) staging5 and microarray-data-based prognostic modeling6 have not effectively distinguished lung cancer subtypes in terms of NSCLC patient survival. Moreover, the prognoses within the same TNM stage vary widely7 and the gene signature is yet limited in coding genes8 and microRNAs.6,9 Evidence provides the primary rationale to develop the lncRNA model for predicting lung cancer survival.
lncRNAs are a class of RNA molecules with more than 200 nucleotides in length and have no evident open reading frames.10 These long molecules play key roles in gene regulation and carcinogenesis including proliferation, adhesion, migration and apoptosis.11 Current non-coding RNA profiling research reveals that lncRNAs are dysregulated among cancers12 and some may serve as promising therapeutic targets.13 For instance, Gutschner et al reported that elevated MALAT1 expression was associated with metastasis in multiple tumor types.14 Previous meta-analysis showed that MALAT1 may have a role in cancer prognosis.15 It is believed that the clinical value of lncRNA is not confined to candidate biomarkers for diagnostic and prognostic purposes. Also, lncRNA expression profiling by RNA sequencing (RNA-seq) might be useful in the classification of various cancers such as lung cancer; it is important for prognostic determinations as well.12
A large number of lncRNAs have been investigated in cancer research.11,16 Some of these lncRNAs are associated with patient survival,17,18 but most of the reports are only supported by clinical survival data from samples within a single institute19 or pooled clinical data with some heterogeneities.20 Given the heterogeneity of LUAD and the susceptibility of non-coding RNA decay, a panel of the lncRNA biomarkers should be more precisely stable for LUAD prognosis determinations. However, using only a single lncRNA would yield unreliable results in predicting cancer survivals. Previous studies have also observed that gene signatures and clinical characteristics among patients are associated with overall survival (OS)21,22 aiming to overcome the limitations of the current TNM staging for predicting clinical outcomes. Several studies have reported that lncRNA expression profiles can be obtained from publicly available microarray data to perform analysis for the development of cancer survival models.23,24 However, RNA-seq-based prognostic lncRNA expression signature for the prediction of LUAD patient survival has not yet been investigated. Though a number of prognostic lncRNA biomarkers for NSCLC have been proposed,25–27 none of them have been successfully applied in real clinics. This is partially due to differences in the acquisition of samples and the usage of different systems for detection. In addition, the populations selected for lncRNA studies of cancer may vary and display inconsistencies. Moreover, the potential role of lncRNA as biomarkers for diagnosis and prognosis is better understood by the patterns of the lncRNA expression profile in the genome rather than as a single lncRNA expression abnormality. Notably, as we mentioned previously, the genomic characteristics of LUAD and squamous cancer distinguish greatly,4 and previous studies identifying the signature pattern of NSCLC may be modified further by separate analysis of each cancer subtype.
To construct a reliable prognostic lncRNA signature that could improve the current staging system for predicting LUAD survival, we identified lncRNAs that can stratify the risk of LUAD recurrence through survival outcomes. RNA-seq data and corresponding clinical data were analyzed to identify lncRNAs that associate with the risk of LUAD recurrence in patients. A panel of key lncRNAs was identified by next generation sequencing technology, which could diagnose one of the major histological subtypes of lung cancer with fairly high specificity and sensitivity. We developed a risk score formula for predicting the OS time of LUAD patients. In summary, the use of the lncRNA signature provided a deeper insight into the parameters associated with LUAD than what is used exclusively for LUAD prognosis.
Materials and methods
Lung adenocarcinoma RNA-seq data from TCGA
Level 3 RNA-seq data (HTSeq-FPKM-UQ) and the corresponding clinical data of 519 LUAD patients were obtained from the public The Cancer Genome Atlas data portal website (http://cancergenome.nih.gov). Clinicopathological parameters including age, gender, smoking history and TNM stage were also assessed. Patients with incomplete clinical data or OS of less than 1 month were excluded from the analysis. No correlations between patients’ gender as well as expression profile and OS were found; after data filtering and exclusions, a total of 462 LUAD samples comprising 250 females and 212 males were enrolled in the model.
Identification of differentially expressed lncRNAs in LUAD and normal lung tissue samples
To identify lncRNAs that are differentially expressed between LUAD and normal lung tissues, the raw counts of TCGA RNA-seq data (HTSeq-Counts) were downloaded for the analysis. Differential expression analysis was performed using the DESeq package in Bioconductor.28 The thresholds for screening the expression differences of lncRNAs were adjusted p-value <0.01 and |log2(fold change)|>2. The log2(fold change) indicates the fold change in the expression of each lncRNA between LUAD and normal lung tissue samples.
Cox regression analysis
First, the RNA-seq expression values of differentially expressed lncRNAs were normalized with log2 transformation. Afterward, the association between lncRNA expression and patient survival was determined by univariate Cox regression analysis. lncRNAs with a p-value less than 0.05 from the univariate Cox regression analysis were used for further mining potential lncRNAs that were associated with OS time, and they were fitted in a multivariate Cox regression analysis. The mathematical model was built based on the Akaike information criterion, which allows determination of the best trade-off between the complexity of model and its goodness of fit.29
Risk score and survival curve
Based on the multivariate Cox regression analysis, a formula (Equation 1) was built to predict the risk score for each patient. In Equation 1, Gi represents expression value of the ith lncRNA and Weighti is the coefficient of each lncRNA from the Cox analysis results (Table 1). According to this risk scoring system, patients were divided into low-risk (< median risk score) and high-risk (> median risk score) groups. Subsequently, the log-rank statistical test was used to determine the differences in survivals between the low-risk and high-risk groups. A Kaplan–Meier OS curve was plotted against the two groups and the hazard ratio was calculated. Cox multivariate analysis was employed to test whether the risk score was independent of potential clinical risk factors including age, gender, smoking history and disease stage. The prognostic performance was measured by calculating the area under the receiver operating characteristic curve.
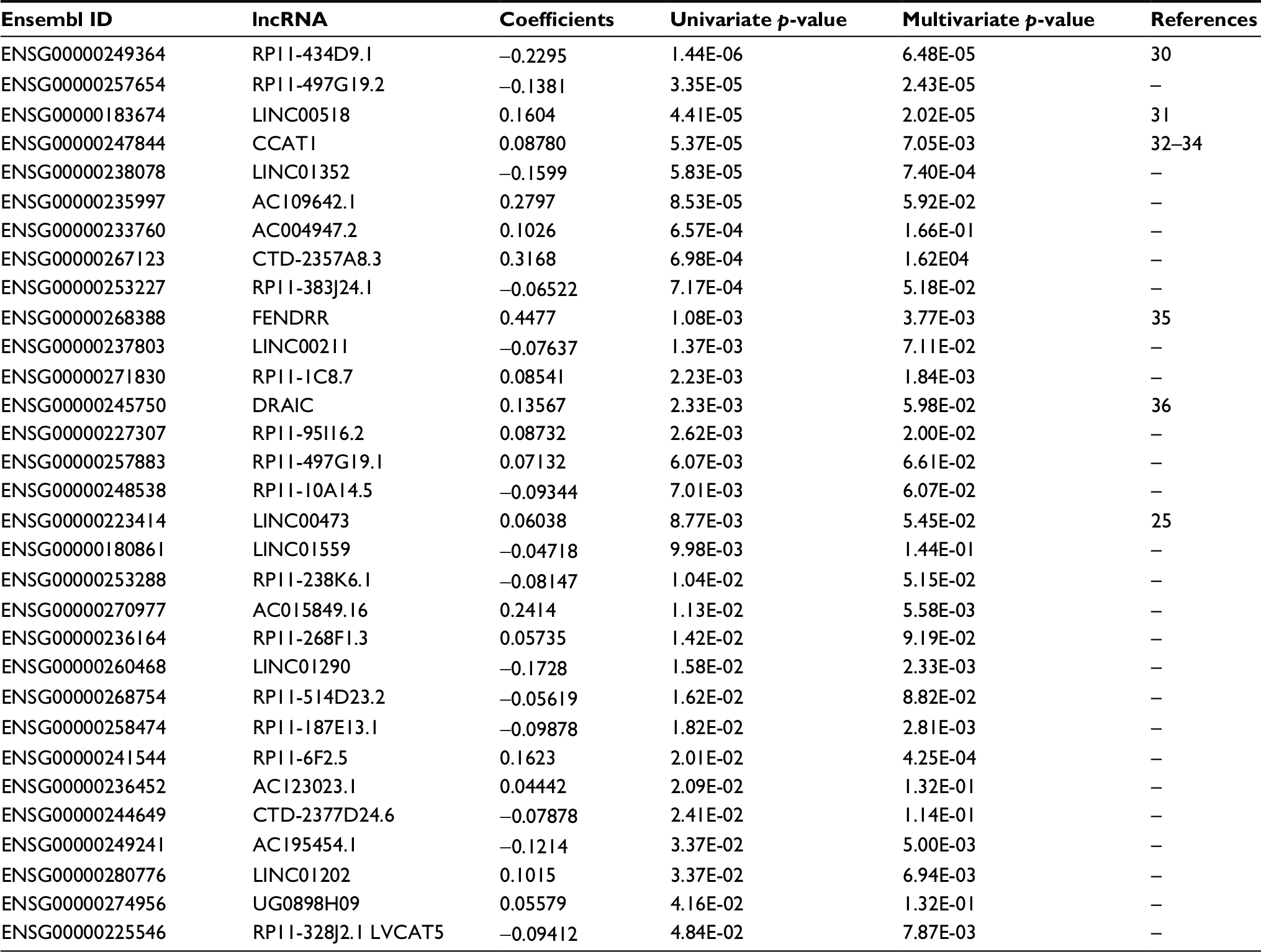 | Table 1 31-lncRNA risk score model Notes: The panel comprised 31 lncRNAs: RP11-434D9.1, RP11-497G19.2, LINC00518, CCAT1, LINC01352, AC109642.1, AC004947.2, CTD-2357A8.3, RP11-383J24.1, FENDRR, LINC00211, RP11-1C8.7, DRAIC, RP11-95I16.2, RP11-497G19.1, RP11-10A14.5, LINC00473, LINC01559, RP11-238K6.1, AC015849.16, RP11-268F1.3, LINC01290, RP11-514D23.2, RP11-187E13.1, RP11-6F2.5, AC123023.1, CTD-2377D24.6, AC195454.1, LINC01202, UG0898H09 and RP11-328J2.1. |
|
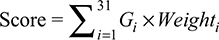
Results
Differentially expressed lncRNAs in LUAD patients
The analysis of the lncRNA expression profiles in both LUAD tissues and normal lung tissues identified a total of 346 differentially expressed lncRNAs, which were used for subsequent survival analyses (Table S1). Compared to normal samples, 249 lncRNAs were overexpressed and 97 lncRNAs were underexpressed in LUAD samples. A cluster dendrogram was generated to ensure that the differentially expressed lncRNAs were good characterizations of LUAD (Figure S1).
The association of lncRNA expressions and OS time
To identify the lncRNAs associated with patient survival in LUAD, univariate Cox regression analysis for the differentially expressed lncRNAs data was assessed. With the significance level threshold of 0.05, a set of 60 lncRNAs was selected (Table S2). These lncRNAs were used in stepwise multivariate Cox regression analysis and 31 lncRNAs were chosen. A cluster dendrogram for these 31 lncRNAs is shown in Figure 1. We conducted a risk score analysis using Equation 1 on the 31 lncRNAs to calculate risk scores for patients. The coefficients of the 31-lncRNA model for determining risk scores are listed in Table 1.
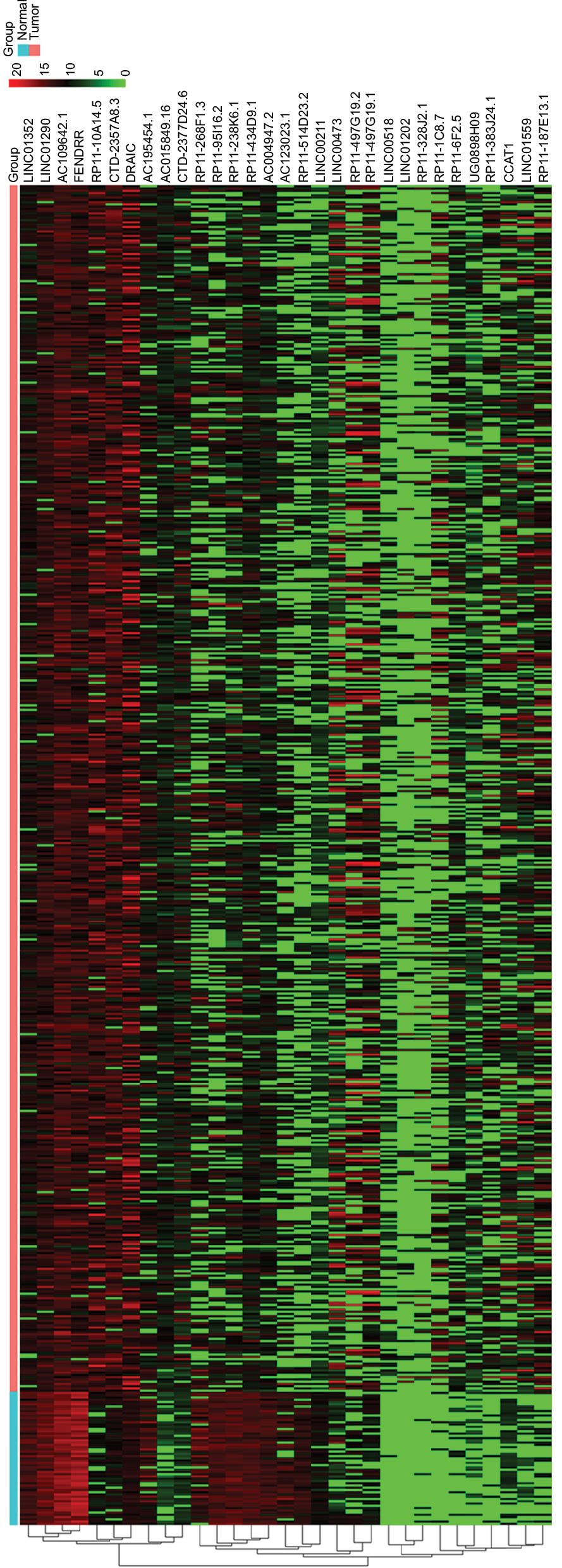 | Figure 1 Hierarchical cluster dendrogram of selected 31 lncRNAs from TCGA LUAD RNA-seq dataset. The left vertical axis shows clusters of lncRNAs. The red rectangular strip in the upper portion of the picture represents normal tissue samples, and the light blue rectangular strip denotes LUAD samples. Red rectangles represent overexpressed genes, and green rectangles represent under-expressed lncRNAs. Black rectangles represent median-expressed lncRNAs. Abbreviations: TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; RNA-seq, RNA sequencing. |
Stage prognostic classifiers
Tumor stage classification was significantly associated with OS of LUAD patients. In order to test whether the 31-lncRNA model is applicable for the prediction of LUAD survival in all stages, we calculated the risk scores for patients in all stages and divided the patients into high-risk and low-risk groups according to the median risk score of patients in all stages (value=1.111). Kaplan–Meier survival curves are displayed in Figure 2, showing that the 31-lncRNA model performs well for LUAD in all stages (p-value=8.508e-11).
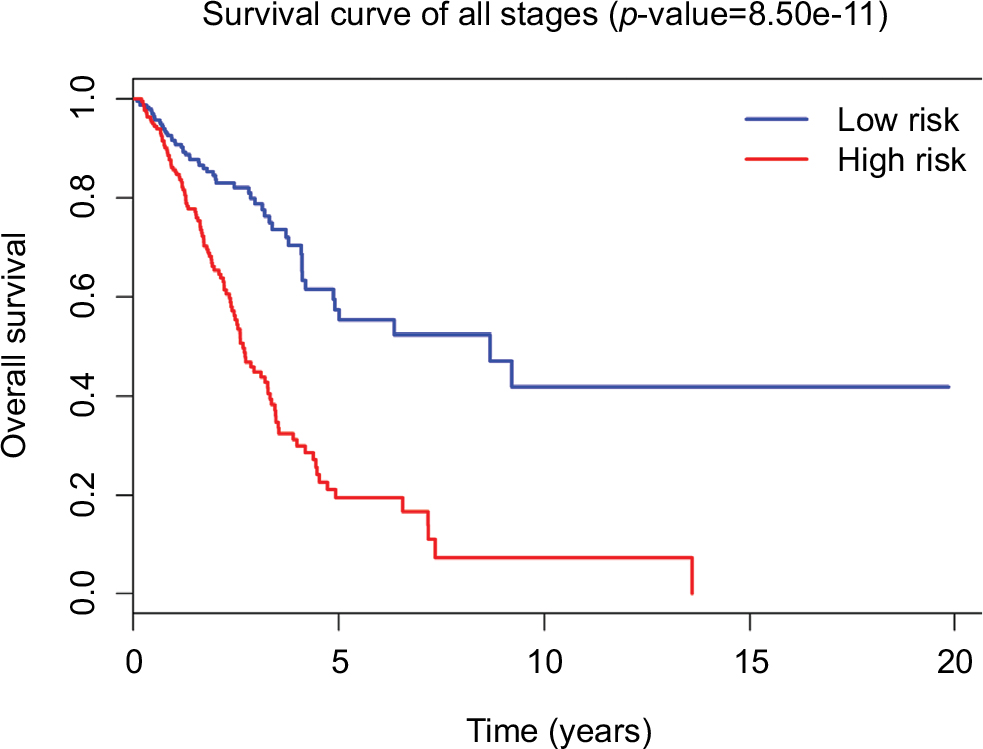 | Figure 2 Survival curves for LUAD patients in all stages using 31-lncRNA model. The differences between the high-risk (n=222) and low-risk (n=240) groups were determined by the log-rank test. Abbreviation: LUAD, lung adenocarcinoma. |
Survival times of low-risk and high-risk groups in stage I
Since tumor stage serves as an important factor that independently affects the survival of LUAD patients, we applied the 31-lncRNA model to stage I patients in order to test the effectiveness of the survival prediction. We divided the patients into high-risk and low-risk groups using the median risk score value 1.111, and found that the high-risk group correlated with poor prognoses for OS (log-rank test p-value 8.917e-13). Five-year OS was 18.9% (95% CI: 10.4%–34.5%) and 89.5% (95% CI: 80.7%–99.2%) for the high-risk and low-risk groups, respectively. The Kaplan–Meier OS curves as well as ROC curves (Figure 3) indicated that the AUC of the 31-lncRNA model was 0.881 (Figure 4), which demonstrated that the 31-lncRNA model has high specificity and sensitivity in predicting the OS time of LUAD patients.37
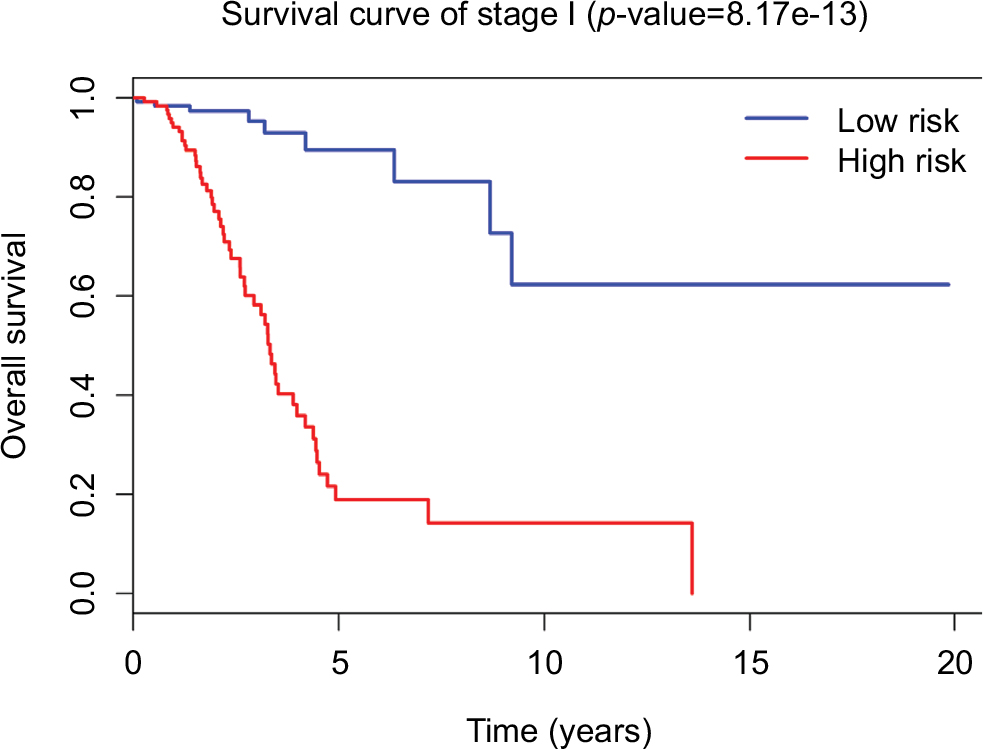 | Figure 3 Survival curves for stage I LUAD patients through the 31-lncRNA model. The differences between the high-risk (n=126) and low-risk (n=126) groups were determined by the log-rank test. Abbreviation: LUAD, lung adenocarcinoma. |
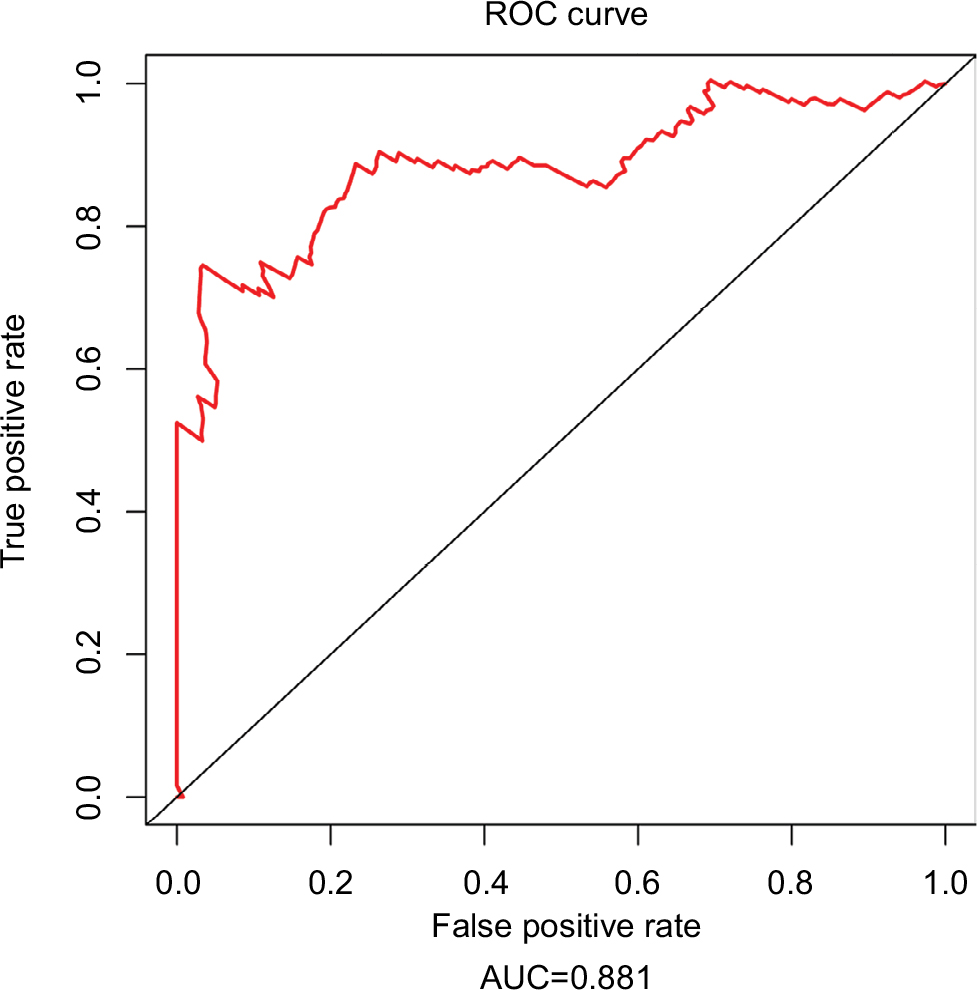 | Figure 4 ROC analysis of the 31-lncRNA model. AUC of the 31-lncRNA model was 0.881. Abbreviations: ROC, receiver operating characteristic; AUC, area under the ROC curve. |
Discussion
In present study, lncRNA expression data and clinical data were obtained from the public TCGA database (HTSeq-FPKM-UQ) of LUAD level 3 RNA-seq. A total of 254 stage I LUAD patients were included. lncRNAs associated with survival were identified by stepwise multivariate Cox regression analysis. Subsequently, an expression pattern of 31 lncRNAs was found to be significantly associated with OS of LUAD patients. The 31-lncRNA panel accurately predicted OS and was applied to conduct a risk score analysis for further investigation of the specificity and sensitivity. ROC analysis results indicate that the statistical power of 31-lncRNA model for high-risk and low-risk patients is formidable. Therefore, the 31-lncRNA signature predicts OS fairly well based on TCGA data sets, which shows its good predictive performance. The AUC was 0.881, which is better than previously reported results.
Some of the lncRNAs that are predicted in our analysis were shown previously to function as potential biomarkers. LINC00473 has been found to correlate with poor NSCLC prognosis.25 Its overexpression is required for the growth and survival of LKB1-inactivated NSCLC cells through CREB-regulated transcription coactivator/CREB-mediated transcription.25 In addition, four lncRNAs including RP11-434D9.1 were found to be differentially expressed in microarray analyses.30 RP11-434D9.1 is correlated with TNBC occurrence, and is a potential biomarker for diagnosis for breast cancer treatment.30 Notably, one lncRNA, CCAT1, was reported to be associated with a variety of solid tumors. Abnormal expression of CCAT1 has been shown in a variety of tumors including lung cancers.34 Abnormally expressed CCAT1 can promote proliferation, migration and invasion in hepatocellular cells,32 gastric carcinoma cells38 and colon cancer cells.39 Currently, some evidences show that CCAT1 acts as a driver lncRNA of malignancy through miRNA sponging. CCAT1 also functions as a molecular sponge for let-7c in docetaxel-resistant LUAD cells,33 and miR-155 in acute myeloid leukemia HL-60 cells. Therefore, CCAT1 may hold an important role in the carcinogenesis of LUAD. Our lncRNA model includes cancer-associated lncRNAs that provide reference values for investigators and give eventuality of explorations in this perspective.
The risk score of the 31-lncRNA signature model was found to have an independent correlation with OS (p-value <0.001) (Table 1). Moreover, we found that one of the clinicopathological parameters of LUAD in the TCGA data, i.e., tumor staging, was significantly associated with OS,5 which was consistent in clinical practice. Furthermore, tumor staging does not efficiently stratify the risk of early stage (stage I and II) LUAD patients. Since this parameter may affect the predictive performance of the model for cancer survival, we further tested the model in stage I LUAD patients. We calculated the risk score for each early stage patient, and the results indicate that the 31-lncRNA signature model is potentially a prognostic classifier for early stage LUAD patients (p-value <0.001). This suggests that patients in stage I and II could be divided into high-risk and low-risk groups by their lncRNA signatures. The results imply that patients with LUAD may benefit from this prognostic signature model, which could determine timeline of adjuvant chemotherapy treatments. There is a speculation that further treatments such as adjuvant therapy will alter the effectiveness of the predictions; however, relevant modifications could be adapted accordingly under the proposed framework.
Using the updated TCGA data (June 2016 version), we were able to obtain 60,483 data points including both protein coding and non-coding genes. Many of these genes are known biomarkers and some are novel genes with survival records. This broadens our scope in gene signature modeling for cancer survival prediction. The prognostic power of the signature model in this study is applied for predicting OS of stage I patients. Moreover, since these lncRNAs might have a predicative role in the outcome of LUAD, further experimental studies to survey the biological roles of these lncRNAs in carcinogenesis are worthwhile to shed new light on specific investigations. Present results show that the current prognostic model is promising and further validations on independent datasets are still needed. In addition, co-regulatory relationship between these 31 lncRNAs will affect the model efficacy, which would be validated furthermore. Based on this work, further analysis such as expression network and co-expression analysis could be applied.
This model is not without limitations. First of all, the data that we could access and analyze are still limited. The TCGA data involved fresh frozen samples, which were collected from top-notch institutions with robust tissue collection systems in place. Thus, only samples that were found to be of very high quality were included. Data in an average setting are expected to be applied by the model to further verify its robustness. For instance, further available data could be applied to analyze the correlation between the lncRNA profile and etiology such as smoking by patients or air pollutant profile of the local area of Guangzhou as well as the expression pattern. With more relevant data, a complete co-expression network could be derived.
Conclusion
In this study, we developed a signature model (Table 1) that is associated with OS in LUAD patients. Patients with a high-risk score from the model have shorter survival time, and this lncRNA panel could help to serve as a prognostic classifier for LUAD. The results of this study suggest that these lncRNAs may play specific roles in the carcinogenesis of LUAD and be of potential prognostic values. Further experiments are needed to verify the connection of 31 lncRNAs to cell survival and apoptosis linking with DNA repair. This model is LUAD specific, and proper adjustments could be made for reference of other disease-associated models.
Availability of data and material
All data generated or analyzed during this study are enclosed in this article and the supplementary information files.
Acknowledgments
This work was supported by Science and Technology Planning Project of Guangdong Province, China (411234349027) , Science and Technology Program of Guangzhou (201803010024) and the Open Project Program of the State Key Laboratory of Respiratory Disease (2014SKLRD-O09). Authors would like to extend their sincere gratitude to Mark Deiparine for editing the manuscript. They also appreciate reviewers’ comments, which helped to improve this presentation. The abstract of this paper and part of the work were presented as a poster in the American Thoracic Society 2017 International Conference in Washington, DC on May 19, 2017 – May 24, 2017.
Author contributions
XL, KH, VYL, YL and AMC analyzed the TCGA data and performed the bioinformatics analysis. XS, HT, HX, XL, YL and JZ analyzed and interpreted the patient data in the TCGA dataset and evaluated the clinical significance. JZ, YL, AMC and XS conceived and designed the study. XS, HT and HX were major contributors in writing the manuscript. All the authors read and approved the final manuscript. All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Tsao MS, Sakurada A, Cutz JC, et al. Erlotinib in lung cancer – molecular and clinical predictors of outcome. N Engl J Med. 2005;353(2):133–144. | ||
Solomon BJ, Mok T, Kim DW, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–2177. | ||
Devarakonda S, Morgensztern D, Govindan R, Genomic alterations in lung adenocarcinoma. Lancet Oncol. 2015;16(7):e342–e351. | ||
Campbell JD, Alexandrov A, Kim J, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48(6):607–616. | ||
Goldstraw P, Chansky K, Crowley J, et al; International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee, Advisory Boards, and Participating Institutions; International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee Advisory Boards and Participating Institutions. The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11(1):39–51. | ||
Chen HY, Yu SL, Chen CH, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356(1):11–20. | ||
Chansky K, Sculier JP, Crowley JJ, Giroux D, Van Meerbeeck J, Goldstraw P; International Staging Committee and Participating Institutions. The International Association for the Study of Lung Cancer Staging Project: prognostic factors and pathologic TNM stage in surgically managed non-small cell lung cancer. J Thorac Oncol. 2009;4(7):792–801. | ||
Kratz JR, He J, Van Den Eeden SK, et al. A practical molecular assay to predict survival in resected non-squamous, non-small-cell lung cancer: development and international validation studies. Lancet. 2012;379(9818):823–832. | ||
Yu SL, Chen HY, Chang GC, et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13(1):48–57. | ||
Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15(1):7–21. | ||
Huarte M. The emerging role of lncRNAs in cancer. Nat Med. 2015;21(11):1253–1261. | ||
Yan X, Hu Z, Feng Y, et al. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015;28(4):529–540. | ||
Mendell JT. Targeting a long noncoding RNA in breast cancer. N Engl J Med. 2016;374(23):2287–2289. | ||
Gutschner T, Hämmerle M, Diederichs S, MALAT1 – a paradigm for long noncoding RNA function in cancer. J Mol Med (Berl). 2013;91(7):791–801. | ||
Shi XS, Li J, Yang RH, et al. Correlation of increased MALAT1 expression with pathological features and prognosis in cancer patients: a meta-analysis. Genet Mol Res. 2015;14(4):18808–18819. | ||
Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391–407. | ||
Ji P, Diederichs S, Wang W, et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003;22(39):8031–8041. | ||
Serghiou S, Kyriakopoulou A, Ioannidis JP. Long noncoding RNAs as novel predictors of survival in human cancer: a systematic review and meta-analysis. Mol Cancer. 2016;15(1):50. | ||
Yuan JH, Yang F, Wang F, et al. A long noncoding RNA activated by TGF-β promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25(5):666–681. | ||
Wei Y, Niu B. Role of MALAT1 as a prognostic factor for survival in various cancers: a systematic review of the literature with meta-analysis. Dis Markers. 2015;2015:164635. | ||
Skrzypski M, Jassem E, Taron M, et al. Three-gene expression signature predicts survival in early-stage squamous cell carcinoma of the lung. Clin Cancer Res. 2008;14(15):4794–4799. | ||
Raz DJ, Ray MR, Kim JY, et al. A multigene assay is prognostic of survival in patients with early-stage lung adenocarcinoma. Clin Cancer Res. 2008;14(17):5565–5570. | ||
Zhou M, Zhao H, Wang Z, et al. Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J Exp Clin Cancer Res. 2015;34:102. | ||
Liu H, Li J, Koirala P, et al. Long non-coding RNAs as prognostic markers in human breast cancer. Oncotarget. 2016;7(15):20584–20596. | ||
Chen Z, Li JL, Lin S, et al. cAMP/CREB-regulated LINC00473 marks LKB1-inactivated lung cancer and mediates tumor growth. J Clin Invest. 2016;126(6):2267–2279. | ||
Han L, Zhang EB, Yin DD, et al. Low expression of long noncoding RNA PANDAR predicts a poor prognosis of non-small cell lung cancer and affects cell apoptosis by regulating Bcl-2. Cell Death Dis. 2015;6: e1665. | ||
Schmidt LH, Spieker T, Koschmieder S, et al. The long noncoding MALAT-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J Thorac Oncol. 2011;6(12):1984–1992. | ||
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. | ||
Yamaoka K, Nakagawa T, Uno T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J Pharmacokinet Biopharm. 1978;6(2):165–175. | ||
Lv M, Xu P, Wu Y, et al. LncRNAs as new biomarkers to differentiate triple negative breast cancer from non-triple negative breast cancer. Oncotarget. 2016;7(11):13047–13059. | ||
Rambow F, Job B, Petit V, et al. New functional signatures for understanding melanoma biology from tumor cell lineage-specific analysis. Cell Rep. 2015;13(4):840–853. | ||
Zhu H, Zhou X, Chang H, et al. CCAT1 promotes hepatocellular carcinoma cell proliferation and invasion. Int J Clin Exp Pathol. 2015;8(5):5427–5434. | ||
Chen J, Zhang K, Song H, Wang R, Chu X, Chen L. Long noncoding RNA CCAT1 acts as an oncogene and promotes chemoresistance in docetaxel-resistant lung adenocarcinoma cells. Oncotarget. 2016;7(38):62474–62489. | ||
Xin Y, Li Z, Shen J, Chan MT, Wu WK. CCAT1: a pivotal oncogenic long non-coding RNA in human cancers. Cell Prolif. 2016;49(3):255–260. | ||
Xu TP, Huang MD, Xia R, et al. Decreased expression of the long non-coding RNA FENDRR is associated with poor prognosis in gastric cancer and FENDRR regulates gastric cancer cell metastasis by affecting fibronectin1 expression. J Hematol Oncol. 2014;7:63. | ||
Sakurai K, Reon BJ, Anaya J, Dutta A. The lncRNA DRAIC/PCAT29 locus constitutes a tumor-suppressive nexus. Mol Cancer Res. 2015;13(5):828–838. | ||
Davis J, Goadrich M. The relationship between precision-recall and ROC curves. In: Proceedings of the 23rd International Conference on Machine Learning; June 25–29, 2006; Pittsburgh, PA: ACM. | ||
Yang F, Xue X, Bi J, et al. Long noncoding RNA CCAT1, which could be activated by c-Myc, promotes the progression of gastric carcinoma. J Cancer Res Clin Oncol. 2013;139(3):437–445. | ||
He X, Tan X, Wang X, et al. C-Myc-activated long noncoding RNA CCAT1 promotes colon cancer cell proliferation and invasion. Tumour Biol. 2014;35(12):12181–12188. |
Supplementary materials
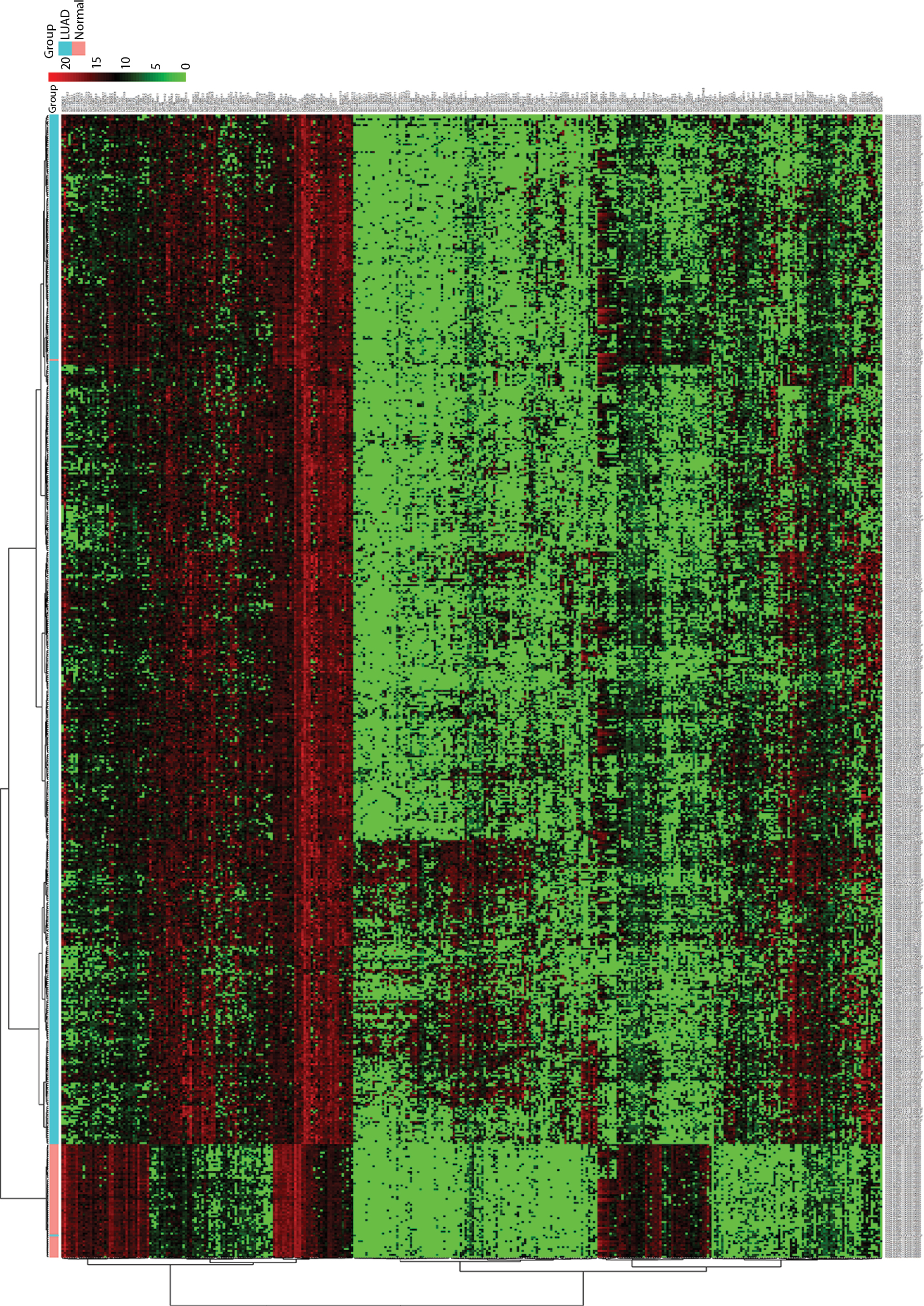 | Figure S1 Hierarchical cluster dendrogram of all differentially expressed lncRNAs from TCGA LUAD RNA-seq dataset. The horizontal axis shows clusters of samples, and the left vertical axis shows clusters of lncRNAs. The red rectangular strip in the upper portion of the picture represents normal tissue samples, and the light blue rectangular strip denotes LUAD samples. Red rectangles represent overexpressed genes, and green rectangles represent underexpressed lncRNAs. Black rectangles represent median-expressed lncRNAs. Abbreviations: TCGA, The Cancer Genome Atlas; LUAD, lung adenocarcinoma; RNA-seq, RNA sequencing. |
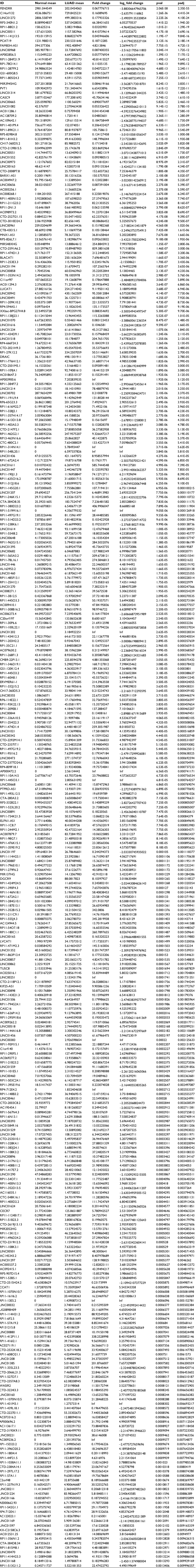 | Table S1 Differentially expressed lncRNAs Abbreviations: LUAD, lung adenocarcinoma; pval, p-value; padj, adjusted p-value; Inf, infinity. |
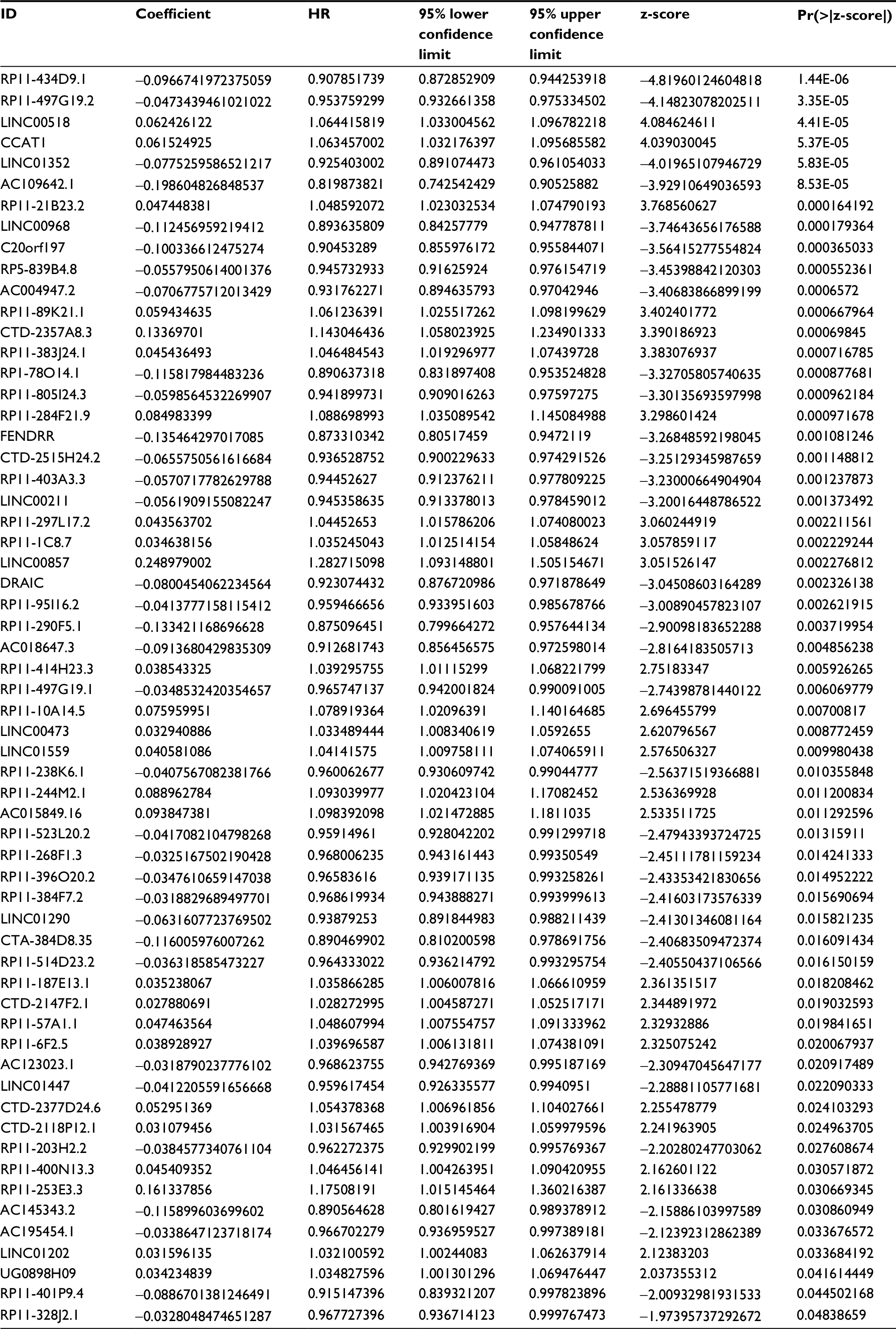 | Table S2 Univariate Cox regression result Abbreviations: HR, hazard ratio; Pr, probability. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.