")
Back to Journals » HIV/AIDS - Research and Palliative Care » Volume 15
An Evaluation on the Role of Non-Coding RNA in HIV Transcription and Latency: A Review
Authors Ramirez PW, Pantoja C , Beliakova-Bethell N
Received 21 December 2022
Accepted for publication 24 February 2023
Published 14 March 2023 Volume 2023:15 Pages 115—134
DOI https://doi.org/10.2147/HIV.S383347
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Olubunmi Akindele Ogunrin
Peter W Ramirez,1 Christina Pantoja,1 Nadejda Beliakova-Bethell2,3
1Department of Biological Sciences, California State University, Long Beach, CA, USA; 2VA San Diego Healthcare System and Veterans Medical Research Foundation, San Diego, CA, USA; 3Department of Medicine, University of California, San Diego, CA, USA
Correspondence: Nadejda Beliakova-Bethell, University of California San Diego, 9500 Gilman Dr, La Jolla, CA, 92093, USA, Tel +858-552-8585; x7189, Fax +1-858 552 7445, Email [email protected]
Abstract: The existence of latent cellular reservoirs is recognized as the major barrier to an HIV cure. Reactivating and eliminating “shock and kill” or permanently silencing “block and lock” the latent HIV reservoir, as well as gene editing, remain promising approaches, but so far have proven to be only partially successful. Moreover, using latency reversing agents or “block and lock” drugs pose additional considerations, including the ability to cause cellular toxicity, a potential lack of specificity for HIV, or low potency when each agent is used alone. RNA molecules, such as microRNAs (miRNAs) and long non-coding RNAs (lncRNAs) are becoming increasingly recognized as important regulators of gene expression. RNA-based approaches for combatting HIV latency represent a promising strategy since both miRNAs and lncRNAs are more cell-type and tissue specific than protein coding genes. Thus, a higher specificity of targeting the latent HIV reservoir with less overall cellular toxicity can likely be achieved. In this review, we summarize current knowledge about HIV gene expression regulation by miRNAs and lncRNAs encoded in the human genome, as well as regulatory molecules encoded in the HIV genome. We discuss both the transcriptional and post-transcriptional regulation of HIV gene expression to align with the current definition of latency, and describe RNA molecules that either promote HIV latency or have anti-latency properties. Finally, we provide perspectives on using each class of RNAs as potential targets for combatting HIV latency, and describe the complexity of the interactions between different RNA molecules, their protein targets, and HIV.
Keywords: HIV, HIV latency, micro RNA, long non-coding RNA, HIV transcripts, gene expression regulation
Plain Language Summary
Human Immunodeficiency Virus (HIV) remains a global concern. Although antiretroviral therapy (ART) significantly increases the life expectancy of people with HIV by eliminating all active forms of the virus, HIV persists in a silent form, called “latency”, in certain types of immune cells. This silent infection is invisible to the immune system, resistant to ART and represents the main barrier to an HIV cure. Approaches to target HIV latency include reactivating (“shock”) and then eliminating (“kill”) cells with latent HIV, permanently silencing “block and lock” HIV in a latent state, or gene editing. While these approaches are promising, they often suffer from a lack of specificity for HIV, cellular toxicity, or low potency. An alternative, or complementary, strategy is to target latently infected cells using RNA-based approaches. Two major types of regulatory RNA molecules exist: micro RNAs and long non-coding RNAs. Their activity is usually cell, tissue, and disease-type specific; therefore, RNA targeting represents an attractive strategy to purge or silence latent HIV infection. Here, we highlight current research on the role of cellular and viral micro RNAs and long non-coding RNAs in the regulation of HIV gene activity. We then discuss how each of these RNAs may be potentially targeted to combat HIV latency.
Introduction
In the present era of combination antiretroviral therapy (cART), the latent cellular reservoir of HIV is recognized as the major barrier to a cure.1 This latent reservoir was initially defined as CD4+ T cells bearing quiescent proviruses that retain the capacity to produce infectious particles.2,3 However, since blocks to RNA splicing, nuclear export, and translation prevent the formation of infectious particles, the current definition of latency does not require complete proviral quiescence.4 Therefore, in this review, we will address the regulation of HIV transcription and the post-transcriptional steps of the replication cycle by expression of host and viral RNAs. Additionally, while CD4+ T cells represent the main reservoir of latently infected cells, monocytes and macrophages are becoming increasingly recognized as additional sources of latent viral reservoirs.5 This review focuses primarily on the roles of RNA molecules in HIV regulation of HIV latency in CD4+ T cells and lymphocytic cell lines; however, observations of their roles in monocytes, macrophages, and promonocytic cell lines are also discussed.
Non-coding RNAs, including micro RNAs (miRNAs) and long non-coding RNAs (lncRNAs), are becoming increasingly recognized as important regulators of gene expression. Generation of miRNAs initiates in the nucleus, where primary miRNA transcripts are processed by the ribonuclease DROSHA into 70–100 nucleotide (nt) long precursor miRNAs (pre-miRNAs).6 Following nuclear export, pre-miRNAs are further processed by the ribonuclease DICER to generate ~22 nt long miRNAs. These miRNAs are loaded into the RNA-induced silencing complex (RISC), which guides them to specific mRNAs using perfect or partial base pairing. Downregulation of target mRNAs is achieved by miRNA-mediated degradation7 or translational repression.8 Additionally, miRNAs can enter the nucleus and either induce9,10 or repress11 gene transcription. LncRNAs are non-coding transcripts greater than 200 nt in length that regulate gene expression at the level of transcription and/or post-transcriptionally by forming RNA-DNA hybrids, via formation of secondary structures, by serving as decoys for proteins and regulatory miRNAs, by guiding proteins to their targets, or serving as scaffolds for protein complexes.12,13 Both miRNAs and lncRNAs may also regulate gene expression indirectly, for example, by targeting transcription factors. Furthermore, the HIV genome encodes several regulatory short RNAs including miRNAs, and lncRNAs.
Clarifying the mechanisms by which RNA molecules regulate HIV gene expression is needed in order to combat HIV latency. This is particularly important, given that several promising approaches have been tested and proven to be only partially successful. One such approach is the “shock and kill” treatment strategy, which has been envisioned as a controlled induction of virus reactivation in the presence of cART to reveal latently infected cells for immune system recognition and destruction. Several functional classes of small-molecule latency reversing agents (LRAs) including epigenetic modifiers, chromatin modulators, signaling effectors, and transcriptional elongation modulators have been identified in low- and high-throughput screens.14 Many have been tested in clinical trials, but the success of these strategies has been limited.15–18 Another promising approach is “block and lock”, designed for long-term silencing of the HIV provirus.19 Several small-molecule compounds have “block and lock” properties;20–23 however, it remains questionable whether complete proviral inhibition using these compounds can be achieved long term. Another important consideration with using LRAs or “block and lock” drugs as therapeutic agents is the potential presence of side effects such as cellular toxicities, lack of specificity for HIV, or low potency when each agent is used alone.24 Finally, genome editing to excise the HIV provirus out of the host genome is another tantalizing approach. Several tools have been used to achieve excision of the HIV provirus, including zinc finger nucleases (ZNFs),25 transcription activator-like effector nucleases (TALENs),26 engineered endonucleases,27 or the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) system.28 While these strategies are promising, they also have limitations, including the potential for off-target editing, the inability to edit all viral quasispecies in a given person with HIV, the development of resistance to genome editing, and challenges with specifically editing latently infected cells.29
Using an RNA-based approach to achieve HIV latency reversal or silencing may be more promising as these strategies may be more specific to latently infected cells. Indeed, miRNAs and lncRNAs are more tissue and cell-type specific than protein coding genes.30–32 RNAs encoded by the HIV genome are expressed exclusively in HIV-infected cells and may present the best targets for HIV latency reversal or silencing. Furthermore, investigating the regulatory roles of RNA molecules will likely reveal redundancies and discrepancies in their function on known protein regulators of HIV expression. Finally, using an RNA-based approach to combat HIV latency has precedence: the FDA-approved drug Patisiran uses RNA interference (RNAi) to reduce the production of a mutated form of the transthyretin protein to treat hereditary transthyretin amyloidosis.33 Here, we provide a summary of the roles of miRNAs, lncRNAs and HIV RNA in the regulation of HIV gene expression and highlight strategies to target the HIV latent reservoir.
Methods
Literature searches were conducted in PubMed using keywords (eg HIV, HIV latency, microRNAs, miRNAs, long non-coding RNAs, lncRNAs, and regulatory RNAs). Papers specifically related to transcriptional or post-transcriptional regulation of HIV latency were selected for the review. Both validated and hypothetical roles of RNA molecules in HIV latency control were discussed. Figures were created in Biorender.com.
Role of miRNAs Encoded in the Human Genome in the Regulation of HIV Latency
Mechanisms of action of miRNAs to regulate HIV expression include acting on protein-coding genes that function as HIV regulators, or directly targeting HIV RNA (Figure 1). MiRNAs that target mRNAs of HIV activator proteins, such as transcription factors, may regulate “deep” proviral latency and facilitate complete quiescence. Alternatively, miRNAs may cause degradation of mRNAs of HIV repressor proteins and have anti-latency properties. MiRNAs that act directly on HIV RNA may regulate post-transcriptional latency, for example, by causing inhibition of viral protein production. Below, we provide a comprehensive list of miRNAs that act via these mechanisms. For the summary of this section, please refer to Table 1.
 |
Table 1 Summary of miRNAs That Promote or Antagonize HIV Latency |
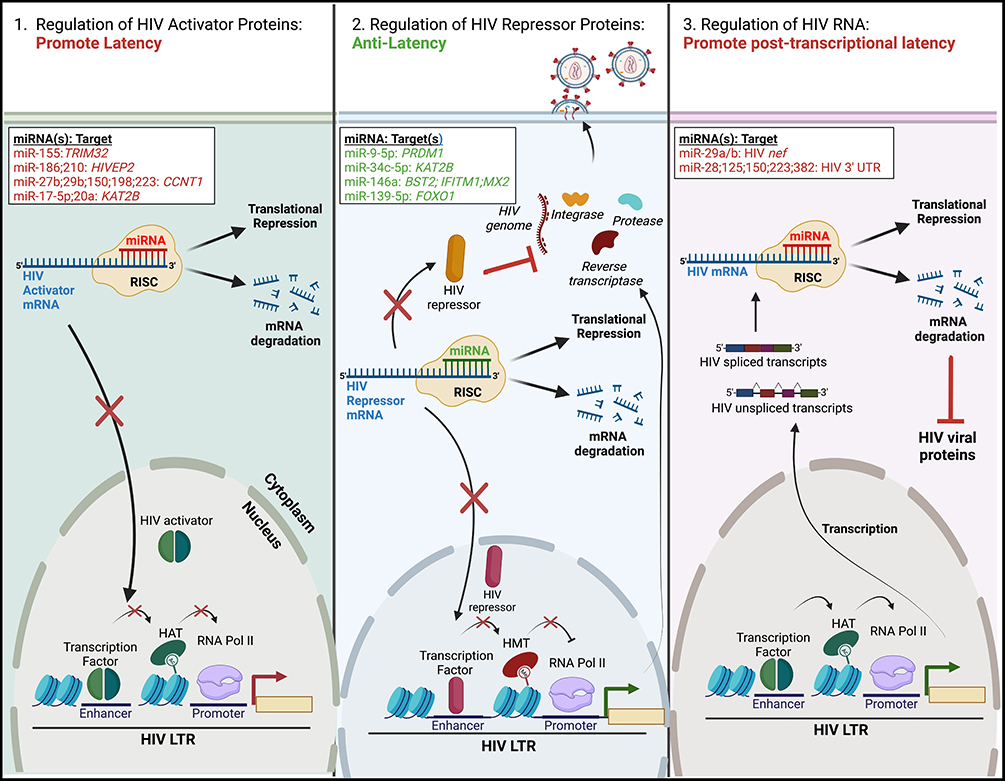 |
Figure 1 Regulation of HIV latency by cellular miRNAs. MiRNAs can regulate HIV latency via three distinct mechanisms. All rely on the binding of a cellular miRNA, in complex with the RNA-induced silencing complex (RISC), to the 3’ end of cellular or viral mRNAs. This either leads to translational repression or mRNA degradation. (Left) Some miRNAs reduce the levels of HIV activator proteins (ie transcriptional activators), leading to maintenance of the HIV latent reservoir. (Middle) Alternatively, other miRNAs decrease the expression of HIV repressor proteins, such as transcriptional silencers or restriction factors. This facilitates activation of the proviral LTR and latency reversal. (Right) MiRNAs may also promote latency post-transcriptionally via the repression of HIV spliced or unspliced transcripts, leading to reduced levels of HIV viral proteins. Specific cellular miRNAs and their cellular or viral mRNA target(s) are listed; miRNAs/target(s) that promote latency are highlighted in red, while those that have anti-latency properties are highlighted in green. The figure was created in Biorender.com. Abbreviations: BST2, bone-marrow stromal antigen 2; CCNT1, cyclin T1; FOXO1, forkhead box O1; HIVEP2, HIVEP zinc finder 2; IFITM1, interferon induced transmembrane protein 1; KAT2B, lysine acetyltransferase 2B; LTR, long terminal repeat; MX2, MX dynamin-like GTPase 2; PRDM1, PR/SET Domain 1; RNA Pol II, RNA polymerase II; TRIM32, tripartite motif containing 32; UTR, untranslated region. |
Regulators of HIV Activator Proteins
A number of miRNAs act to negatively regulate HIV activator proteins; thus, their action promotes HIV latency establishment or maintenance of the provirus in an inactive state (Figure 1, left). In a leukemic T cell line latently infected with HIV encoding green fluorescent protein (GFP) and containing a frameshift-mutation in the envelope gene (termed J-Lat 5A8), miR-155 targeted tripartite motif containing 32 (TRIM32).34TRIM32 stimulates nuclear factor kappa light-chain enhancer of activated B cells (NF-kB) signaling, a central pathway that regulates viral gene transcription from the HIV promoter, long terminal repeat (LTR).35,36 As a result of TRIM32 downregulation by miR-155, NF-kB signaling was suppressed, leading to reduced activity of the HIV LTR. MiRNAs miR-186 and miR-210 were upregulated by HIV infection in the human T cell lymphoblastic lymphoma (Sup-T1) cell line.37 The mRNA target of these miRNAs is HIVEP zinc finder 2 (HIVEP2), an RNA encoding an HIV enhancer binding protein. Its expression was induced by mitogen and phorbol ester treatment, suggesting that it may act in HIV reactivation from latently infected cells.38 Thus, downregulation of HIVEP2 by miR-186 and miR-210 would be predicted to result in viral suppression.
Several miRNAs were reported to act via interfering with cellular cofactors of the HIV transactivator protein Tat, thus inhibiting HIV mRNA elongation and promoting latency. Overexpression of miRNAs miR-27b, miR-29b, miR-150 and miR-223 in primary CD4+ T cells resulted in reduced levels of Cyclin T1 (encoded by CCNT1), a component of the positive transcription elongation factor (p-TEFb) complex.39 These miRNAs had higher expression in resting compared to activated primary CD4+ T cells, where levels of Cyclin T1 are low.39 Cyclin T1 mRNA was shown to be a direct target of miR-27b; however, miR-29b, miR-150 and miR-223 likely acted indirectly39 to reduce expression of Cyclin T1 and promote HIV latency. Interestingly, a different miRNA, miR-198, acted to promote HIV latency via the reduction of Cyclin T1 expression in the promonocytic cell line Mono Mac 6.40 Another cofactor of Tat regulated by miRNAs is p300/CBP-associated factor (PCAF),41 also known as lysine acetyltransferase 2B (KAT2B). MiR-17-5p and miR-20a targeted the 3’ untranslated region (UTR) of PCAF, leading to PCAF translational inhibition. Overexpression of these miRNAs in T lymphocytic (Jurkat) and promonocytic (U1) cell lines and peripheral blood mononuclear cells (PBMCs) isolated from persons with HIV resulted in the inhibition of HIV replication.41 While these miRNAs were not directly tested in HIV latency reversal, their ability to modulate PCAF similar to the action of miR-27b via Cyclin T1 suggests they may play a role in promoting latency.
Regulators of HIV Repressor Proteins
Conversely, the downregulation of a number of miRNAs was reported to promote integration and establishment of latent reservoirs or facilitate stable latent HIV infection. These miRNAs negatively regulate HIV repressor proteins and therefore have anti-latency properties (Figure 1, middle). One example of such miRNA is miR-9-5p. In a T-cell lymphoma cell line HUT78, this miRNA negatively regulated expression of B lymphocyte-induced maturation protein-1 (BLIMP-1, encoded by PR/SET Domain 1, PRDM1),42 which inhibits HIV transcription via binding to the HIV LTR directly43 or acts indirectly via recruitment of histone deacetylases (HDACs).44 Conversely, silencing of miR-9-5p in primary CD4+ T cells45 and its downregulation by transforming growth factor beta 1 in primary differentiated human bronchial epithelial cells46 resulted in an increase of expression levels of BLIMP-1, promoting the establishment of the latent HIV reservoir. MiR-9-5p may have clinical relevance because its expression was lower in PBMCs from elite controllers (ECs) compared to viremic long-term non-progressors (LTNPs).47
Another example is miR-34c-5p. Expression of this miRNA was elevated in activated CD4+ T cells but was downregulated during HIV infection. In a leukemic T cell line (Jurkat) engineered to stably express miR-34c-5p, downregulation of miR34c-5p was associated with a reduction in HIV proteins, but an increase in integrated proviral DNA levels,48 suggesting that this miRNA negatively affects the establishment of the HIV latent reservoir. Furthermore, the targets of miR-34c-5p were enriched for proteins with functions in the HIV replication cycle, of which six were enhancers and two inhibitors of viral replication.48 Surprisingly, enhancers were downregulated as the result of overexpression of miR-34c-5p, including transcriptional activator KAT2B, which was experimentally validated in this study. The authors argued that despite the downregulation of HIV transcriptional activators, the combined impact of miR-34c-5p expression on multiple cellular pathways created a favorable environment for HIV replication48 rather than a latent proviral state.
CRISPR-Cas9 depletion of miR-146a in a leukemic T cell line (MT2) infected with HIV led to the downregulation of HIV mRNA and the upregulation of multiple HIV restriction factors, such as MxB (also known as MX dynamin-like GTPase 2, MX2), interferon (IFN) induced transmembrane protein 1 (IFITM1) and Tetherin (also known as bone marrow stromal cell antigen 2, BST2). Furthermore, in a T lymphocyte cell line (Jurkat-C11) containing a replication-incompetent latent HIV genome expressing enhanced GFP,25 abrogation of miR-146a expression resulted in suppression of proviral reactivation by the LRA suberoylanilide hydroxamic acid.49 Finally, miR-139-5p was upregulated in the JLat 10.6 cell line model of HIV latency by treatment with plasma extracellular vesicles.50 These leukemic T cells contain a molecular clone of HIV encoding GFP in place of nef and a frameshift mutation in env.51 As a result of miR-139-5p upregulation, forkhead box O1 (FOXO1) expression was reduced, while the proportion of cells expressing the GFP reporter, indicative of proviral activation, was increased.50 Knockdown or chemical inhibition of FOXO1 was previously demonstrated to result in reactivation of latent HIV proviruses in primary CD4+ T cells;52 thus, the mechanism of viral reactivation in JLat 10.6 cells is likely the interaction of miR-139-5p with its gene target, FOXO1.50
Direct Regulators of HIV RNA
Several miRNAs have been experimentally confirmed to act by directly targeting HIV mRNA and repress HIV replication post-transcriptionally (Figure 1, right). One of the miRNAs that performs such a function is miR-29a. Its functional site is located in a highly conserved region of the 3′ untranslated region (UTR) of the HIV genome. In promonocytic (U1, a derivative of U937) and T cell (J1.1, derived from Jurkat T cells) line models of HIV latency with low levels of basal HIV RNA expression, overexpression of miR-29a further reduced HIV replication, and its knockdown resulted in modest HIV reactivation from latency.53 Using HEK 293 and Jurkat T cell lines transfected with either a nef expression vector or an HIV molecular clone, Ahluwalia et al demonstrated that miR-29a and miR-29b acted to reduce the levels of Nef protein, proposing this as a mechanism for the reduction in HIV replication.54 However, it was not clear from these studies whether other HIV proteins, or mRNA itself, were reduced due to mRNA degradation. An elegant study by Nathans et al used reciprocal mutagenesis analysis in the HEK 293 T cell line to demonstrate the direct interaction of miR-29a and HIV RNA, which resulted in enhanced association of HIV RNA interactions with the RISC complex.55 They further demonstrated that HIV mRNA is associated with processing body (P body) proteins, and that disruption of P bodies resulted in the increase of HIV replication. P bodies localize to the cytoplasm and are composed of ribonucleoprotein complexes primarily associated with post-transcriptional regulation.56 Consistent with post-transcriptional functions of P bodies, the study by Nathans et al showed that regulation of HIV replication in P bodies occurred via a translation inhibition mechanism, as levels of HIV proteins were reduced, but levels of HIV mRNA were unaffected by P body disruption.55
In a report that used primary CD4+ T cells, 5 miRNAs were identified with a likely function in promoting HIV latency: miR-125b, miR-150, miR-28, miR-223, and miR-382.57 Binding at the 3′ end of HIV RNA, these miRNAs could inhibit the translation of almost all HIV-encoded proteins, including Tat and Rev.57 Downregulation of Tat and Rev proteins could further contribute to enhancing viral latency. Inhibition of these five miRNAs did not result in the increased expression of either unspliced or spliced HIV RNA, only HIV proteins, suggesting a role in translational regulation.57 Remarkably, this study demonstrated that inhibition of these five miRNAs could reactivate latent provirus both in vitro and using samples from people with HIV ex vivo.57
MiRNA Perspectives
Studies that provide evidence on the role of miRNAs in HIV gene expression regulation, and particularly, latency, point to an overall complexity of regulation of HIV by miRNAs. Examples provided here show that the miRNA regulation of host factors that control HIV expression is multi-faceted. Expression of a single miRNA, miR-34c-5p, provided a favorable environment for HIV replication48 despite targeting HIV activators such as KAT2B; conversely, targeting KAT2B by miR-17-5p and miR-20a resulted in the inhibition of HIV replication.41 These observations suggest that the expression levels of each individual miRNA in a given latently infected cell must be important for the overall activity of the HIV LTR. The cellular environment also likely plays a role in HIV gene regulation. For example, even though inhibition of miR-125b, miR-150, miR-28, miR-223, and miR-382 regulated HIV reactivation from latency when cells from people with HIV were used,57 these observations were not confirmed with models of latency established in vitro.58 These models of latency were established in the presence of chemokines or cytokines, such as C–C Motif Chemokine Ligand 19 or interleukin 7;58 therefore, the cellular environment in these models could differ from resting CD4+ T cells obtained from people with HIV. Furthermore, contradictory relationships were observed between the roles of miRNAs evaluated in vitro and their expression levels in clinically interesting populations such as HIV ECs. These individuals maintain undetectable viral loads (less than 50 RNA copies/mL) in the absence of ART.59 Within ECs, several miRNAs had lower levels of expression compared to viremic LTNPs: miR-150, 155–5p, 29a, 146a, and 9–5p.47 In vitro studies summarized above suggest that miR-150, 155–5p, and 29a promote latency, while 146a and 9–5p antagonize latency. Therefore, low expression levels of 146a and 9–5p, but not miR-150, 155–5p, and 29a, would be consistent with the HIV EC phenotype, where provirus is suppressed.
Overall, the results from many studies give us hope that manipulating miRNA expression can facilitate changes in HIV expression levels (such as reactivation from latency). However, this may not always be the case, and the results should be interpreted with caution and validated in several experimental systems. For example, López-Huertas et al identified several miRNAs differentially expressed in latently infected cells: miR-98-5p, miR-4516, and miR-7974; however, their inhibition did not result in quantifiable changes in HIV gene expression.58 Finally, the role played by any individual miRNA may be cell type-dependent. For example, miR-4697-3p was upregulated in latently infected, but not activated CD4+ T cells or during productive infection.60 Its predicted mRNA target is colony-stimulating factor 2 (CSF2).60 This protein was initially shown to negatively regulate HIV replication at a post-DNA synthesis step in macrophages,61 suggesting a potential role in latency maintenance. However, in primary CD4+ T cells this gene was upregulated by activators of the protein kinase C pathway.62 Expression levels of CSF2 were elevated in conjunction with HIV reactivation, suggesting a potential role of CSF2 as an HIV activator in CD4+ T cells. Evaluation of miRNA expression in conjunction with the expression of protein-coding host factors and activity of the HIV LTR at the single cell level is the next logical step that would help tie the results of these studies together.
Role of Human Long Non-Coding RNAs in the Regulation of HIV Latency
LncRNAs are generally greater than 200 nucleotides long and were previously characterized as “dark matter” or transcriptional noise.63 However, lncRNAs are now appreciated to affect many biological processes, including gene expression, oncogenesis, and pathogenesis. Functionally, lncRNAs serve as molecular “sponges”, binding to ribonucleoprotein complexes to aid in gene activation, repression, and chromatin modification.12 LncRNAs may regulate HIV latency at either the transcriptional or posttranscriptional level (Figure 2). Below we describe their mechanisms in detail. For the summary of this section, please refer to Table 2.
 |
Table 2 Summary of lncRNAs That Promote or Antagonize HIV Latency |
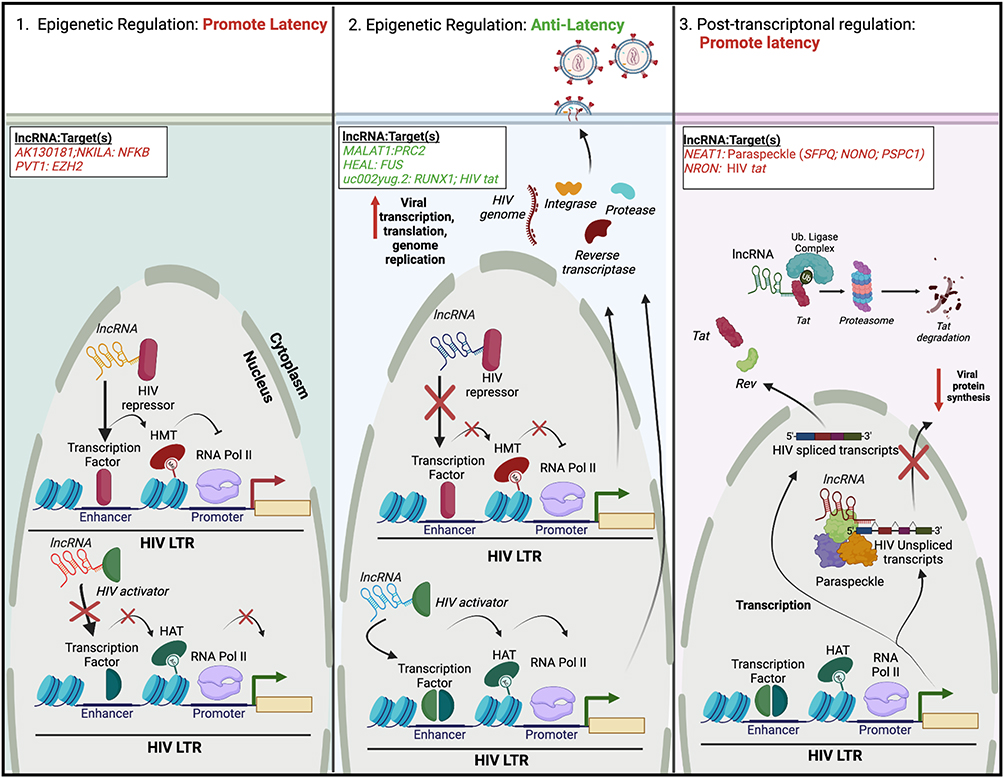 |
Figure 2 Regulation of HIV latency by cellular lncRNAs. Similar to miRNAs, lncRNAs may facilitate the regulation of HIV latency via three mechanisms, two of which exist at the epigenetic level. (Left) LncRNAs can bind and/or recruit HIV repressors to the HIV 5′ LTR. This leads to repressive epigenetic modifications, such as histone methylation. Alternatively, lncRNAs can inhibit the binding/recruitment of HIV activators to the HIV 5′ LTR. Both cases would lead to transcriptional repression and latency maintenance. (Middle) Alternatively, some lncRNAs may either inhibit (HIV repressors) or promote (HIV activators) the recruitment of protein complexes to the HIV-1 5′ LTR, resulting in gene activation, and reactivation of HIV from latency. (Right) Other lncRNAs may promote latency post-transcriptionally. In one scenario, lncRNAs can associate with nuclear proteins to form paraspeckles, or complexes that regulate mRNA export. Binding of unspliced HIV transcripts to paraspeckles can inhibit their nuclear export, decreasing viral protein synthesis. Alternatively, lncRNAs can link viral regulatory proteins (Tat) to the ubiquitin proteasome system, facilitating their premature degradation. Specific lncRNAs and their cellular or viral protein target(s) are listed; lncRNAs/targets that promote latency are highlighted in red, while those that have anti-latency properties are highlighted in green. The figure was created in Biorender.com. Abbreviations: EZH2, enhancer of zeste 2 polycomb repressive complex 2 subunit; HAT, histone acetyl transferase; HEAL, HIV-enhanced lncRNA; HMT, histone methyl transferase; LTR, long terminal repeat; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; NEAT1, nuclear paraspeckle assembly transcript 1; NF-kB, nuclear factor kappa light-chain enhancer of activated B cells; NKILA, NF-kB-interacting lncRNA; NONO, non-POU domain containing octamer binding; NRON, nuclear factor of activated T cells; PRC2, polycomb repressive complex 2; PSPC1, paraspeckle component 1; PVT1, plasmacytoma variant translocation 1; RUNX1, RUNX family transcription factor 1; SFPQ, splicing factor proline and glutamine rich; RNA Pol II, RNA polymerase II; Ub, ubiquitin. |
Epigenetic Regulation Promoting HIV Latency
Some lncRNAs act to negatively regulate the epigenetic status of the HIV LTR. Therefore, their expression maintains HIV in a transcriptionally silent state, promoting latency (Figure 2, left). Inhibiting the lncRNA AK130181 in HIV latently infected Jurkat and primary CD4+ T cells or resting CD4+ T cells from people with HIV on cART led to viral reactivation upon T cell activation.64 The NF-kB-interacting lncRNA (NKILA), initially identified as a tumor suppressor that negatively regulates NF-kB signaling,65 inhibited the replication of diverse HIV clones and clinical subtypes in the HEK 293 T cell line.66 Mechanistically, both AK130181 and NKILA inhibited LTR-driven gene transcription in a NF-kB dependent manner.64,66 NF-kB (p50/p65) binding to the HIV LTR facilitated the recruitment of histone acetyltransferases (HATs), promoting HIV transcription and activation. Expression of NKILA blocked the recruitment of p65 to the HIV LTR, impeding HIV activation. Conversely, silencing NKILA promoted the reactivation of HIV within latently infected T cell lines.66 Interestingly, levels of NKILA are inversely correlated with HIV replication in clinical samples, and overexpressing NKILA in cells from people with HIV decreased proviral reactivation upon stimulation with phytohemagglutinin.66
Other lncRNAs may possibly promote latency by recruiting and/or stabilizing transcriptional repressors. Recently, RNA sequencing (RNA-Seq) identified plasmacytoma variant translocation 1 (PVT1) as being upregulated in two T cell models of HIV latency.60 PVT1 has been implicated in many cases of human disease, mainly oncogenesis.67,68 In the setting of hepatocellular carcinoma (HCC), PVT1 increased expression of the transcriptional repressor enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), leading to HCC propagation and the inhibition of apoptosis.69 Because EZH2 has been implicated in the regulation of HIV latency,70 PVT1 may act to repress HIV transcription via the recruitment and/or activation of EZH2. However, the precise role of PVT1 in this process remains to be determined.
Epigenetic Regulation Promoting HIV Activation
Conversely, the expression of some lncRNAs may serve to activate HIV transcription and replication (Figure 2, middle). The lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was upregulated in an RNA-Seq analysis of HIV-infected CD4+ T cells.71 Reducing expression of MALAT1 led to a decrease in HIV LTR-driven gene transcription and viral replication. By associating with the polycomb repressive complex 2 (PRC2), MALAT1 inhibited EZH2 from binding to the HIV promoter. Collectively, this resulted in the inhibition of the methylation of histone 3 on lysine 27, altering the epigenetic status of the HIV LTR to promote active gene transcription. Treating HIV latently infected cells with LRAs resulted in the induction of MALAT1 expression, while MALAT1 expression decreased in people with HIV on cART.71 Thus, expression of MALAT1 is positively correlated with active HIV replication. Similarly, the lncRNA HIV-enhanced lncRNA (HEAL) was also upregulated upon HIV infection, particularly within myeloid-derived macrophages (MDMs), microglia (macrophages found in the brain), and T cells.72 In contrast to NKILA, HEAL acted as a broad regulator in promoting, rather than inhibiting, the replication of a diverse range of HIV strains and tropisms.72 HEAL was associated with the RNA-binding protein FUS and together this complex promoted active HIV transcription and replication via two distinct mechanisms. First, the HEAL-FUS complex could bind to the HIV promoter, recruiting the HAT p300. This led to histone H3K27 acetylation and p-TEFb enrichment. Second, the HEAL-FUS complex enhanced cyclin-dependent kinase 2 (CDK2) gene expression by being enriched at the CDK2 promoter.72 Importantly, knockout of HEAL via CRISPR/Cas9 inhibited the return of active HIV replication within T cells and microglia after stopping azidothymidine treatment in vitro.72
The effect of an lncRNA on HIV replication may be indirect, via altering the expression of HIV activator and repressor proteins. For example, the lncRNA uc002yug.2 enhanced HIV replication and promoted HIV LTR activity and the reactivation from latency within T cell lines and primary CD4+ T cells from people with HIV.73 Uc002yug.2 facilitated these functions by either 1) upregulating the expression of the HIV activator Tat or 2) downregulating mRNA levels of cellular RUNX family transcription factor 1 (RUNX1) isoforms, particularly HIV repressors RUNX1b and −1c, in a cell type-dependent manner.73
Post-Transcriptional Regulation Promoting HIV Latency
In contrast to lncRNAs that regulate HIV transcription at an epigenetic level, some lncRNAs contribute to the maintenance of HIV proviral DNA post-transcriptionally (Figure 2, right). A profiling of 83 disease-related lncRNAs within HIV-infected T cells identified the lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1) as being dysregulated during HIV infection.74 HIV infection increased overall levels of NEAT1, while silencing of NEAT1 increased HIV production via a decrease in paraspeckle formation.74 Paraspeckles are subnuclear bodies composed of the core proteins: splicing factor proline and glutamine rich (SFPQ, also known as PSF), non-POU domain containing octamer binding (NONO, also known as P54NRB) and paraspeckle component 1 (PSPC1) that associate with NEAT1 to regulate gene expression via nuclear retention of certain RNAs.75 Some unspliced HIV transcripts contain cis-acting instability elements that are bound by paraspeckle proteins,76 impeding their nuclear export and stability.77 Interestingly, the knockdown of NEAT1 increased the Rev-dependent nuclear export of unspliced viral mRNAs.74 Furthermore, because knockout of NEAT1 in CD4+ T cell lines increased HIV replication and NEAT1 was downregulated upon T cell activation in primary CD4+ T cells and resting PBMCs,78 NEAT1 likely contributes to overall antiviral immunity. The exact role of NEAT1 in regulating HIV latency, however, remains undefined.
In contrast to NEAT1, the noncoding repressor of nuclear factor of activated T cells (NRON) contributes to HIV latency via two distinct mechanisms. NRON was discovered via lncRNA profiling in two human T cell lines, where its expression was modulated following HIV infection and replication.79 In one model, the early viral accessory protein Nef reduced NRON expression, while the late viral accessory protein Vpu increased NRON expression levels. How exactly Nef and Vpu modulate NRON expression remains unclear, though reducing expression of NRON led to an increase in HIV replication in a nuclear factor of activated T cells (NFAT)-dependent manner and both Nef and Vpu modulated NFAT activity indirectly through their effects on NRON.79 In another model, NRON induced the degradation of the viral transactivator protein Tat by specifically linking the protein to components of the ubiquitin/proteasome system.80 Moreover, reducing expression of NRON in combination with LRA treatment, specifically HDAC inhibitors, drastically increased viral reactivation within HIV latently infected primary CD4+ T cells.80
Finally, in MDMs, RNA-seq identified the lncRNA negative regulator of IFN response (NRIR) as being upregulated upon either HIV infection or treatment with Type I, II or III IFNs.81 NRIR was initially discovered in a lncRNA screen within primary hepatocytes treated with Type I IFN.82 Knockdown of NRIR reduced Hepatitis C virus infection and led to the upregulation of various IFN stimulated genes.82 This suggests that NRIR may dampen the IFN response against HIV within macrophages. Exactly how NRIR contributes to HIV latency, however, is still unclear.
LncRNA Perspectives
Similar to miRNAs, lncRNAs participate in the regulation of HIV expression at different levels and present potential gene targets to achieve HIV LTR repression or reactivation. Some reported modes of action may suggest specific anti-latency strategies. For example, knockdown of AK130181 and NKILA or overexpression of MALAT1, HEAL, or uc002yug.2 would result in HIV reactivation from latency, while overexpression of AK130181 and NKILA and knockdown of MALAT1, HEAL or uc002yug.2 might lead to suppression of the HIV LTR. Due to multiple lncRNAs interacting with the same protein (eg MALAT1,71 NEAT183,84 and PVT169,85 all interact with EZH2), the functions of these lncRNAs on the HIV LTR may be redundant or opposite. Like with miRNAs, it is important to consider the level of expression of individual lncRNAs in any given latently infected cell to better understand the balance that dictates a certain HIV transcriptional outcome.
Because lncRNAs can serve as miRNA sponges, lncRNA/miRNA interactions likely contribute to the complexity of HIV expression regulation. Indeed, growth arrest-specific transcript 5 (GAS5) was reported to be a sponge for miR-873.86 While miR-873 promoted HIV replication, the effect of GAS5 was inhibitory, in line with GAS5 acting as a competing endogenous RNA for miR-873.86 To our knowledge, there have been no reports on the role of lncRNA/miRNA interactions in specifically regulating HIV latency. However, some interactions can be hypothesized. For example, PVT1 has been widely investigated as a miRNA sponge in multiple cancers and other non-HIV related studies. In these studies, a number of miRNAs that promote HIV latency were shown to be sponged by PVT1: miR-186,87–89 miR-27b,90 miR-17-5p,91,92 miR-20a,93 and miR-125b.94 Since these miRNAs promote HIV latency, targeting their interaction with PVT1 may result in driving the provirus into a deeper silent state.
Targeting lncRNAs for HIV cure approaches does not require that they be knocked down or knocked out. Instead, identifying sequences by which an lncRNA interacts with a protein or a miRNA would allow the introduction of precise mutations using genome editing such as CRISPR/Cas9. Unlike mutations in protein-coding genes, these lncRNA mutations will not result in the disruption of their overall function. We pose that such strategies are more likely to have less off-target or toxic effects, compared to complete abrogation of lncRNA expression.
Role of HIV-Encoded RNAs in the Regulation of HIV Latency
There is growing evidence that abortive transcription occurs from the HIV genome generating short transcripts; that miRNAs are encoded in the HIV genome, and that transcription from the HIV genome can also occur in the antisense orientation (Figure 3). Below we review in detail how these different HIV RNA species contribute to the regulation of HIV expression and latency. For the summary of this section, please refer to Table 3.
 |
Table 3 Summary of RNAs Encoded in the HIV Genome That Promote or Antagonize HIV Latency |
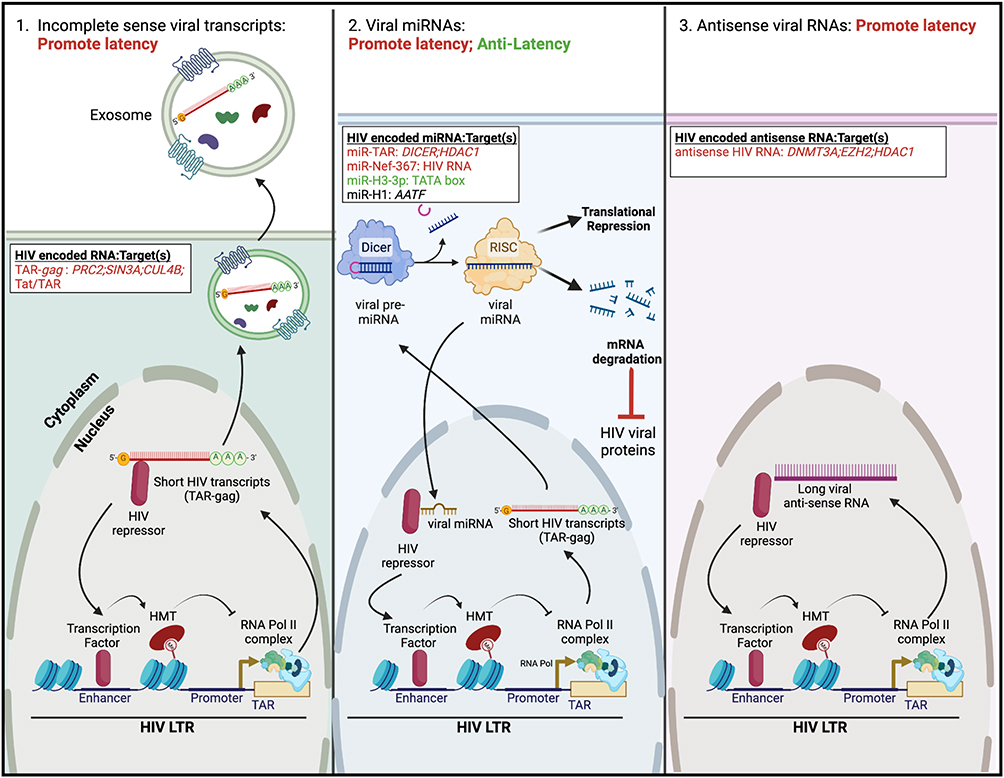 |
Figure 3 Regulation of HIV latency by viral RNAs. (Left) In some instances, viral transcription from the HIV LTR produces short non-coding HIV transcripts (termed TAR-gag). These viral transcripts can either be packaged into exosomes or recruit HIV repressor proteins to facilitate latency maintenance. (Middle) TAR-gag encodes viral pre-miRNAs that are processed by the cellular protein Dicer to form mature viral miRNAs. These viral miRNAs either facilitate protein degradation of HIV viral proteins or associate with HIV repressor proteins to mediate transcriptional silencing of the HIV LTR. Three additional virally encoded miRNAs have variable function in regulating latency. (Right) The HIV genome may transcribe antisense RNA. Like short HIV transcripts, these regulatory RNAs associate with HIV repressors to induce viral transcriptional silencing. Specific non-coding HIV RNAs and their target(s) are listed; HIV RNAs/targets that promote latency are highlighted in red, those that have anti-latency properties are highlighted in green, and those whose function in HIV latency is still unclear are highlighted in black. The figure was created in Biorender.com. Abbreviations: AATF, apoptosis antagonizing transcription factor; CUL4B, cullin 4B; HDAC, histone deacetylase; DNMT3A, DNA methyltransferase 3 alpha; EZH2, enhancer of zeste 2 polycomb repressive complex 2 subunit; PRC2, polycomb repressive complex 2; SIN3A, SIN3 transcription regulator family member A; TAR, trans-activator response element. |
Incomplete Sense HIV Transcripts
When HIV provirus is present in a latent state, transcription initiation may occur in a substantial proportion of cells; however, RNA polymerase II pauses in different places along and downstream the LTR.95 In some people with HIV with cART-suppressed viremia, only the short HIV transcripts were detected96 and proposed as a biomarker of latent infection.97 Four different short transcript species were identified, found to be packaged into exosomes from infected premyeloid line OM10.1 and the latently infected Jurkat cell derivative, J1.1 (Figure 3, left), and not to produce any proteins.98 Because these transcripts included parts of the gag region, they were termed TAR-gag98 (that is, spanning the trans-activator response element (TAR) and gag). A later study by the same group demonstrated that in J1.1 cells these non-coding RNAs were found in several complexes that play an inhibitory role in HIV transcription: PRC2 (EZH2 subunit), SIN3 transcription regulator family member A (SIN3A), cullin 4B (CUL4B), and Tat/TAR.99 Via these interactions, these short RNA transcripts performed several functions including epigenetic regulation of proviral DNA, histone modifications, and degradation of Tat.99 Thus, there appears to be a feedback loop, whereby in the latent state most of the transcription initiation results in incomplete transcripts, and these incomplete transcripts further promote HIV latency (Figure 3, left). In these experiments, however, HIV transcription elongation was inhibited using a Tat mimetic peptide (F07#13); in these conditions, production of TAR-gag increased due to the enhancement in RNA polymerase II pausing. It is unclear from this study whether such interactions are also present but not detectable in HIV latently infected cells not treated with F07#13 or whether they are the result of TAR-gag overexpression. Further studies will be needed to evaluate the role of incomplete sense HIV transcripts in promoting HIV latency.
HIV-Encoded miRNAs
Deep sequencing analysis demonstrated that multiple small RNAs were expressed from the HIV genome.100 It is quite possible that through the expression of miRNA HIV can regulate its own expression. Several miRNAs encoded by the HIV genome were identified that function to regulate HIV latency. Of these, miR-TAR (Figure 3, middle) and miR-Nef-367 function to downregulate HIV expression and promote latency; miR-H3-3p has anti-latency properties, and hiv1-miR-H1 induces multiple cellular responses some of which are beneficial and some antagonistic to latency maintenance.
MiR-TAR was hypothesized to serve as a source of viral miRNAs because of the resemblance of the TAR sequence with DICER substrates.101 Indeed, Klase et al demonstrated binding of DICER to TAR, cleavage of the TAR stem loop by DICER, and generation of TAR-derived miRNA.101 In a similar study, miRNA processing from both the left and right arms of TAR was reported, generating miR-TAR-5p and miR-TAR-3p.102 Despite the extensive secondary structure of TAR, miRNA produced by DICER from TAR was able to reduce expression of proteins under the control of the LTR promoter though to a lesser extent compared to siRNA designed to structurally poor segments within the protein coding genes.101 Inhibitory effects mediated by miR-TAR-3p were more pronounced compared to its miR-TAR-5p counterpart.102 In addition to RISC-mediated inhibition, TAR miRNA was also shown to recruit HDAC1 to the HIV LTR, resulting in chromatin remodeling and suppression of HIV gene expression at the transcriptional level.101 Furthermore, miR-TAR had a much broader effect on gene expression in cells infected with HIV, shifting the balance between apoptosis and survival and creating a cellular environment favorable for the maintenance of the latent reservoir.103,104 In a later study, the source of miR-TAR was validated to be incomplete HIV transcripts generated as a result of non-processive transcription; thus, the formation of these miRNAs can occur without the cleavage of the HIV genomic RNA.105 Although the majority of studies were performed in vitro or using cell lines, there is evidence that miR-TAR is produced in the course of HIV infection in both primary CD4+ T cells105 and macrophages.106
Another latency-promoting miRNA, miR-Nef-367, is encoded in the nef region, and was shown to directly suppress HIV RNA expression by either affecting RNA stability or mRNA translation.107 More intriguingly, this miRNA could also act by the mechanism of transcriptional interference from the 5′ LTR.108 Since double-stranded mutated nef RNA derived from LTNPs with low to undetectable levels of viremia interfered with HIV replication,109 the authors postulated that functions of nef via the RNAi pathway may allow persistently low pathogenic or latent HIV infection.107
The anti-latency miRNA, miR-H3-3p, was first predicted computationally, with subsequent experimental validation.110 This miRNA is located in the coding sequence of the HIV reverse transcriptase. Overexpression of this miRNA in the HEK 293 T cell line transfected with an env-defective HIV clone resulted in substantial enhancement of viral production while introducing mutations to disrupt its secondary structure resulted in the reduction of viral production.110 Furthermore, miR-H3-3p was shown to activate transcription from the HIV LTR via targeting the TATA-box, a property that was independent of either TAR or Tat.110 When small RNAs targeting the HIV TATA-box were synthesized and transfected into CD4+ T cells from people with suppressed HIV viremia, viral-associated RNA could be detected in supernatants 72 hours post-transfection, consistent with the idea that these RNAs could reactivate proviruses from latency.110
Hiv1-miR-H1 is perhaps the most intriguing case among HIV-encoded miRNAs. Encoded in the HIV LTR, this miRNA was shown to downregulate the expression of the apoptosis antagonizing transcription factor (AATF), which was accompanied by significant downregulation of the MYC proto-oncogene, BCL2 apoptosis regulator, β-amyloid at the translational level and pro-apoptotic WT1 regulator (PAWR, also known as Par-4) and DICER at the transcriptional level in human PBMCs.111,112 Moreover, the target scanning algorithm in miRanda identified human miR-149 as a potential target of hiv1-miR-H1.111 In turn, a study to identify predicted miRNA targets in the HIV genome identified the vpr gene as a potential target of miR-149.113 Presumably, targeting miR-149 by hiv1-miR-H1 would facilitate elevated levels of Vpr in cells. Interestingly, both AATF and Vpr were reported to bind p300,114,115 with this interaction facilitating transcriptional activation of the HIV provirus.115 Because AATF was downregulated by hiv1-miR-H1, and Vpr is predicted to be upregulated, the action of hiv1-miR-H1 would likely result in competing regulatory processes at the HIV LTR. Identification of a novel miRNA encoded in the AATF sequence, predicted to target both HIV hiv1-miR-H1 and vpr,116 adds another layer of complexity. Further studies will be needed to validate these predictions and determine the precise mode of action of hiv1-miR-H1.
HIV-Encoded Antisense RNAs
Identification of short antisense RNAs encoded by the HIV genome dates to the early nineties.117,118 Early studies showed that HIV antisense RNA had an inhibitory role in HIV replication.119 Even though protein production was demonstrated from the antisense HIV RNA,118–120 inhibitory properties of the antisense RNA were protein-independent.119 A more recent study identified several transcription start sites in the antisense strand spanning the U3 region of the 3′ LTR, nef, and env genes, and a polyA sequence in the pol region,121 indicating the existence of a long RNA encoded by the antisense strand. Later, a more detailed mapping of all antisense transcripts was conducted, identifying splice sites and transcription termination sites.122 Among the 7 different transcripts identified, previously reported short117 and long121 antisense transcripts were validated.122 The long antisense transcript was shown to localize mainly to the nucleus and play a role in inhibiting HIV replication.122 The study by Saayman et al characterized the longest antisense HIV RNA. Spanning nearly the entire HIV genome, including the 5′ LTR in antisense orientation, this antisense HIV RNA lacks a polyA tail and is thus non-coding in nature.123 This was the only study to evaluate the role of antisense HIV RNA in the regulation of latency and not just HIV replication. Using cell line models of HIV latency, the authors demonstrated a direct role of this RNA in the epigenetic regulation of HIV expression (Figure 3, right). The antisense HIV RNA recruited chromatin remodeling complexes consisting of proteins encoded by DNA methyltransferase 3 alpha (DNMT3A), EZH2, and HDAC1, thus inducing histone modifications and altered chromatin state at the HIV LTR.123
Non-Coding HIV RNA Perspectives
Non-coding HIV RNAs are perhaps the most promising targets to achieve an HIV cure, as they are 100% specific to cells that are infected with HIV. Growing evidence suggests that the HIV LTR is not entirely quiescent during latency,124 and that expressed viral transcripts have a great degree of heterogeneity.125–128 Therefore, strategies based on non-coding HIV RNA targeting should be investigated for both proviral reactivation and long-term silencing. However, the effect of protein partners shared by non-coding HIV RNAs and cellular RNA molecules remains a concern. For instance, the protein encoded by EZH2 interacts with several cellular lncRNAs, including MALAT1,71 NEAT1,83,84 and PVT1;69,85 yet antisense HIV RNA can also bind to the EZH2 protein.123 These observations are consistent with the idea that targeting a single non-coding RNA may not be feasible to achieve full suppression or reactivation of the HIV provirus, similar to what we know from targeting single protein-coding genes. Despite this limitation, we believe that targeting HIV latency via regulatory RNAs may be achieved with higher specificity and less toxicity compared to current protein-based strategies.
Conclusions
Even though regulatory RNAs represent an attractive opportunity for HIV cure research due to high cell and tissue specificity, the available literature describing identified interactions between regulatory RNA molecules, HIV, and protein regulators of HIV gene expression demonstrates the high complexity of these interactions. It may therefore be challenging to identify a small enough set of RNA targets needed to achieve an HIV cure. To our knowledge, no comprehensive screens of lncRNA or miRNA have been conducted to identify what specific regulatory RNA molecules play a role in HIV latency control or reactivation. Therefore, we hypothesize that more regulatory RNA players will be discovered. Even when all such molecules are identified, the important question relies on how to select the best RNA targets that regulate HIV expression. We hypothesize that the best RNA targets are those that have the fewest interaction partners regulating HIV and do not compete with other molecules for the same interaction partners. In the best-case scenario, one regulatory RNA would have a single interaction partner, either an HIV activator, repressor, or HIV RNA (Figure 4, scenario 1 (top panel)). Alternatively, a regulatory RNA may have several interaction partners (Figure 4, scenario 2 (middle panel)), or several regulatory RNAs may have the same interaction partner (Figure 4, scenario 3 (bottom panel)). In the latter cases, careful examination of all RNA regulators and their mechanisms of action will be needed. With the best RNA targets selected, several strategies may be feasible to inhibit their function. One strategy is conducting a screen for small-molecule compounds to disrupt specific interactions of RNAs with protein regulators of HIV expression. In the case of lncRNAs, multiple methods of inhibiting their expression and/or function are currently being explored in cancer therapy, including post-transcriptional degradation, gene editing, and steric hindrance. These methods can be adapted to the area of infectious diseases and combatting HIV latency.129 In sum, targeting RNA expression regulators remains a promising strategy for an HIV cure, but much remains to be done before a viable RNA-based strategy can be designed.
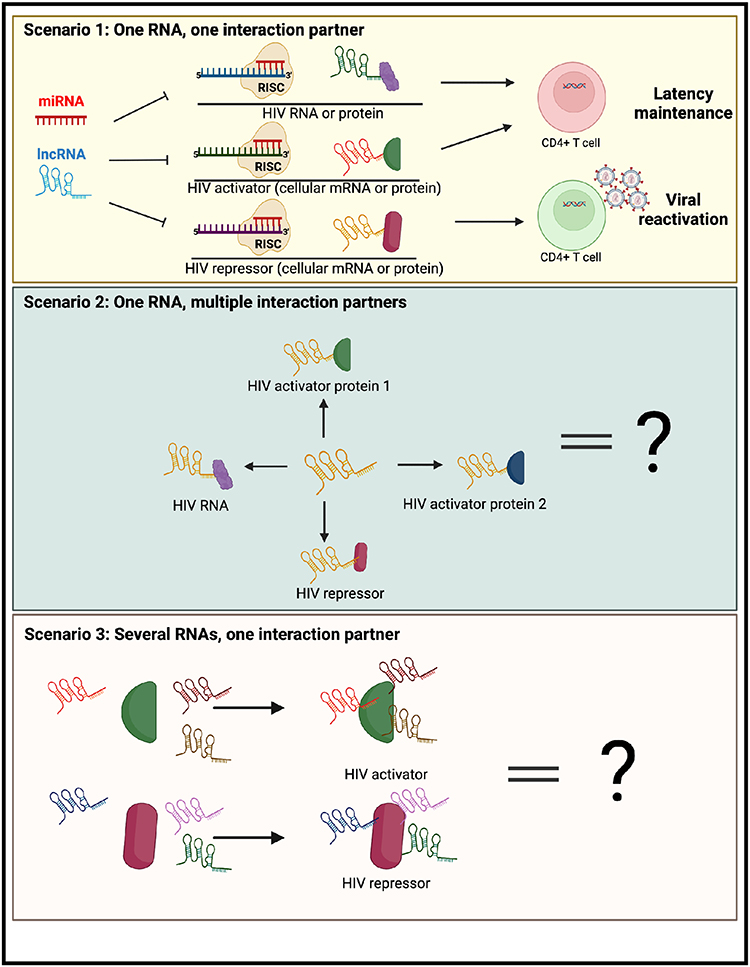 |
Figure 4 Summary of different scenarios for therapeutic targeting of regulatory RNAs for an HIV cure. (Top) The best-case scenario for an HIV cure would be the identification of a regulatory RNA, preferably a viral RNA, that has one interaction partner (HIV RNA, HIV activator or HIV repressor). Targeting of this RNA could lead to latency reversal and/or long-term proviral silencing. (Middle) Alternatively, a regulatory RNA may have diverse functions with different types of HIV regulators. (Bottom) Finally, regulators of HIV (activators/repressors) may interact with multiple regulatory RNAs, and thus disrupting their function may necessitate novel approaches. The figure was created in Biorender.com. |
Abbreviations
AATF, apoptosis antagonizing transcription factor; BLIMP-1, B lymphocyte-induced maturation protein-1; BST2, bone marrow stromal cell antigen 2; cART, combination anti-retroviral therapy; Cas9, CRISPR-associated protein 9; CCNT1,Cyclin T1; CDK2, cyclin-dependent kinase 2; CRISPR, clustered regularly interspaced short palindromic repeats; CSF2, colony-stimulating factor 2; CUL4B, cullin 4B; DNMT3A, DNA methyltransferase 3 alpha; EC, elite controller; EZH2, enhancer of zeste 2 polycomb repressive complex 2 subunit; FOXO1, forkhead box O1; GAS5, growth arrest-specific transcript 5; GFP, green fluorescent protein; HATs, histone acetyltransferases; HCC, hepatocellular carcinoma; HDACs, histone deacetylases; HEAL, HIV-enhanced lncRNA; HIVEP2, HIVEP zinc finder 2; IFITM1, interferon induced transmembrane protein 1; IFN, interferon; KAT2B, lysine acetyltransferase 2B; lncRNAs, long non-coding RNAs; LRAs, latency reversing agents; LTNP, long-term non-progressor; LTR, long terminal repeat; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; MDMs, myeloid-derived macrophages; miRNAs, micro RNAs; MX2, MX dynamin-like GTPase 2; NEAT1, nuclear paraspeckle assembly transcript; 1NFAT, nuclear factor of activated T cells; NF-kB, nuclear factor kappa light-chain enhancer of activated B cells; NKILA, NF-kB-interacting lncRNA; NONO, non-POU domain containing octamer binding; NRIR, negative regulator of interferon response; NRON, nuclear factor of activated T cells; nt, nucleotide; PAWR, pro-apoptotic WT1 regulator; PBMCs, peripheral blood mononuclear cells; P body, processing body; PCAF, p300/CBP-associated factor; PRC2, polycomb repressive complex 2; PRDM1, PR/SET Domain 1; pre-miRNAs, precursor miRNAs; PSPC1, paraspeckle component 1; p-TEFb, positive transcription elongation factor; PVT1, plasmacytoma variant translocation 1; RISC, RNA-induced silencing complex; RNAi, RNA interference; RNA-Seq, RNA sequencing; RUNX1, RUNX family transcription factor 1; SFPQ, splicing factor proline and glutamine rich; SIN3A - SIN3 transcription regulator family member A; TALEN, transcription activator-like effector nuclease; TAR, trans-activator response element; TRIM32, tripartite motif containing 32; UTR, untranslated region; ZNF, zinc finger nuclease.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
PWR was supported by Departmental startup funds from California State University Long Beach (CSULB) and a New Investigator Grant (GF00610932) from the California State University Program for Education and Research in Biotechnology (CSUPERB). CP was supported by award T32GM138075 from the National Institute of General Medical Sciences (NIGMS). NBB was supported by an R21 AI162159 from the National Institutes of Health.
Disclosure
The authors declare no competing interests in this work.
References
1. Tyagi M, Bukrinsky M. Human immunodeficiency virus (HIV) latency: the major hurdle in HIV eradication. Mol Med. 2012;18(7):1096–1108. doi:10.2119/molmed.2012.00194
2. Eisele E, Siliciano RF. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity. 2012;37(3):377–388. doi:10.1016/j.immuni.2012.08.010
3. Chun T-W, Carruth L, Finzi D, et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature. 1997;387(6629):183–188. doi:10.1038/387183a0
4. Pasternak AO, Berkhout B. What do we measure when we measure cell-associated HIV RNA. Retrovirology. 2018;15(1):13. doi:10.1186/s12977-018-0397-2
5. Chitrakar A, Sanz M, Maggirwar SB, Soriano-Sarabia N. HIV latency in myeloid cells: challenges for a cure. Pathogens. 2022;11(6):611. doi:10.3390/pathogens11060611
6. King VM, Borchert GM. MicroRNA expression: protein participants in microRNA regulation. In: Huang J, Borchert GM, Dou D, editors. Bioinformatics in MicroRNA Research. New York: Springer New York; 2017:27–37.
7. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi:10.1038/nature09267
8. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12(2):99–110. doi:10.1038/nrg2936
9. Huang V, Place RF, Portnoy V, et al. Upregulation of cyclin B1 by miRNA and its implications in cancer. Nucleic Acids Res. 2012;40(4):1695–1707. doi:10.1093/nar/gkr934
10. Place RF, Li LC, Pookot D, Noonan EJ, Dahiya R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc Natl Acad Sci U S A. 2008;105(5):1608–1613. doi:10.1073/pnas.0707594105
11. Younger ST, Corey DR. Transcriptional gene silencing in mammalian cells by miRNA mimics that target gene promoters. Nucleic Acids Res. 2011;39(13):5682–5691. doi:10.1093/nar/gkr155
12. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43(6):904–914. doi:10.1016/j.molcel.2011.08.018
13. Hung T, Chang HY. Long noncoding RNA in genome regulation: prospects and mechanisms. RNA Biol. 2010;7(5):582–585. doi:10.4161/rna.7.5.13216
14. Hashemi P, Sadowski I. Diversity of small molecule HIV-1 latency reversing agents identified in low- and high-throughput small molecule screens. Med Res Rev. 2020;40(3):881–908. doi:10.1002/med.21638
15. Rasmussen TA, Lewin SR. Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents? Curr Opin HIV AIDS. 2016;11(4):394–401. doi:10.1097/COH.0000000000000279
16. Archin NM, Liberty AL, Kashuba AD, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487(7408):482–485. doi:10.1038/nature11286
17. Elliott JH, Wightman F, Solomon A, et al. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathog. 2014;10(11):e1004473. doi:10.1371/journal.ppat.1004473
18. Rasmussen TA, Tolstrup M, Brinkmann CR, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a Phase 1/2, single group, clinical trial. Lancet HIV. 2014;1(1):e13–e21. doi:10.1016/S2352-3018(14)70014-1
19. Vansant G, Bruggemans A, Janssens J, Debyser Z. Block-and-lock strategies to cure HIV infection. Viruses. 2020;12(1):84. doi:10.3390/v12010084
20. Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio. 2015;6(4):e00465–e00465. doi:10.1128/mBio.00465-15
21. Lu Y, Bohn-Wippert K, Pazerunas PJ, Moy JM, Singh H, Dar RD. Screening for gene expression fluctuations reveals latency-promoting agents of HIV. Proc Nat Acad Sci U S A. 2021;118(11):e2012191118. doi:10.1073/pnas.2012191118
22. Kyei GB, Meng S, Ramani R, et al. Splicing factor 3B subunit 1 interacts with HIV Tat and plays a role in viral transcription and reactivation from latency. mBio. 2018;9(6). doi:10.1128/mBio.01423-18
23. Mukim A, Smith DM, Deshmukh S, Qazi AA, Beliakova-Bethell N. A camptothetin analog, topotecan, promotes HIV latency via interference with HIV transcription and RNA splicing. J Virol. 2023;31:e0163022. doi:10.1128/jvi.01630-22
24. Maina EK, Adan AA, Mureithi H, Muriuki J, Lwembe RM. A review of current strategies towards the elimination of latent HIV-1 and subsequent HIV-1 cure. Curr HIV Res. 2020;19(1):14–26.
25. Qu X, Wang P, Ding D, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41(16):7771–7782. doi:10.1093/nar/gkt571
26. Ebina H, Kanemura Y, Misawa N, et al. A high excision potential of TALENs for integrated DNA of HIV-based lentiviral vector. PLoS One. 2015;10(3):e0120047. doi:10.1371/journal.pone.0120047
27. Karpinski J, Chemnitz J, Hauber I, et al. Universal Tre (uTre) recombinase specifically targets the majority of HIV-1 isolates. J Int AIDS Soc. 2014;17(4 Suppl 3):19706. doi:10.7448/IAS.17.4.19706
28. Kaminski R, Chen Y, Fischer T, et al. Elimination of HIV-1 genomes from human T-lymphoid cells by CRISPR/Cas9 gene editing. Sci Rep. 2016;6(1):22555. doi:10.1038/srep22555
29. Panfil AR, London JA, Green PL, Yoder KE. CRISPR/Cas9 genome editing to disable the latent HIV-1 provirus. Front Microbiol. 2018;9:3107. doi:10.3389/fmicb.2018.03107
30. Derrien T, Johnson R, Bussotti G, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22(9):1775–1789. doi:10.1101/gr.132159.111
31. Cabili MN, Trapnell C, Goff L, et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25(18):1915–1927. doi:10.1101/gad.17446611
32. Liang Y, Ridzon D, Wong L, Chen C. Characterization of microRNA expression profiles in normal human tissues. BMC Genom. 2007;8(1):166. doi:10.1186/1471-2164-8-166
33. Kristen AV, Ajroud-Driss S, Conceição I, Gorevic P, Kyriakides T, Obici L. Patisiran, an RNAi therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis. Neurodegener Dis Manag. 2019;9(1):5–23. doi:10.2217/nmt-2018-0033
34. Ruelas DS, Chan JK, Oh E, et al. MicroRNA-155 reinforces HIV latency. J Biol Chem. 2015;290(22):13736–13748. doi:10.1074/jbc.M115.641837
35. Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326(6114):711–713. doi:10.1038/326711a0
36. Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Proc Natl Acad Sci U S A. 1989;86:2336–2340. doi:10.1073/pnas.86.7.2336
37. Modai S, Farberov L, Herzig E, Isakov O, Hizi A, Shomron N. HIV-1 infection increases microRNAs that inhibit Dicer1, HRB and HIV-EP2, thereby reducing viral replication. PLoS One. 2019;14(1):e0211111. doi:10.1371/journal.pone.0211111
38. Nomura N, Zhao MJ, Nagase T, et al. HIV-EP2, a new member of the gene family encoding the human immunodeficiency virus type 1 enhancer-binding protein. Comparison with HIV-EP1/PRDII-BF1/MBP-1. J Biol Chem. 1991;266(13):8590–8594. doi:10.1016/S0021-9258(18)93015-2
39. Chiang K, Sung T-L, Rice AP. Regulation of cyclin T1 and HIV-1 replication by microRNAs in resting CD4(+) T lymphocytes. J Virol. 2012;86(6):3244–3252. doi:10.1128/JVI.05065-11
40. Sung TL, Rice AP. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009;5(1):e1000263. doi:10.1371/journal.ppat.1000263
41. Triboulet R, Mari B, Lin Y-L, et al. Suppression of microRNA-silencing pathway by HIV-1 during virus replication. Science. 2007;315(5818):1579–1582. doi:10.1126/science.1136319
42. Seddiki N, Phetsouphanh C, Swaminathan S, et al. The microRNA-9/B-lymphocyte-induced maturation protein-1/IL-2 axis is differentially regulated in progressive HIV infection. Eur J Immunol. 2013;43(2):510–520. doi:10.1002/eji.201242695
43. Kaczmarek Michaels K, Natarajan M, Euler Z, Alter G, Viglianti G, Henderson AJ. Blimp-1, an intrinsic factor that represses HIV-1 proviral transcription in memory CD4+ T cells. J Immunol. 2015;194(7):3267–3274. doi:10.4049/jimmunol.1402581
44. Yu J, Angelin-Duclos C, Greenwood J, Liao J, Calame K. Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol. 2000;20(7):2592–2603. doi:10.1128/MCB.20.7.2592-2603.2000
45. Thiele S, Wittmann J, Jäck H-M, Pahl A. miR-9 enhances IL-2 production in activated human CD4+ T cells by repressing Blimp-1. Eur J Immunol. 2012;42(8):2100–2108. doi:10.1002/eji.201142203
46. Chinnapaiyan S, Dutta RK, Nair M, Chand HS, Rahman I, Unwalla HJ. TGF-β1 increases viral burden and promotes HIV-1 latency in primary differentiated human bronchial epithelial cells. Sci Rep. 2019;9(1):12552. doi:10.1038/s41598-019-49056-6
47. Ayala-Suárez R, Díez-Fuertes F, Calonge E, et al. Insight in miRNome of long-term non-progressors and elite controllers exposes potential RNAi role in restraining HIV-1 infection. J Clin Med. 2020;9(8). doi:10.3390/jcm9082452
48. Amaral AJ, Andrade J, Foxall RB, et al. miRNA profiling of human naive CD4 T cells links miR‐34c‐5p to cell activation and HIV replication. EMBO J. 2017;36(3):346–360. doi:10.15252/embj.201694335
49. Teng Y, Luo M, Yu T, et al. CRISPR/Cas9-mediated deletion of miR-146a enhances antiviral response in HIV-1 infected cells. Genes Immun. 2019;20(4):327–337. doi:10.1038/s41435-018-0036-x
50. Okoye I, Xu L, Oyegbami O, et al. Plasma extracellular vesicles enhance HIV-1 infection of activated CD4(+) T cells and promote the activation of latently infected J-Lat10.6 cells via miR-139-5p transfer. Front Immunol. 2021;12:697604. doi:10.3389/fimmu.2021.697604
51. Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J. 2003;22(8):1868–1877. doi:10.1093/emboj/cdg188
52. Roux A, Leroy H, De Muylder B, et al. FOXO1 transcription factor plays a key role in T cell-HIV-1 interaction. PLoS Pathog. 2019;15(5):e1007669. doi:10.1371/journal.ppat.1007669
53. Patel P, Ansari MY, Bapat S, Thakar M, Gangakhedkar R, Jameel S. The microRNA miR-29a is associated with human immunodeficiency virus latency. Retrovirology. 2014;11:108. doi:10.1186/s12977-014-0108-6
54. Ahluwalia JK, Khan SZ, Soni K, et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology. 2008;5:117. doi:10.1186/1742-4690-5-117
55. Nathans R, Chu C-Y, Serquina AK, Lu -C-C, Cao H, Rana TM. Cellular microRNA and P-bodies modulate host-HIV-1 interactions. Mol Cell. 2009;34(6):696–709. doi:10.1016/j.molcel.2009.06.003
56. Luo Y, Na Z, Slavoff SA. P-bodies: composition, properties, and functions. Biochemistry. 2018;57(17):2424–2431. doi:10.1021/acs.biochem.7b01162
57. Huang J, Wang F, Argyris E, et al. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007;13:1241–1247. doi:10.1038/nm1639
58. López-Huertas MR, Morín M, Madrid-Elena N, et al. Selective miRNA modulation fails to activate HIV replication in in vitro latency models. Mol Ther Nucleic Acids. 2019;17:323–336. doi:10.1016/j.omtn.2019.06.006
59. Woldemeskel BA, Kwaa AK, Blankson JN. Viral reservoirs in elite controllers of HIV-1 infection: implications for HIV cure strategies. EBioMedicine. 2020;62:103118. doi:10.1016/j.ebiom.2020.103118
60. Trypsteen W, White CH, Mukim A, et al. Long non-coding RNAs and latent HIV – a search for novel targets for latency reversal. PLoS One. 2019;14(11):e0224879. doi:10.1371/journal.pone.0224879
61. Matsuda S, Akagawa K, Honda M, Yokota Y, Takebe Y, Takemori T. Suppression of HIV replication in human monocyte-derived macrophages induced by granulocyte/macrophage colony-stimulating factor. AIDS Res Hum Retroviruses. 1995;11(9):1031–1038. doi:10.1089/aid.1995.11.1031
62. Vemula SV, Maxwell JW, Nefedov A, et al. Identification of proximal biomarkers of PKC agonism and evaluation of their role in HIV reactivation. Antiviral Res. 2017;139:161–170. doi:10.1016/j.antiviral.2016.11.014
63. Yamada K, Lim J, Dale JM, et al. Empirical analysis of transcriptional activity in the Arabidopsis genome. Science. 2003;302(5646):842–846. doi:10.1126/science.1088305
64. Li H, Chi X, Li R, Ouyang J, Chen Y. A novel lncRNA, AK130181, contributes to HIV-1 latency by regulating viral promoter-driven gene expression in primary CD4(+) T cells. Mol Ther Nucleic Acids. 2020;20:754–763. doi:10.1016/j.omtn.2020.04.011
65. Liu B, Sun L, Liu Q, et al. A cytoplasmic NF-κB interacting long noncoding RNA blocks IκB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27(3):370–381. doi:10.1016/j.ccell.2015.02.004
66. Wang H, Liu Y, Huan C, et al. NF-κB-interacting long noncoding RNA regulates HIV-1 replication and latency by repressing NF-κB signaling. J Virol. 2020;94(17). doi:10.1128/JVI.01057-20
67. Guan Y, Kuo WL, Stilwell JL, et al. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin Cancer Res. 2007;13(19):5745–5755. doi:10.1158/1078-0432.CCR-06-2882
68. Carramusa L, Contino F, Ferro A, et al. The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. J Cell Physiol. 2007;213(2):511–518. doi:10.1002/jcp.21133
69. Guo J, Hao C, Wang C, Li L. Long noncoding RNA PVT1 modulates hepatocellular carcinoma cell proliferation and apoptosis by recruiting EZH2. Cancer Cell Int. 2018;18:98. doi:10.1186/s12935-018-0582-3
70. Tripathy MK, McManamy MEM, Burch BD, Archin NM, Margolis DM. H3K27 demethylation at the proviral promoter sensitizes latent HIV to the effects of vorinostat in ex-vivo cultures of resting CD4+ T cells. J Virol. 2015;89:8392–8405. doi:10.1128/JVI.00572-15
71. Qu D, Sun -W-W, Li L, et al. Long noncoding RNA MALAT1 releases epigenetic silencing of HIV-1 replication by displacing the polycomb repressive complex 2 from binding to the LTR promoter. Nucleic Acids Res. 2019;47(6):3013–3027. doi:10.1093/nar/gkz117
72. Chao T-C, Zhang Q, Li Z, et al. The long noncoding RNA HEAL regulates HIV-1 replication through epigenetic regulation of the HIV-1 promoter. mBio. 2019;10(5):e02016–02019. doi:10.1128/mBio.02016-19
73. Huan C, Li Z, Ning S, Wang H, Yu X-F, Zhang W. Long noncoding RNA uc002yug.2 activates HIV-1 latency through regulation of mRNA levels of various RUNX1 isoforms and increased Tat expression. J Virol. 2018;92(9). doi:10.1128/JVI.01844-17
74. Zhang Q, Chen C-Y, Yedavalli VS, Jeang K-T. NEAT1 long noncoding RNA and paraspeckle bodies modulate HIV-1 posttranscriptional expression. mBio. 2013;4(1):e00596–00512. doi:10.1128/mBio.00596-12
75. Bond CS, Fox AH. Paraspeckles: nuclear bodies built on long noncoding RNA. J Cell Biol. 2009;186(5):637–644. doi:10.1083/jcb.200906113
76. Zolotukhin AS, Michalowski D, Bear J, et al. PSF acts through the human immunodeficiency virus type 1 mRNA instability elements to regulate virus expression. Mol Cell Biol. 2003;23(18):6618–6630. doi:10.1128/MCB.23.18.6618-6630.2003
77. Schwartz S, Felber BK, Pavlakis GN. Distinct RNA sequences in the gag region of human immunodeficiency virus type 1 decrease RNA stability and inhibit expression in the absence of rev protein. J Virol. 1992;66(1):150–159. doi:10.1128/jvi.66.1.150-159.1992
78. Liu H, Hu P-W, Couturier J, Lewis DE, Rice AP. HIV-1 replication in CD4+ T cells exploits the down-regulation of antiviral NEAT1 long non-coding RNAs following T cell activation. Virology. 2018;522:193–198. doi:10.1016/j.virol.2018.07.020
79. Imam H, Shahr Bano A, Patel P, Holla P, Jameel S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci Rep. 2015;5(1):8639. doi:10.1038/srep08639
80. Li J, Chen C, Ma X, et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing Tat protein degradation. Nat Commun. 2016;7(1):11730. doi:10.1038/ncomms11730
81. Schynkel T, Szaniawski MA, Spivak AM, et al. Interferon-mediated long non-coding RNA response in macrophages in the context of HIV. Int J Mol Sci. 2020;21(20):7741. doi:10.3390/ijms21207741
82. Kambara H, Niazi F, Kostadinova L, et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res. 2014;42(16):10668–10680. doi:10.1093/nar/gku713
83. Chen Q, Cai J, Wang Q, et al. Long noncoding RNA NEAT1, regulated by the EGFR pathway, contributes to glioblastoma progression through the WNT/β-catenin pathway by scaffolding EZH2. Clin Cancer Res. 2018;24(3):684–695. doi:10.1158/1078-0432.CCR-17-0605
84. Wang Q, Liu L, Zhang S, et al. Long noncoding RNA NEAT1 suppresses hepatocyte proliferation in fulminant hepatic failure through increased recruitment of EZH2 to the LATS2 promoter region and promotion of H3K27me3 methylation. Exp Mol Med. 2020;52(3):461–472. doi:10.1038/s12276-020-0387-z
85. Zhou Q, Chen J, Feng J, Wang J. Long noncoding RNA PVT1 modulates thyroid cancer cell proliferation by recruiting EZH2 and regulating thyroid-stimulating hormone receptor (TSHR). Tumor Biol. 2016;37(3):3105–3113. doi:10.1007/s13277-015-4149-9
86. Chen L, Chen L, Zuo L, et al. Short communication: long noncoding RNA GAS5 inhibits HIV-1 replication through interaction with miR-873. AIDS Res Hum Retroviruses. 2018;34(6):544–549. doi:10.1089/aid.2017.0177
87. Chang Z, Cui J, Song Y. Long noncoding RNA PVT1 promotes EMT via mediating microRNA-186 targeting of Twist1 in prostate cancer. Gene. 2018;654:36–42. doi:10.1016/j.gene.2018.02.036
88. Lan T, Yan X, Li Z, et al. Long non-coding RNA PVT1 serves as a competing endogenous RNA for miR-186-5p to promote the tumorigenesis and metastasis of hepatocellular carcinoma. Tumour Biol. 2017;39(6):1010428317705338. doi:10.1177/1010428317705338
89. Huang T, Liu HW, Chen JQ, et al. The long noncoding RNA PVT1 functions as a competing endogenous RNA by sponging miR-186 in gastric cancer. Biomed Pharmacother. 2017;88:302–308. doi:10.1016/j.biopha.2017.01.049
90. Lu X, Yu Y, Yin F, et al. Knockdown of PVT1 inhibits IL-1β-induced injury in chondrocytes by regulating miR-27b-3p/TRAF3 axis. Int Immunopharmacol. 2020;79:106052. doi:10.1016/j.intimp.2019.106052
91. Wang Z, Zhang Q, Sun Y, Shao F. Long non-coding RNA PVT1 regulates BAMBI to promote tumor progression in non-small cell lung cancer by sponging miR-17-5p. Onco Targets Ther. 2020;13:131–142. doi:10.2147/OTT.S217335
92. Yuan W, Xiong X, Du J, Fan Q, Wang R, Zhang X. LncRNA PVT1 accelerates LPS-induced septic acute kidney injury through targeting miR-17-5p and regulating NF-κB pathway. Int Urol Nephrol. 2021;53(11):2409–2419. doi:10.1007/s11255-021-02905-8
93. Huang F, Chen W, Peng J, et al. Retracted article: lncRNA PVT1 triggers Cyto-protective autophagy and promotes pancreatic ductal adenocarcinoma development via the miR-20a-5p/ULK1 axis. Mol Cancer. 2018;17(1):98. doi:10.1186/s12943-018-0845-6
94. Li X, Zhang Z, Jiang H, et al. Circular RNA circPVT1 promotes proliferation and invasion through sponging miR-125b and activating E2F2 signaling in non-small cell lung cancer. Cell Physiol Biochem. 2018;51(5):2324–2340. doi:10.1159/000495876
95. Jadlowsky JK, Wong JY, Graham AC, et al. Negative elongation factor is required for the maintenance of proviral latency but does not induce promoter-proximal pausing of RNA polymerase II on the HIV long terminal repeat. Mol Cell Biol. 2014;34(11):1911–1928. doi:10.1128/MCB.01013-13
96. Lin X, Irwin D, Kanazawa S, et al. Transcriptional profiles of latent human immunodeficiency virus in infected individuals: effects of Tat on the host and reservoir. J Virol. 2003;77(15):8227–8236. doi:10.1128/JVI.77.15.8227-8236.2003
97. Adams M, Sharmeen L, Kimpton J, et al. Cellular latency in human immunodeficiency virus-infected individuals with high CD4 levels can be detected by the presence of promoter-proximal transcripts. Proc Nat Acad Sci U S A. 1994;91:3862–3866. doi:10.1073/pnas.91.9.3862
98. Barclay RA, Schwab A, DeMarino C, et al. Exosomes from uninfected cells activate transcription of latent HIV-1. J Biol Chem. 2017;292(28):11682–11701. doi:10.1074/jbc.M117.793521
99. Pinto DO, Scott TA, DeMarino C, et al. Effect of transcription inhibition and generation of suppressive viral non-coding RNAs. Retrovirology. 2019;16(1):13. doi:10.1186/s12977-019-0475-0
100. Schopman NC, Willemsen M, Liu YP, et al. Deep sequencing of virus-infected cells reveals HIV-encoded small RNAs. Nucleic Acids Res. 2012;40(1):414–427. doi:10.1093/nar/gkr719
101. Klase Z, Kale P, Winograd R, et al. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol Biol. 2007;8:63. doi:10.1186/1471-2199-8-63
102. Ouellet DL, Plante I, Landry P, et al. Identification of functional microRNAs released through asymmetrical processing of HIV-1 TAR element. Nucleic Acids Res. 2008;36(7):2353–2365. doi:10.1093/nar/gkn076
103. Klase Z, Winograd R, Davis J, et al. HIV-1 TAR miRNA protects against apoptosis by altering cellular gene expression. Retrovirology. 2009;6(1):18. doi:10.1186/1742-4690-6-18
104. Ouellet DL, Vigneault-Edwards J, Létourneau K, et al. Regulation of host gene expression by HIV-1 TAR microRNAs. Retrovirology. 2013;10(1):86. doi:10.1186/1742-4690-10-86
105. Harwig A, Jongejan A, van Kampen AH, Berkhout B, Das AT. Tat-dependent production of an HIV-1 TAR-encoded miRNA-like small RNA. Nucleic Acids Res. 2016;44(9):4340–4353. doi:10.1093/nar/gkw167
106. Li L, Feng H, Da Q, et al. Expression of HIV-encoded microRNA-TAR and its inhibitory effect on viral replication in human primary macrophages. Arch Virol. 2016;161(5):1115–1123. doi:10.1007/s00705-016-2755-5
107. Omoto S, Ito M, Tsutsumi Y, et al. HIV-1 nef suppression by virally encoded microRNA. Retrovirology. 2004;1(1):44. doi:10.1186/1742-4690-1-44
108. Omoto S, Fujii YR. Regulation of human immunodeficiency virus 1 transcription by nef microRNA. J Gen Virol. 2005;86(3):751–755. doi:10.1099/vir.0.80449-0
109. Yamamoto T, Omoto S, Mizuguchi M, et al. Double-stranded nef RNA interferes with human immunodeficiency virus type 1 replication. Microbiol Immunol. 2002;46(11):809–817. doi:10.1111/j.1348-0421.2002.tb02768.x
110. Zhang Y, Fan M, Geng G, et al. A novel HIV-1-encoded microRNA enhances its viral replication by targeting the TATA box region. Retrovirology. 2014;11(1):23. doi:10.1186/1742-4690-11-23
111. Kaul D, Ahlawat A, Gupta SD. HIV-1 genome-encoded hiv1-mir-H1 impairs cellular responses to infection. Mol Cell Biochem. 2009;323(1–2):143–148. doi:10.1007/s11010-008-9973-4
112. Kaul D, Khanna A. Evidence and nature of a novel miRNA encoded by HIV-1. Proc Indian Nat Sci Acad. 2006;72(2):91–95.
113. Hariharan M, Scaria V, Pillai B, Brahmachari SK. Targets for human encoded microRNAs in HIV genes. Biochem Biophys Res Commun. 2005;337(4):1214–1218. doi:10.1016/j.bbrc.2005.09.183
114. Leister P, Burgdorf S, Scheidtmann KH. Apoptosis antagonizing transcription factor AATF is a novel coactivator of nuclear hormone receptors. Signal Transduct. 2003;3(1–2):17–25. doi:10.1002/sita.200300020
115. Felzien LK, Woffendin C, Hottiger MO, Subbramanian RA, Cohen EA, Nabel GJ. HIV transcriptional activation by the accessory protein, VPR, is mediated by the p300 co-activator. Proc Natl Acad Sci U S A. 1998;95(9):5281–5286. doi:10.1073/pnas.95.9.5281
116. Kaul D, Hussain A. Cellular AATF gene encodes a novel miRNA that can contribute to HIV-1 latency. Indian J Biochem Biophys. 2009;46(3):237–240.
117. Michael NL, Vahey MT, d’Arcy L, et al. Negative-strand RNA transcripts are produced in human immunodeficiency virus type 1-infected cells and patients by a novel promoter downregulated by Tat. J Virol. 1994;68(2):979–987. doi:10.1128/jvi.68.2.979-987.1994
118. Vanhée-Brossollet C, Thoreau H, Serpente N, D’Auriol L, Lévy JP, Vaquero C. A natural antisense RNA derived from the HIV-1 env gene encodes a protein which is recognized by circulating antibodies of HIV+ individuals. Virology. 1995;206(1):196–202. doi:10.1016/S0042-6822(95)80034-4
119. Tagieva NE, Vaquero C. Expression of naturally occurring antisense RNA inhibits human immunodeficiency virus type 1 heterologous strain replication. J Gen Virol. 1997;78(Pt 10):2503–2511. doi:10.1099/0022-1317-78-10-2503
120. Ludwig L, Ambrus J, Krawczyk K, et al. Human immunodeficiency virus-type 1 LTR DNA contains an intrinsic gene producing antisense RNA and protein products. Retrovirology. 2006;3(1):80. doi:10.1186/1742-4690-3-80
121. Landry S, Halin M, Lefort S, et al. Detection, characterization and regulation of antisense transcripts in HIV-1. Retrovirology. 2007;4(1):71. doi:10.1186/1742-4690-4-71
122. Kobayashi-Ishihara M, Yamagishi M, Hara T, et al. HIV-1-encoded antisense RNA suppresses viral replication for a prolonged period. Retrovirology. 2012;9(1):38. doi:10.1186/1742-4690-9-38
123. Saayman S, Ackley A, Turner AW, et al. An HIV-encoded antisense long noncoding RNA epigenetically regulates viral transcription. Mol Ther. 2014;22(6):1164–1175. doi:10.1038/mt.2014.29
124. Wiegand A, Spindler J, Hong FF, et al. Single-cell analysis of HIV-1 transcriptional activity reveals expression of proviruses in expanded clones during ART. Proc Natl Acad Sci U S A. 2017;114(18):E3659–E3668. doi:10.1073/pnas.1617961114
125. Yukl SA, Kaiser P, Kim P, et al. HIV latency in isolated patient CD4+ T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci Transl Med. 2018;10(430). doi:10.1126/scitranslmed.aap9927
126. Ishizaka A, Sato H, Nakamura H, et al. Short intracellular HIV-1 transcripts as biomarkers of residual immune activation in patients on antiretroviral therapy. J Virol. 2016;90(12):5665–5676. doi:10.1128/JVI.03158-15
127. Kaiser P, Joshi SK, Kim P, et al. Assays for precise quantification of total (including short) and elongated HIV-1 transcripts. J Virol Methods. 2017;242:1–8. doi:10.1016/j.jviromet.2016.12.017
128. Lassen KG, Bailey JR, Siliciano RF. Analysis of human immunodeficiency virus type 1 transcriptional elongation in resting CD4ȡ + T cells in vivo. J Virol. 2004;78(17):9105–9114. doi:10.1128/JVI.78.17.9105-9114.2004
129. Boliar S, Russell DG. Lnc(ing)RNAs to the “shock and kill” strategy for HIV-1 cure. Mol Ther Nucleic Acids. 2021;23:1272–1280. doi:10.1016/j.omtn.2021.02.004
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.