")
Back to Journals » Open Access Journal of Clinical Trials » Volume 11
An evaluation of informed consent comprehension by adult trial participants in South Africa at the time of providing consent for clinical trial participation and a review of the literature
Authors Burgess LJ , Gerber B, Coetzee K, Terblanche M, Agar G, Kotze TJ
Received 28 June 2017
Accepted for publication 17 February 2019
Published 8 July 2019 Volume 2019:11 Pages 19—35
DOI https://doi.org/10.2147/OAJCT.S145068
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Lesley Jean Burgess,1,2 Berna Gerber,3 Kathleen Coetzee,1 Marli Terblanche,1 Gareth Agar,1 Theunis JvW Kotze2
1TREAD Research CC, Cardiology Unit, Department of Medicine, Stellenbosch University and Tygerberg Hospital, Parow, South Africa; 2University of Liverpool/Laureate Online Education, Liverpool, UK; 3Division of Speech-Language and Hearing Therapy, Faculty of Medicine and Health Sciences, Stellenbosch University Stellenbosch, South Africa
Introduction: The informed consent process is a fundamental part of clinical trials and is driven by both a legal and ethical agenda. The process may be seriously compromised if trial participants sign the informed consent document without fully understanding its contents. In developing countries such as South Africa, this concern is important due to the potential vulnerability of these patients and their risk for research exploitation.
Aim: To evaluate the understanding of 11 important components and concepts related to clinical research by adult trial participants in a developing country at the time of providing consent for trial participation.
Methods: 46 consecutive adult patients who qualified and consented to being enrolled in ongoing cardiovascular risk clinical trials at TREAD Research in the Western Cape, South Africa, were included in this study. After giving informed consent, participants were subjected to both a close-ended (self-report) and an open-ended method (descriptive narrative) to assess their understanding of various components and concepts related to clinical research pertaining to the initial informed consent document. The descriptive narrative was recorded and then later transcribed and assessed by two independent assessors.
Results: There was a marked difference between the two methodologies used to assess patient comprehension of the various components. With the exception of concepts voluntariness and right to withdraw, trial participants’ understanding of the informed consent document was poor – especially with regard to the following concepts: randomization, risks, placebo and blinding. Higher levels of comprehension were obtained for the participant self-reports and lower levels for the narrative descriptions.
Conclusion: The participant comprehension at this site was poor, and the process for taking informed consent subsequently needs to be modified so as to improve informed consent comprehension.
Keywords: clinical trials, informed consent comprehension
Introduction
Informed consent, “an autonomous authorization by individuals of a medical intervention or of involvement in research”,1 is fundamental to the clinical trial process. Lidz maintains that “true” informed consent includes the concepts of voluntarism, capacity, disclosure, understanding and decision.2
There are a number of regulatory and ethical guidelines highlighting the need for prospective trial participants to understand the information imparted during the informed consent process; these guidelines include the Nuremburg code,3 the Belmont Report,4 45 CFR 46 & 21 CFR 50,5 the Council for International Organization of Medical Science (CIOMS) guidelines6 and the Declaration of Helsinki.7 However, the reality is that many prospective trial participants sign this document without fully understanding the contents thereof.8–11 Informed consent may be seriously compromised in these instances.
To date, the majority of interest in informed consent comprehension in developing countries has been sparked by ethicists questioning the potential vulnerability of patients and their risk of research exploitation.12–14 Emanuel et al have identified “quality of the informed consent process” as a major concern.14 Patients may agree to participate in clinical trials so as to obtain some of the associated benefits of clinical trials without truly understanding the relevant facts and implications.15 In South Africa, these benefits include free access to medical care and medication and access to the obligatory trial participant remuneration as mandated by the South African regulatory authority, namely the Medicines’ Control Council (MCC).16,17 This problem may be further exacerbated by the fact that, in sponsor-initiated clinical trials conducted in South Africa, the informed consent form is often relatively complex and dictated by the requirements of developed countries.18 Ultimately, however, it is the investigators’ responsibility to ensure that trial participants sign the informed consent document and understand the contents.19
There have been numerous studies conducted which have evaluated patients’ understanding of informed consent issues in clinical trials, as summarized by Falagas et al.11 However, there are relatively few publications from South Africa and other developing countries.18,20–30 Notably, previous studies performed in these countries have mainly been performed in vaccine, HIV or epidemiological trials. Being community based, these trials lack the global perspective of the large, pharmaceutically sponsored multinational clinical trials and focus on national issues pertaining to developing countries. In addition, the study by Moodley was conducted in a healthy elderly patient population 4–12 months after completing the trial; these factors may, accordingly, also affect patient recall and understanding.18 A number of other trials such as those conducted by Joubert et al and Manafa et al were also conducted at between 2 and 12 months after consenting to the initial trial.21,30
The current study included trial participants with chronic lifestyle-based diseases related to cardiovascular heart disease who had consented to participate in nonvaccine, non-HIV clinical trials at TREAD Research, a private research unit based in Tygerberg Hospital, a tertiary-care, academic hospital in the Western Cape of South Africa. In all cases, the trial sponsors were large multinational pharmaceutical companies and the clinical research program was part of the companies’ multinational drug development pipeline. In addition, the assessment was done soon after signing informed consent for a clinical trial, thus giving a better indication of the participants’ understanding at the time of signing consent. The knowledge gained from this study will be useful in identifying areas of concern with regard to the informed consent process in developing countries such as South Africa.
Objective
To evaluate and compare the understanding (perceived and actual) of 11 components and concepts related to clinical research by adult trial participants in South Africa at the time of providing consent for trial participation in cardiovascular risk clinical trials by the use of:
- A closed-ended assessment (self-report) and
- An open-ended assessment (descriptive narrative)
Materials and methods
This is a prospective study involving consecutive adult patients who qualified and consented to being enrolled in ongoing cardiovascular risk clinical trials at TREAD Research. All clinical trials were approved by both the Stellenbosch University’s Health Research Ethics Committee (SHREC) and the South African MCC. In addition, this research study’s proposal was approved by both the SHREC and the University of Liverpool. Identical English/Afrikaans documents were determined so and approved by SHREC.
Study participants
All trial participants signed an additional consent to participate in this study and fulfilled the following criteria:
- Aged 18 years or older
- Able to speak and read English and/or Afrikaans fluently
- No history of any language disorders
- No history of grade repetition at school.
Data collection
The study doctors working at TREAD Research took consent from the trial participants for the enrolling cardiovascular risk clinical trial. All doctors had a minimum of 8 years’ clinical trial experience and had been trained to take informed consent according to the site’s standard operating procedures. After signing initial consent, these doctors then consented the trial participants to participate in the proposed research study.
Participants were subjected to both a close-ended (self-report) and an open-ended (descriptive narrative) assessment to determine their understanding of various components and concepts pertaining to the initial informed consent document. In keeping with previous studies conducted in this field, and in order to satisfy Lidz’ definition of “true” informed consent,2 the following 11 components and concepts were selected: Research, Voluntariness, The right to withdraw, Risks associated with the trial, Benefits associated with the trial, Side effects associated with the trial, Blinding, Placebo, Randomization, Ethics committee, and Compensation. The self-report assessment was administered first so that the participants’ assessment of their own understanding was not influenced by their experiences with the descriptive narrative assessment.
For the close-ended assessment, each participant was asked to indicate for each of the 11 components and concepts whether his/her understanding was “good enough” or whether (s)he understood “very little or nothing” about the concept. The open-ended assessment involved participants being asked to describe their participation in research, as they would explain it to a friend. Standardized prompts were developed to elicit a narrative from participants if little or no information was provided spontaneously. A digital audio recorder was used to record the participants’ responses to the open-ended tasks, namely the descriptive narrative. These recordings were then transcribed and the patients’ responses were analyzed. Oral consent for use of the audio recorder was obtained from the study participants.
Data analysis
The data collected through the descriptive narrative was transcribed and then rated by 2 blinded reviewers. When the 2 reviewers differed in their assessment, they met and discussed each discordant result on a case-by-case basis. A score of “good enough understanding” versus “little or no understanding” for the various trial components and other research-related concepts was awarded accordingly. For the self-report, participants indicated their own level of comprehension.
Statistical analysis
Nonparametric statistics were performed due to the relatively small sample size. The following procedures will be conducted:
- Comparisons were made of the participants’ understanding of the listed 11 trial components above across the two different methods of assessment. This was performed using version 7 of the STATISTICA data analysis software system.
- Cross-tabulation was performed and the Chi-square test used to determine the level of interaction between the results obtained from the “self-report” components and the corresponding “narrative evaluation” of the same item.
The hypotheses tested:
1. H0: there is no difference between the proportion of self-reported understanding and the narrative understanding in each of 11 traits 2. H1: there is a difference between the proportion of self-reported understanding and the narrative understanding in each of 11 traits 3. H0: there is no difference between the means of the summative scores for self-reported understanding and for narrative understanding 4. H1: there is a difference between the means of the summative scores for self-reported understanding and for narrative understanding
Statistical methodology
McNemar’s tests for paired binary proportions were used to compare the proportions between self-reported and narrative understanding.
Nonparametric Wilcoxon signed ranks test was used to compare the paired scores of self-reported understanding with narrative understanding scores.
Results
Although 47 participants signed consent for this study, one participant withdrew consent prior to completing the self-report. The remaining 46 participants were included in the study; however, the recording of the descriptive narrative was interrupted due to equipment failure for one participant and the last four answers in the narrative section were lost. Informed consents from 3 different cardiovascular risk trials that were recruiting at the time were used to examine the informed consent comprehension. All trials had a similar design, namely multinational, randomized controlled trial with a placebo comparator arm, and the readability of the respective consent forms according to the Flesch–Kincaid Grade Level ranged from 10.0 to 12.3. The number of participants enrolling in each study was 14, 14 and 18, respectively.
The demographics of the included trial participants were as follows:
- Gender: 20 males (43.5%) versus 26 females (56.5%)
- Age: Mean 53,8 years (range, 33–77 years)
- Home language: 80.4% Afrikaans (n=37) versus 19.6% English (n=9)
- Education: 85% of the participants had at least 6 years of schooling (n=39), while 43.5% had at least 10 years of schooling (n=20). The level of education was not specified in the remaining 7 trial participants.
According to the self-report questionnaires, the concepts of right to withdraw and voluntariness were best understood with 98% (n=45) and 96% (n=44) of the study population, respectively, claiming to have a “good enough understanding”. The concepts of blinding and placebo were the most poorly understood at 30% (n=14) and 37% (n=17), respectively, followed by ethics committee and randomization at 41% (n=19) and 50% (n=23), respectively. The remaining concepts were, according to the self-reports, fairly well understood, ranging from 74% to 80%.
The results of the participants’ narratives demonstrated that the concepts of voluntariness and right to withdraw were best understood with 96% (n=44) and 93% (n=43) of the study population, respectively, being found to have a “good enough understanding” of these concepts. Only 13% (n=6) and 17% (n=8) of the patient population understood the concepts of randomization and risks, respectively, while 20% (n=9) of the participants understood each of the concepts placebo and blinding, and 22% (n=10) understood the concept ethics committee. The remaining concepts were adequately understood by 52–65% of the participants.
Overall, the self-report and narrative yielded similar results for the concepts of voluntariness and right to withdraw. However, although 80% of participants reported understanding the concepts of risks, benefits and research, the narrative revealed that the actual understanding of these concepts was much lower at 17%, 52% and 57%, respectively. These results are summarized in Figure 1.
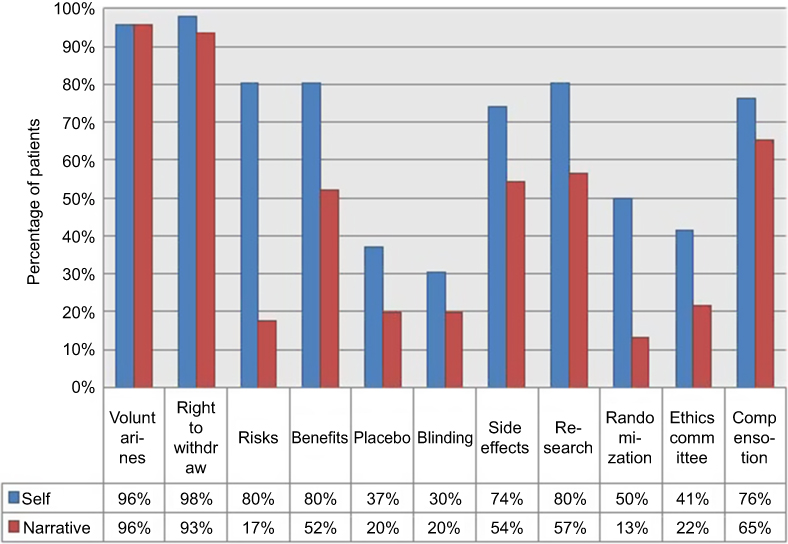 |
Figure 1 Comparison of overall patient comprehension: Self-report versus descriptive narrative. |
Based on the self-report questionnaire, the median score for comprehension of all concepts was 72.7% (n=8), interquartile range 54.5–81.8% (n=6–9) – as illustrated in Figure 2. Of note, 1 patient reported to not have understood any concepts, while 7 patients claimed to have understood all the concepts. The corresponding result for the narrative description was 36.4% (n=4), interquartile range 27.3–63.6% (n=3–7) – as indicated in Figure 3. Only 1 trial participant was shown to have understood all concepts according to the descriptive narrative.
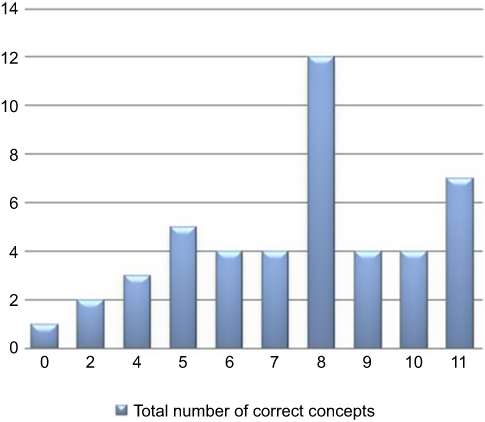 |
Figure 2 Distribution of total comprehension scores according to the self-report. |
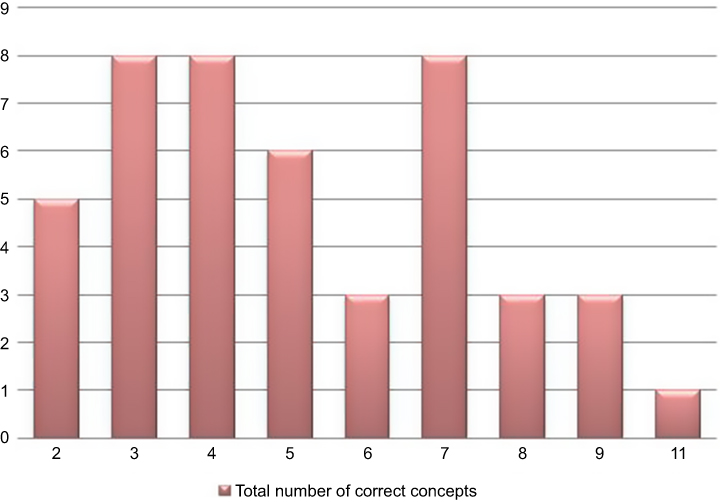 |
Figure 3 Distribution of total comprehension scores according to the descriptive narrative. |
As indicated by the p-values in Table 1, all components except voluntariness, right to withdraw, blinding and compensation were significantly differently understood in self-reporting compared with narrative report. In the above mentioned 4 components, there was no statistical difference in the two methods of report. Examination of the cross-tabulations shows that in all 7 components which showed a statistically different understanding between self-report and narrative, understanding was over-reported in self-report.
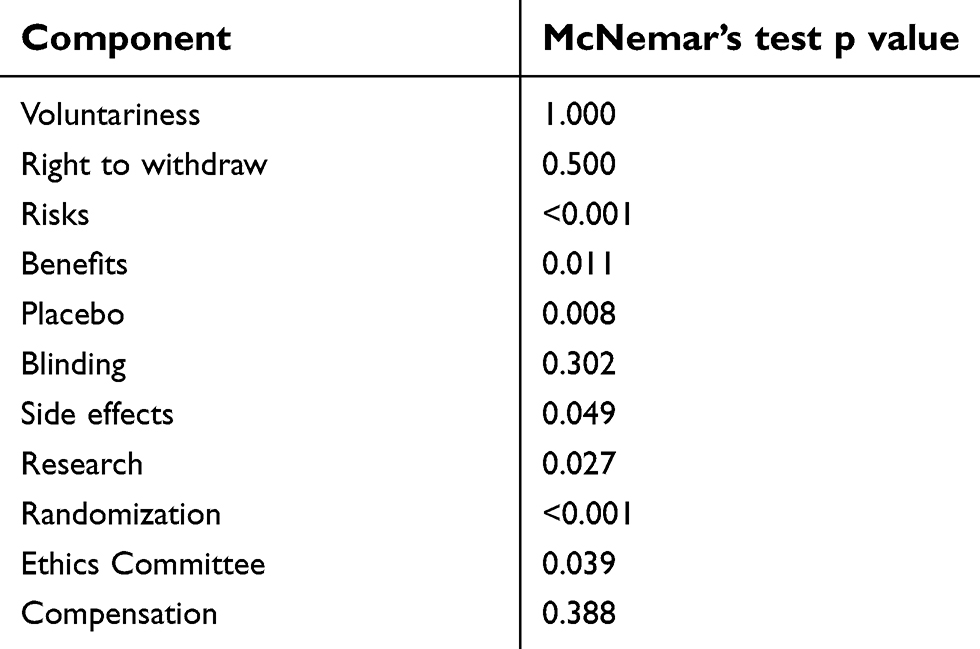 |
Table 1. McNemar’s test p values for each component comparison |
Table 2 displays the descriptive statistics for self-report and narrative summative scores and Figure 4 displays boxplots of self-report and narrative report scores. Table 2 and Figure 4 show that the median for self-report score as well as the interquartile range was higher than for narrative scores. A related samples Wilcoxon signed ranks test gave a test statistic of 83.0 with p<0.001. Therefore, the null hypothesis of equivalent scores between self-report and narrative report was rejected. We conclude that the scores were significantly lower on narrative report.
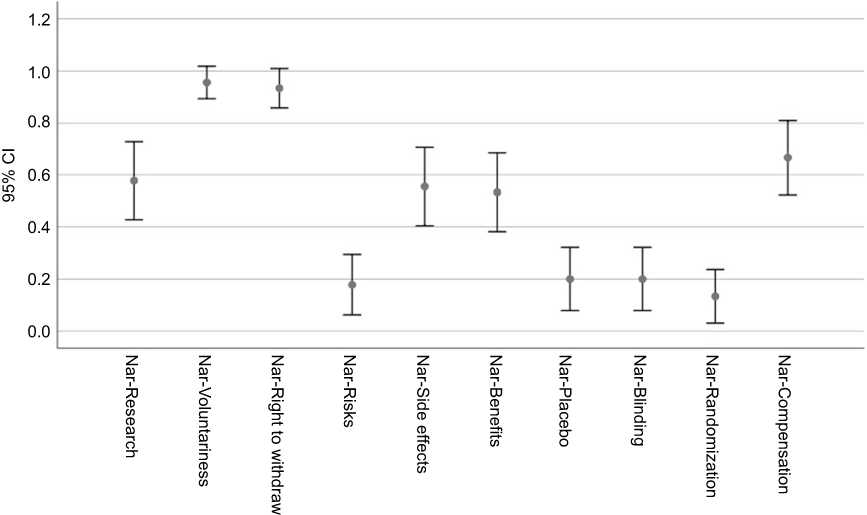 |
Figure 4 McNemar’s test p-values for each component comparison. |
 |
Table 2. Descriptive statistics for self-report and narrative summative scores |
The error bars (95% CIs) around the proportion estimates are shown in Figures 5 and 6, respectively, separately for the self-report and narrative components.
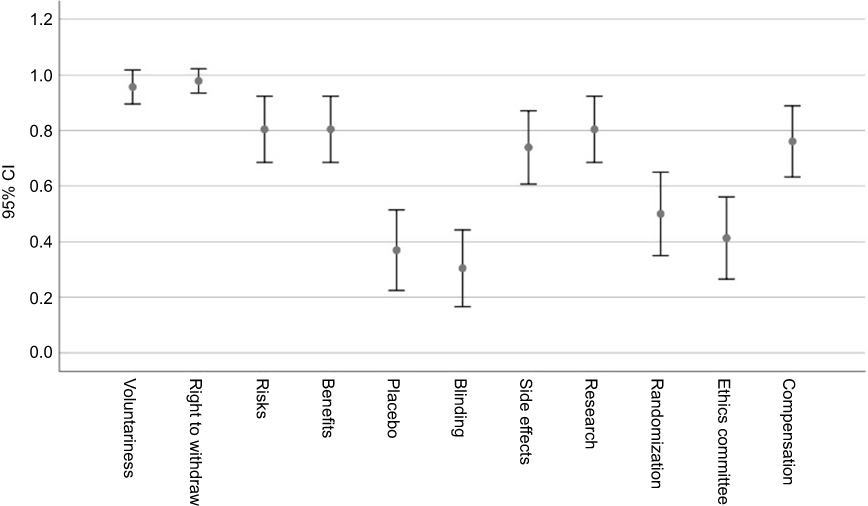 |
Figure 5 Descriptive statistics for self-report and narrative summative scores. |
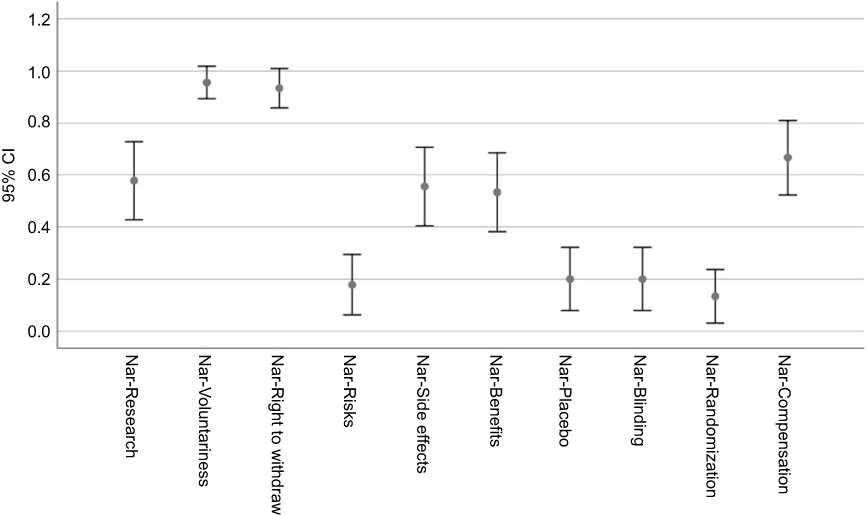 |
Figure 6 Boxplots of self-report and narrative report scores. |
Discussion
Although the various ethical guidelines mandate that prospective trial participants must comprehend the information given to them prior to signing the informed consent form, concern has been raised about the participant’s comprehension of this information – especially in developing countries.11 Making an accurate assessment of a participant’s understanding of the informed consent is complex and requires the conduct of a test of understanding of the basic elements of consent as well as trial related concepts and procedures.18 However, distinguishing between comprehension and recall (memory) of trial information can be equally difficult, and may be affected by time. Most researchers believe that testing the informed consent comprehension of trial participants is important; however, it appears that this is seldom done.22 Possible reasons for this include inconsistency with regard to the mechanism for doing so, as well as uncertainty with regard to dealing with trial participants who fail. Should these trial participants be refused trial enrollment? Should researchers continue to repeat these concepts until all participants meet the passing grade? What would the ramifications be if such a paticipant was enrolled in a clinical trial and then suffered a trial injury, and how would this affect potential liability?
A number of comprehension tests have been developed to make this assessment. However, the diverse study methodologies employed in these tests have created difficulties in interpretation. For instance, Guarino measured comprehension by questioning patients on how well they perceived their comprehension to be.31 Other studies have utilized tools (including patient narratives, multiple choice questions and interviews) to develop a more independent assessment of patient understanding.11 A number of standardized comprehension tests have been proposed. However, most of these, including the well-known MacArthur Competence Assessment Tool, have been developed for cognitively impaired adults.32–34
Instruments for noncognitively impaired individuals include:
- The Deaconess Informed Consent Comprehension Test (DICCT): Initially designed to provide objective understanding in anti-infective clinical trials, this test employs US federal consent requirements and was developed using mainly female respondents, aged 36, with 2 years’ college education.35 It comprises a 14-item open-ended, generic structured interview questionnaire format and is written at an 8th grade reading level.
- The Quality of Informed Consent (QuIC) questionnaire: This is also based on US federal consent requirements and is designed to provide both a subjective and objective understanding of oncology clinical trials; accordingly, “cancer-specific” language is used throughout.36 The respondents were mainly white, well-educated people over the age of 55 years. The test is written at a 12th grade reading level with a 34-item questionnaire containing 2 sections:
- Section A has 20 objective questions with a 3-point response scale (disagree/unsure/agree) and
- Section B has 14 subjective questions.
In order to utilize this test, researchers are required to take missing data into account and write scoring algorithms for the statistical program to be used for data analysis.37
- The Brief Informed Consent Protocol (BICEP): Based on a combination of published empirical literature together with recommendations from an advisory group, the BICEP is aimed at determining satisfaction with the quality of the informed consent process, as well as assessing common therapeutic misconceptions.38 There are 12 structured, open-ended interview questions.10
- Buccini has proposed 2 instruments, namely the Basic Investigator Questionnaire (BIQ) and the Modular Informed Consent Comprehension Assessment (MICCA), for assessing informed consent comprehension in noncognitively impaired trial participants.10 The MICCA assesses understanding of essential informed consent concepts, while the BIQ functions as a companion instrument to assess trial-specific details.
Most of these tests are relatively easy to administer, require minimal training and can be performed in about 7–12 mins. However, they suffer from a number of drawbacks, including insufficient testing with regard to reliability and validity, lack of generalizability, lack of detail with regard to administration and interpretation of the tests and poorly defined psychometric properties.10,18,37
Readability of informed consent documents and education of trial participants
The readability of the 3 informed consent forms for the concurrent cardiovascular risk trials ranged from 10.0 to 12.3, according to the Flesch–Kincaid Grade Level. Minimal details with regard to the corresponding formal education of the trial participants were collected; however, it appears to be incongruent to the readability of the consent forms, with 85% of the participants having 6 or more years of formal schooling and 43.5% having 10 or more years. This is in keeping with previous studies conducted at TREAD Research where it was demonstrated that:
- During the period 2000 to 2009, the mean ± SD Flesch–Kincaid Grade Level for informed consents used in the unit was 12.13±1.8 (range, 8.3–14.9)39 and
- Only 53.8% of the study population seen in the unit had more than 8 years of formal schooling.16
The high readability scores seen in the informed consent documents utilized in this setting are not unique and are in keeping with those seen in other settings, including Australia and the US.10,40,41 In addition, nearly 50% of The Australians and Americans are reported to have literacy skills at or below an 8th grade reading level.10
Buccini regards readability formulae as an indirect method of measuring informed consent comprehension and advises that ethics committees should take cognizance of this when approving informed consent documents.10 Readability is highlighted as a criteria for the documents submitted to the SHREC; the checklist stresses that the researcher must ensure that “simple, clear language has been used (Maximum Grade 8 reading level) and all medical and technical terms have been explained”.42 The continued ethics approval of documents with readability scores in excess of grade 8 suggests that neither the researchers nor the ethics committee members are promoting and/or enforcing this requirement. There may be a number of possible reasons behind this practice:
- Already most informed consents used at TREAD Research are in excess of 15–20 pages. Attempts to improve the readability, for example by restricting polysyballic vocabulary, explaining technical terms and limiting sentences to less than 12 words,43 are likely to result in lengthier informed consent documents, as demonstrated by many researchers to date.42,44 The low literacy skill of the research population is likely to be further challenged by this.
- Documents forwarded to investigators and ethics committees are usually in traditional paper-based format. Thus, unless the informed consent documents are typed out and processed by an applicable software programme, the readability assessment is more than likely being made subjectively. As Buccini points out, most ethics committee members hold postgraduate degrees and tend to read and write at a grade 15 level or higher, making it difficult for them “to identify documents that are actually written at an 8th grade reading level”.10 Research ethics committees are advised to standardize their policy with regard to readability of informed consent forms – perhaps by insisting that researchers measure and declare the readability grade level of an informed consent at the time of submission. Another recommendation would be to involve lay people (defined as someone not involved in medical, legal or scientific work and not affilitated to the institution) to make this assessment, as recommended by the South African Good Clinical Practice Guidelines.45
Despite the importance of readability, it must be emphasized that readability and comprehension are not synonymous, and there is very little empirical evidence to suggest that decreasing the readability of these forms will have any significant impact on comprehension.46,47 In addition, there appears to be conflict with regard to the choice of readability formulae. Hochhauser analyzed a 23-page informed consent document using 17 different readability formulae and demonstrated that the readability scores ranged from 11.5 to 16.37 The incorporation of the Flesch Reading Ease and Flesch–Kincaid formulae into the Microsoft Word™ software package has resulted in these becoming the most commonly used formulae. However, the Gunning Fog and SMOG indices may, in fact, be more appropriate for the purpose of informed consent documents due to the inclusion of complicated medical, legal, scientific and bureaucratic language. These indices tend to give higher scores than the other readability formulae.37
Language and cultural barriers
Due to the language and cultural profile of the study populations in this study, prospective trial participants were given the option to give informed consent and complete the self-report and descriptive narrative in either Afrikaans (80.4% of the participants) or English (19.6%). None of the patients incorporated in this study had any other language as their mother tongue. In addition, all trial staff at TREAD Research are fluent in both English and Afrikaans; interpreters were thus not required. Thus, it is unlikely that language impacted on the results of this study.
In contrast, the situation in many other research units in South Africa is probably very different. In South Africa, there are 11 official languages. The 2011 census in South Africa demonstrated that Afrikaans is the mother tongue of 13.5% of the population and English 9.6%. The remaining 76.9% of the population speak one of the South African black indigenous languages, in particular isiZulu (22.7%), isiXhosa (16%), Setswana (8%) and Sesotho (7.6%).48 This may give rise for the need for interpreters in these research units and the problems associated therewith – for example, the possibility that the interpretation is inaccurate and/or that the interpreters may omit essential information “in order to protect the patient from the harsh reality”.49
Another problem recognized with black indigenous languages is the fact that formal writing is a relatively new phenomenon, and orthographics and vocabulary are still being standardized by piercing together spoken dialects.50 Pandiya reports a similar problem in India where literal translations from English do not capture the true meaning of concepts or phrases and the nuances of the original document changes on translation.51 The net impact of this is that readers often struggle to understand written language, even fluent speakers. This often manifests in these trial participants requesting English versions of consent forms in South Africa; a similar phenomenon has been observed by Chaisson in Botswana.22
Understanding of voluntariness and right to withdraw
Both the participants’ self-report and the descriptive narrative demonstrated that the terms voluntariness and right to withdraw were well understood by most trial participants in this study. These findings are supported by Falagas et al.11 Manafa et al, however, demonstrated that while most trial participants understood the concept of right to withdraw, many regarded withdrawing from a trial as disrespectful or leading to a loss of benefits.21
Findings of previous studies conducted in South Africa, however, also question this level of comprehension:
- Abdool Karim demonstrated that informed consent was “truly informed but not truly voluntary”, with 88% of the participants stating that they “felt compelled to participate”.23
- Joubert et al demonstrated that trial participants were not informed and, despite the fact that the participants believed that they had voluntarily consented for the trial, they were “clearly aware of the lack of alternative sources of care”.24
It must be noted, however, that both of these studies were conducted as sub-studies to trials investigating vertical perinatal transmission of HIV and included pregnant, HIV-positive women in a public hospital. Not only can these be regarded as a particularly vulnerable group of patients, but these trials were conducted at a time when the political regime in South Africa was publically denying the pathogenesis and therapeutic options of HIV patients.52
A previous paper published by TREAD Research examined the motivation for clinical trial participation and demonstrated that altruism and learning about their condition were the leading reasons for participants to volunteer for clinical trials, even though benefits such as free access to medical care and medication also played a role.16 These findings are in keeping with studies conducted in a number of other countries, including the US,53,54 Romania55 and Taiwan.56 Financial gain has often been cited as a possible reason motivating trial participants to volunteer for clinical trials in South Africa – especially in light of the obligatory trial participant remuneration mandated by the South African regulatory authority, namely the MCC.16,17 This was not identified as a significant motivating factor in this unit. This may partly be due to the nature of the trials conducted in this unit and the location of the unit, together with the fact that the standard operating procedure for this unit with regard to patient remuneration discourages monetary values from being mentioned in the informed consent and rather follows a policy of participant remuneration based on distance traveled.16
This appears to be in contrast to the grant-funded, community-based programmes being conducted mainly for HIV where reports have emerged of participants attempting to enroll at multiple research units in an attempt to access the obligatory participant remuneration (personal communication). The problem has become so rife that sites have started investigating measures to curb the practice, such as the use of fingerprint and retinal scanners. Financial gain has also been cited as the leading motivating factor for trial participation at a unit in Brazil.57
TREAD Research is an independent, dedicated trial unit where patients are invited to participate by independent recruiters as opposed to their routine health care providers. This, together with the fact that patients still receive their basic medical care for their underlying conditions from their routine health care provider during the course of the clinical trial, may assist in their understanding that this is a voluntary activity.
Understanding of side effects, risks and benefits
In this study, the concept of side effects of the study drug was understood by 54% of the participants (even though 74% of participants reported understanding the concept). However, it is equally important that prospective trial participants understand that there are other potential risks associated with clinical trials, including risks resulting from study procedures. Despite the fact that 80% of trial participants reported understanding the concept of risks and benefits, only 17% understood the concept of risks and 52% the concept of benefits according to their descriptive narrative reports. Manafa et al and Chaisson et al demonstrated similar findings and reports that although most trial participants could mention at least one potential benefit of the study, many did not even recall that there were potential risks.36,38 Furthermore, most of the participants who did understand the concept of risk could not remember any of them.
The apparent bias with regard to the comprehension of risks and benefits is not a new phenomenon.58,59 It appears to be common within the field of oncology and much of the reasoning behind this appears to be based on denial and/or emotional factors. Gattellari reported that 80% of oncology patients who had been informed that they had incurable cancer interpreted this to mean that they had a chance of cure, whilst 15% reported this to mean that they had at least a 75% chance of a cure.60 Overall, oncology patients appear to have unrealistic optimism that enrolling in oncology trials will provide a miracle cure.61–63
The issue of risks and benefits is fundamental to clinical research and is addressed by most of the research guidelines:
- The Nuremburg Code of 1947 states that “the degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment”;3
- Beneficence is one of the 3 major ethical principles identified by the Belmont Report of 1979;4
- Guideline 8 of the CIOMs guidelines states that “for all biomedical research involving human subjects, the researcher must ensure that there is a reasonable balance of potential benefits and risks”;6
- The Declaration of Helsinki states that “medical research in human subjects may only be conducted if the importance of the research outweighs the inherent risk and burdens to the research subject”7 and
- Principle 8 of ICH-GCP guidelines stipulate that “research involving humans should be continued only if the benefit-risk profile remains favourable”.64
It is thus concerning that such a relatively small number of clinical trial participants appear to understand this concept.11 Turney argues that “apparent ignorance or misunderstanding of scientific explanation may represent a resistance to information that runs counter to one’s beliefs or reduces feelings of control over one’s own life”.65 Additionally, it has been reported that the level of comprehension with regard to benefits was higher when the clinician communicated this information than when the participant merely read this in the consent document (81% vs 35%). Researchers must be cautious about overemphasizing benefits over risks in an attempt to encourage trial participation.
Understanding of randomization, placebo and blinding
The current study demonstrated that the concepts of randomization, placebo, and blinding were poorly understood with an actual comprehension of 13%, 20% and 20%, respectively. Even the participant self-reported understanding of these concepts was relatively poor, being 50%, 37%, and 30%, respectively.
The particularly poor understanding of the concept of randomization is in keeping with the findings of Featherstone and Donovan.66 Erroneous lay interpretations have largely been credited for this confusion. Ellis et al conducted a study in Australia and demonstrated that nearly 75% of the recruited oncology participants believed that their physician would make sure that they received the best treatment.67 Snowdon et al demonstrated a similar poor comprehension for this concept while interviewing parents of critically ill infants.68 This lack of understanding was frequently accompanied by anger and confusion from the parents who desired the best treatment option for their infant.
Patients are unaccustomed to situations where treatment decisions are “based on chance” and “blinded” rather than being in their best interest – in other words, therapeutic misconception or equipoise.2,68 In addition, most participants fail to comprehend and/or recall that a placebo arm involves receiving no active treatment.18 Stead et al maintain that “for many respondents, it was not simply that they did nor fully understand the concepts of placebo, random allocation, and double blinding- they disliked and resisted them“.9 It is thus not surprising that these concepts score unfavorably in the majority of studies from both developed and developing countries.11,18,28,29
Perceived versus actual understanding
The difference in comprehension between the self-reported versus the narrative methods was significant – especially with regard to the concepts of benefits (19 of the 37 participants who reported that they understood this concept, in fact, did when assessed by the narrative description) and side effects (21 of the 34 participants who reported that they understood this concept did). The overconfidence of the study participants in this study was also manifested in the median scores for overall comprehension – the self-report revealed median understanding of 70% (interquartile range 50–80%), while the corresponding result for the narrative description was 45% (interquartile range 30–65%).
The difference between perceived versus actual understanding has been documented before; up to 90% of the people demonstrate that they are overconfident about themselves and report having above-average knowledge or skills.69 This phenomenon has also been documented in the DICCT study where 70% of patients reported having a thorough understanding of the informed consent information.35
Other problems associated with assessing informed consent comprehension
In this study, patient comprehension was measured at the time of giving consent. Assessing informed consent comprehension may be complicated by the fact that it requires a distinction to be made between understanding of the various trial related concepts and procedures, and participant recall.15 A number of the previous studies in this field have only assessed patient comprehension after a significant period – in the case of Manafa et al,21 only 12 months after initial trial consent was given. The problem being that recall, especially in a relatively uneducated patient population, is bound to affect patient comprehension.
Hirchhorn (2004) also highlights the importance of the patient’s attention span on the comprehension of the informed consent document. It is estimated that the average adult has an attention span of <20 mins – although a human’s continuous attention span may be as little as 8 s.70,71 The current tendency for informed consents to consist of 15–20 pages raises further questions with regard to informed consent comprehension by the average potential trial participant. No formal testing of potential participant’s attention span was incorporated into this study. However, according to the standard operating procedures for the site, potential trial participants are encouraged to have a friend or family member present during the informed consent process and, if necessary, to take the document home with them to discuss with friends and/or family members at their leisure.
Ideas to improve patient comprehension
A number of mechanisms have been suggested in order to improve comprehension of the informed consent form.11,72–76 The results of such suggestions have shown varying degrees of efficacy; however, it is difficult to compare these suggestions – mainly due to inconsistencies among reporting techniques.72 According to Hochhauser, at the very best such interventions demonstrate limited improvement (about 12%).37
Hocchauser summarizes the strategies that should be adopted henceforth:
- The provision of templates for informed consent forms by regulatory agencies which comply with the recommended 6th to 8th grade readability recommendation
- The promotion and support of consent form comprehension and recall studies which are based on “psychometrically sound instruments, research methods and statistical analyses”
- The recognition that tailor-made consent forms and processes should be designed to meet the diverse needs of the patient population, especially cultural and ethnic diversities
- Revision of regulatory and ethical consent guidelines.37
While Hochhauser’s recommendations are valid, research units, especially those based in developing countries, need to start exploring distinct options and strategies in order to improve informed consent comprehension amongst the research participants. Possible strategies include:
- Formulation and adoption of standard operating procedures for the informed consent process which reflect the needs of the patient population:
- Understand the language and cultural barriers
- Introduce methods to reduce the readability of informed consent documents
- Provide highlighters to the trial participants in order to enable them to identify those concepts which they do not immediately understand
- Emphasize the time and opportunity afforded to each potential trial participant for discussion: According to Chaisson et al, one of the most effective remedies for improving comprehension is the inclusion of more discussion time in the process itself.22
- Encourage participants to voice their understanding of certain concepts explained to them in the informed consent document: Baer et al suggest using prompts such as “Please tell me what I have just said to you” or “What will you tell your son when he calls this evening?”77
- The introduction of educational aids in the waiting area and/or consulting rooms – such as:
- The complimentary “speaking book”, sponsored by Pfizer, the World Medical Association, the Steve Biko Centre for Bioethics and the South African Medical Association, entitled “What it means to be part of a clinical trial” which explains these (and other) important trial concepts and which is directly aimed at “African populations with low literacy rates”78
- Educational videos/DVDs, such as those provided by the Centre for Information and Study on Clinical Research Participation (CISCRP), which explain trial processes and informed consent concepts.79
- More recently, Mytrus® has explored the use of iPad technology in the informed consent process.80 The application utilizes nontext mediums such as visual imagery and animation, as well as interactive graphics, to assist prospective trial participants in understanding more difficult terms and concepts related to clinical trials and informed consent. An additional advantage of this technology is the potential incorporation of self-direction – for example, the inclusion of nonlinear text such as hypertext which would enable participants “to click or hover over a term in an informed consent document, which would then present that participant with a definition”.81
A randomized trial was conducted at Sutter Health’s California Pacific Medical Centre Research Institute in San Francisco to assess the application and demonstrated that participant comprehension increased by 24% when using the iPad application compared to when using traditional paper-based inform consents (namely, 76% vs 52%). Participant comprehension was assessed by means of a quiz within 24 hrs of providing informed consent.80 A similar prototype application is being explored by 3 oncologists in the US within a clinical trial for a node-positive breast cancer.82
- Ongoing education of all investigators taking consent in the unit, in particular raising their awareness with regard to those concepts of informed consent that are especially poorly understood – such as risks, randomization, blinding and placebo: Baer et al suggest that not only should trial personnel taking consent assess the participant’s level of understanding, but also document how this assessment was made.77 Assessing comprehension can be difficult. This is routinely done during the consent process at TREAD Research and documented in the source documents of the trial participant. However, to a large extent, researchers are relying on the participant to confirm whether or not he/she understands the process. As demonstrated in this and other studies, participants frequently overestimate their level of understanding. Suggestions to improve this assessment include observing the participant’s body language and engagement during the informed consent process – including the nature of the questions asked.77 The use of Question Prompt Lists, namely a structured list of questions, within the context of oncology trials has encouraged participants to ask questions and become more involved with the informed consent process.83,84 Universal prompt lists are also available on the CISCRP website.85
- Possible inclusion of a simple assessment of comprehension – such as a simple multiple-choice questionnaire and/or true/false questionnaire.10 Although such tests provide a less sensitive method of assessing comprehension than asking prospective trial participants to voice their understanding in their own words, it would still be useful in identifying participants who do not understand fundamental trial concepts.25,73 Miller et al demonstrated that comprehension could be statistically increased within a sample of Tibetan women with no or little formal education by asking comprehension questions at the end of each section.35 These participants can then be offered an opportunity to be re-trained on those concepts that are poorly understood.
- The use of patient advocates: Not only have patient advocates been useful in providing feedback with regards to the readability of the informed consent form, but they can also provide guidance in:
Conclusion
The ongoing trend toward globalization in the clinical research arena has resulted in increasingly higher numbers of patients from poorer developing countries (including South Africa) being included in clinical trials, including pharmaceutical-sponsored, multinational, randomized controlled trials for diseases that were previously considered first world diseases (eg, cardiovascular risk factors).12,86 As a result, approximately 40% of all clinical trials were being conducted in these developing countries in 2005, compared to approximately 10% in 1991.87 This trend is likely to continue given that the FDA has called for strategies to increase the participation of “racially and ethnically diverse communities” in clinical trials.88 Although there are many positive factors associated with this shift, there are a number of potential risks associated with such a threat, including the risk of increased litigation. A number of ethicists have voiced their concern with regard to the potential exploitation of these vulnerable patients.13,14 In particular, Emanuel et al have identified the quality of the informed consent process as being a major concern.14 This study confirmed that this concern was not misplaced – even in a well-established unit with well-trained, experienced clinical investigators and comprehensive standard operating procedures.
Being a researcher in a developing country, it is important to understand the deficits associated with the informed consent process and be able to identify areas of concern with regard to the informed consent process in trial participants in developing countries such as South Africa. In this way, clinical researchers and research units will be able to work toward eliminating, or at least minimizing, these problems. In the context of this trial, the following concepts were particularly poorly understood: randomization, risks, placebo, blinding, and ethics committee, being understood by only 13–22% of the research participants.
The research site at which this study was conducted is a well-established unit with ongoing training programmes in place and well-documented standard operating procedures with regard to informed consent processes. Current standard operating procedures for the unit include the investigator noting in the patient source notes that he/she is satisfied that the patient understands the contents of the informed consent. The investigators that conducted the consent processes for the purposes of this study have more than 8 years’ research experience each. Despite these precautions and observations, the participant rate of comprehension was poor and the process for taking informed consent subsequently needs to be modified.
Discussions are currently ongoing at the site on the exact nature of the modifications required. At this stage, the following modifications are in the process of being implicated at the site:
- Assessment of the readability level of informed consent forms at the time of ethics submission and the formulation of guidelines to simplify the level if above an 8th grade reading level
- Establishment of a patient advocacy group that is representative of the site patient demographics to discuss the findings of this study and to brainstorm various solutions: this will include discussing ways in which the difficult research concepts can be conveyed to prospective trial participants in a more meaningful manner and the possible development and use of Question Prompt Lists.
- The introduction of educational aids in the waiting area, including:
- The complimentary “speaking book”, entitled “What it means to be part of a clinical trial”:78 unfortunately this publication is only available in English and there are no immediate plans to translate this into any of the South African languages in the immediate future
- The educational DVD provided by CISCRP: This aid is only available in English and is limited by the fact that it was developed in the context of an oncology trial unit in the US;79
- Modification of the standard operating procedure for the informed consent to incorporate more discussion time with the patients and to introduce the concept of prompts to encourage feedback by trial participants and
- Retraining of all trial personnel on the important concepts that are routinely poorly understood by trial participants.
Once these processes have been implemented, this study will be repeated in another group of 45–50 patients in order to assess whether the proposed tools have been effective.
Acknowledgments
I would like to thank the investigators, study coordinators and patients at TREAD Research for their participation in this research project.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Beauchamp JL, Childress JF. Principles of Biomedical Ethics.
2. Lidz CW. The therapeutic misconception and our models of competency and informed consent. Behav Sci Law. 2006;24:535–546. doi:10.1002/bsl.700
3. Katz J. The nuremberg code and the nuremburg trial; a reappraisal. J Am Med Assoc. 1996;276(20):1662–1666.
4. US Department of Health and Human Services. The belmont report [Online]. Available from: http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html.
5. US Department of Health and Human Services. Institutional review board written procedures: guidance for institutions and IRBs (2018) [Online]. Available from: https://www.hhs.gov/ohrp/regulations-and-policy/guidance/institutional-issues/institutional-review-board-written-procedures/index.html.
6. CIOMS. CIOMS international ethical guidelines for biomedical research involving human subjects [Online] Available from: http://www.recerca.uab.es/ceeah/docs/CIOMS.pdf.
7. World Medical Association (WMA). Declaration of Helsinki [Online]. Available from: http://www.wma.net/en/30publications/10policies/b3/17c.pdf.
8. Yuval R, Halon DA, Merdler A, et al. Patient comprehension and reaction to participating in a double-blind randomized clinical trial (ISIS04) in acute myocardial infarction. Arch Intern Med. 2000;160(8):1142–1146.
9. Stead M, Eadie D, Gordon D, Angus K. “Hello, hello.it’s English I speak!”: a qualitative exploration of patients’ understanding of the science of clinical trials. J Med Ethics. 2005;31:664–669. doi:10.1136/jme.2004.011064
10. Buccini LD Developing an instrument to measure informed consent comprehension in non-cognitively impaired adults [Doctor of Public Health Thesis], School of Health sciences, Faculty of Health and Behavioural Sciences, University of Woolongong
11. Falagas ME, Korbila IP, Giannopoulou KP, Kondilis BK, Peppas G. Informed consent: how much and what do patients understand? Am J Surg. 2009;198:420–435. doi:10.1016/j.amjsurg.2009.02.010
12. Denny CC, Grady C. Clinical research with economically disadvantaged populations. J Med Ethics. 2007;33(7):382–385. doi:10.1136/jme.2006.017681
13. Yusuf S. Clinical research and trials in developing countries. Stat Med. 2002;21:2859–2867. doi:10.1002/sim.1290
14. Emanuel E, Wendler D, Killen J, Grady C. What makes clinical research in developing countries ethical? The benchmarks of ethical research. J Infect Dis. 2004;189:930–937. doi:10.1086/381709
15. Lindegger G, Richter L. HIV vaccine trials: critical issues in informed consent. S Afr J Sci. 2000;96:313–318.
16. Burgess LJ, Sulzer NU, Emanuel SC. Clinical trial remuneration: the patient’s perspective. S Afr Med J. 2008;98(2):95–96.
17. Burgess LJ, Sulzer NU, Hoosain F, et al. Patient’s motivations for participating in cardiovascular clinical trials: a local perspective. Cardiovasc J Afr. 2009;2094:220–223.
18. Moodley K, Pather M, Myer L. Informed consent and participant perceptions of influenza vaccine trials in South Africa. J Med Ethics. 2005;31:727–732. doi:10.1136/jme.2004.009910
19. Dunn LB, Nowrangi MA, Palmer BW, et al. Assessing decisional capacity for clinical research or treatment. A review of instruments. Am J Psychiatry. 2006;163(8):1323–1334. doi:10.1176/ajp.2006.163.8.1323
20. Krosin MT, Klitzman R, Levin B, et al. Problems in comprehension of informed consent in rural and peri-urban Mali, West Africa. Clin Trials. 2006;3(3):306–313. doi:10.1191/1740774506cn150oa
21. Manafa O, Lindegger G, Ijsselmuiden C. Informed consent in an antiretroviral trial in Nigeria. Indian J Med Ethics. 2007;IV(1):26–30.
22. Chaisson LH, Kass NE, Chengata B, et al. Repeated assessments of informed consent comprehension among HIV-infected participants of a three-year clinical trial in Botswana. PLoS One. 2011;6(10):e22696. doi:10.1371/journal.pone.0022696
23. Abdool Karim Q, Abdool Karim SS, Coovadia HM, et al. Informed consent for HIV testing in a South African hospital: how informed and voluntary? Am J Public Health. 1998;93:582–584.
24. Joubert G, Steinberg H, van der Ryst E, et al. Consent for participation in the Bloemfontein Vitamin A trial: how informed and voluntary? Am J Public Health. 2003;93(4):582–584.
25. Lindegger G, Milford C, Slack C, et al. Beyond the checklist. Assessing understanding for HIV vaccine trial participation in South Africa. J Acquired Immune Deficiency Syndrome. 2006;43:560–567.
26. Minnies D, Hawkridge T, Hanekom W, et al. Evaluation of the quality of informed consent in a vaccine field trial in a developing country setting. BMC Med Ethics. 2008;9:15. doi:10.1186/1472-6939-9-15
27. Lynöe N, Sandlund M, Jacobsson L, et al. Informed consent in China: quality of information provided to participants in a research project. Scand J Public Health. 2004;32:472–475.
28. Wang H, Erickson JD, Li Z, Berry RJ. Evaluation of the informed consent process in a randomized controlled trial in China: the sino-US NTD project. J Clin Trials. 2004;15:61–75.
29. Pace C, Emanuel EJ, Chuenyam T, et al. The quality of informed consent in a clinical research study in Thailand. IRB. 2005;27:9–17.
30. Joseph P, Schackman BR, Horwitz R, et al. The use of an educational video during informed consent in an HIV clinical trial in Haiti. J Acquired Immunodeficiency Syndrome. 2006;42:588–591. doi:10.1097/01.qai.0000229998.59869.05
31. Guarino P, Lamping DL, Elbourne D, Carpenter J, Peduzzi P. A brief measure of perceived understanding of informed consent in a clinical trial was validated. J Clin Epidemiol. 2006;59:608–614. doi:10.1016/j.jclinepi.2005.11.009
32. Schmand B, Gouwenberg B, Smit JH, Jonker C. Assessment of mental competency in community-dwelling elderly. Alzheimer Dis Assoc Disord. 1999;13:80–87.
33. Buckels VD, Powlishta KK, Palmer JL, et al. Understanding of informed consent by demented individuals. Neurology. 2003;61:1662–1666.
34. Meulenbrock O, Vernooij-Dassen M, Kessels RPC, et al. Informed consent in dementia research. Legislation, theoretical concepts and how to assess capacity to consent. Eur Geriatr Med. 2010;1(1):58–63. doi:10.1016/j.eurger.2010.01.009
35. Miller CK, O’Donnell DC, Searight HR, Barbarash RA. The deaconness informed consent comprehension test: as assessment tool for clinical research subjects. Pharmacotherapy. 1996;16(5):872–878.
36. Joffe S, Cook EF, Cleary PD, Clark JW, Weeks JC. Quality of informed consent in cancer clinical trials: a cross-sectional survey. Lancet. 2001;358:1772–1777. doi:10.1016/S0140-6736(01)06805-2
37. Hochhauser M. Consent comprehension in the 21st century: what is missing? Drug Inf J. 2008;42:375. doi:10.1177/009286150804200410
38. Sugarmann J, Lavori PW, Boeger M, et al. Evaluating the quality of informed consent. Clin Trials. 2005;2:34–41. doi:10.1191/1740774505cn066oa
39. Terblanche M, Burgess LJ. Examining the readability of patient informed consent forms. Open Access J Clin Trials. 2010;2:157–162. doi:10.2147/OAJCT.S13608
40. Sharp SM. Consent documents for oncology trials: does anybody read these things? J Clin Oncol. 2004;27:570–575. doi:10.1097/01.coc.0000135925.83221.b3
41. Paasche-Orlow MK, Taylor HA, Bruncati FL. Readability standards for informed consent forms as compared with actual readability. N Engl J Med. 2003;348:721–726. doi:10.1056/NEJMsa021212
42. Research Development and Support. Clinical trial checklist for the health rsearch ethics committee at the faculty of medicine and health sciences, Stellenbosch University [Online]; n.d. Available from: http://sun025.sun.ac.za/portal/page/portal/Health_Sciences/English/Centres%20and%20Institutions/Research_Development_Support/Ethics/new_application.
43. Wittenberg KM, Dickler HB. Universal use of short and readable informed consent documents: how do we get there? Creating informed consent documents that inform: a literature review. Association of American Medical Colleges Appendix C, Feb 2007. Available at: http://www.aamc.org/research/clinicalresearch/hdickler-mtgsumrpt53007.pdf. Accessed August 10, 2010.
44. LoVerde ME, Prochazka AV, Byyny RL. Research consent forms: continued unreadability and increasing length. J Gen Intern Med. 1989;4:410–412.
45. South African Department of Health. Guidelines for Good Practice in the Conduct of Clinical Trials with Human Participants in South Africa. Pretoria: Government Printer; 2006.
46. Redish J. Readability formulas have even more limitations than Klare discusses. ACM J Comput Doc. 2000;24(3):132–137. doi:10.1145/344599
47. Coyne CA, XU R R, Raich P, et al. Randomized, controlled trial of an easy-to-read informed consent statement for clinical trial participation: a study of the Eastern cooperative oncology group. J Clin Oncol. 2003;21:836–842. doi:10.1200/JCO.2003.07.022
48. Anonymous. The languages of South Africa. SouthAfrica.info [Online]; 2013. Available from: http://www.southafrica.info/about/people/language.htm.
49. Schenker Y, Wang F, Selig SJ, et al. The impact of language barriers on documentation of informed consent at a hospital on-site interpreter services. J Gen Med. 2007;22(supplement2):295–299.
50. Johnson (2012). Wanted: readers, writers and publishers. The Economist [Online].
51. Pandiya A. Readability and comprehensibility of informed consent forms for clinical trials. Perspect Clin Res. 2010;1(3):98–100.
52. BBC News (2000). Controversy dogs AIDS forum. BBC News [Online].
53. Andresen EL, Wilson KA, Castillo A, Koopman C. Patient motivation for participating in clinical trials for depression: validity of the motivation for clinical trials inventory-depression. Int Clin Psychopharmacol. 2010;25(1):7–16. doi:10.1097/YIC.0b013e328332055c
54. Stunkel L, Grady C. More than the money: a review of the literature examining healthy volunteer motivations. Contemp Clin Trials. 2011;32(3):342–352. doi:10.1016/j.cct.2010.12.003
55. Udrea G, Dumitrescu B, Purcarea M, et al. Patients’ perspectives and motivators to participate in clinical trials with novel therapies for rheumatoid arthritis. J Med Life. 2009;15(2):227–231.
56. Wang LH, Tsai YF, Chen JS, Tsay PK. Intention, needs, and expectations of cancer patients participating in clinical trials. Cancer Nurs. 2011;34(2):117–123. doi:10.1097/NCC.0b013e3181efe1c0
57. Nappo SA, Iafrate GB, Sanchez ZM. Motives for participating in a clinical research trial: a pilot study in Brazil. BMC Public Health. 2013;13:19. doi:10.1186/1471-2458-13-19
58. Lloyd AJ. The extent of patients’ understanding of the risk of treatments. Qual Health Care. 2001;10(SupplI):i14–i18. [Online].
59. Lynöe N, Sandlund M, Jacobsson L, et al. Informed consent in two Swedish prisons: a study of quality of information and reasons for participating in a clinical trial. Med Law. 2001;20(4):519–523.
60. Gattelari M, Butow PN, Tattersall MH. What did the doctor say? Lancet. 1999;353:1713. doi:10.1016/S0140-6736(98)09449-5
61. Cheng JD, Hitt J, Koczwara B, et al. Impact of quality of life on patient expectations regarding Phase I clinical trials. J Clin Oncol. 2000;18:421–428. doi:10.1200/JCO.2000.18.2.421
62. Brody BA, McCullough LB, Sharp RR. Consensus and controversy in clinical research ethics. J Am Med Assoc. 2005;294:1411–1444. doi:10.1001/jama.294.11.1411
63. Kardinal CA, Sanders JB. Altruism: a form of hope for patients with advanced cancer. J Clin Oncol. 2010;28(15 Supplement):e19559.
64. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), ICH Guideline E6(R2). Guideline for good clinical practice [Online]; 2016. Available from: http://www.ich.org/LOB/media/MEDIA481.pdf.
65. Turney J. Public understanding of science. Lancet. 1996;347:1087–1090.
66. Featherstone K, Donovan JL. Random allocation of allocation at random? Patient’s perspectives of participation in a randomized controlled trial. Br Med J. 1998;317(7167):1177–1180.
67. Ellis PM, Dowsett SM, Butow PN, et al. Attitudes to randomized clinical trial amongst out-patients attending a medical oncology clinical. Health Expectations. 1999;2:33–43.
68. Snowdon C, Garcia J, Elbourne D. Making sense of randomization; responses of parents of critically ill babies to random allocation of treatment in a clinical trial. Soc Sci Med. 1997;45(9):1337–1355.
69. Camerer CF, Lavallo D. Overconfidence and excess entry: an experimental approach. Am Econ Rev. 1999;89(1):306–318.
70. Hirschhorn W, Ghannam F. Informed consent: an important obligation involving hidden issues. SoCRA Source. 2004;17–20. [Online]. Available from: http://www.socra.org/pdf/200405_Informed_Consent_Obligation.pdf. Accessed April 02, 2013.
71. New York State Council of Health System Pharmacists. Three characteristics to success. J Pharm Pract. 2012;25:622–623. [Online]. Available from: http://jpp.sagepub.com/content/25/6/622.extract. Accessed April 02, 2013.
72. Cohn E, Larson E. Improving participating comprehension in the informed consent process. J Nurs Scholarsh. 2007;39(3):273–280. doi:10.1111/j.1547-5069.2007.00180.x
73. Kripilani S, Bengtzen R, Henderson LE, Jacobson TA. Clinical research in low-literacy populations: using teach-back to assess comprehension of informed consent and privacy information. IRB Ethics Hum Res. 2008;30(2):13–19.
74. Jefford M, Moore R. Improvement of informed consent and the quality of consent documents. Lancet Oncol. 2008;9(5):485–493. doi:10.1016/S1470-2045(08)70128-1
75. Rousanville DB, Hunkele K, Eaton CJ, et al. Making consent more informed: preliminary results from a multiple-choice tests among probation-referred marijuana users entering a rando-mized clinical trial. J Am Acad Psychiatry Law. 2008;36(3):354–359.
76. Ridpath JR, Wiese CJ, Greene SM. Looking at research consent forms through a participant-centred lens: the PRISMS readability toolkit. Am J Health Promotion. 2009;23(6):371–373. doi:10.4278/ajhp.080613-CIT-94
77. Baer AR, Good M, Shapiro L. A new look at informed consent for cancer clinical trials. J Oncol Pract. 2011;7(4):267–270. doi:10.1200/JOP.2011.000347
78. Anonymous. Understanding clinical trials with a “Speaking Book” [Online]; 2009. Available from: http://www.pfizer.com/files/research/research_clinical_trials/ClinicalTrials_SpeakingBook_030209.pdf.
79. National Center for Advancing Translational Sciences. “Clinical |research studies and you” [Online]; n. d. Available from: http://www.ncats.nih.gov/research/cts/clinical-trials/research-and-you/studies-and-you.html.
80. Mansell P (2012). Mytrus launches iPad informed consent application. Online PharmaTimes.
81. Gossen R. Electronic informed consent: possibilities, benefits, and challenges [Online]; 2012. Available from: http://rebarinteractive.com/electronic-informed-consent-introduction/.
82. Butcher L. Health care IT for oncology: informed-consent app. Oncol Time. 2012;34(21):27. doi:10.1097/01.COT.0000422993.49579.25
83. Brown RF, Shuk E, Butow P, Edgerson S, Tattersall MHN, Ostroff JS. Identifying patient information needs about cancer clinical trials using a question prompt list. Patient Edu Couns. 2011;84(1):69–77. doi:10.1016/j.pec.2010.07.005
84. Brown RF, Bylund CL, Li Y, Edgerson S, Butow P. Testing the utility of a cancer clinical trial specific Question Prompt List (QPL-CT) during oncology consultations. Patient Edu Couns. 2012;88(2):311–317. doi:10.1016/j.pec.2012.02.009
85. Centre for information and study on clinical research participation; n. d. Available from: http://www.ciscrp.org.
86. Thiers FA, Sinskey AJ, Berndt ER. Trends in the globalization of clinical trials. Nat Rev Drug Discov. 2008;7(1):13–14. doi:10.1038/nrd2441
87. Petryna A. When Experiments Travel; Clinical Trials and the Global Search for Human Subjects. USA: Princeton University Press; 2009.
88. US Food and Drug Administration. Participation of certain population subsets in clinical drug trials: request for comment. Fed Regist. 2009;74(8). [Online]. Available from http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=2009_register&docid=fr13ja09-60. Accessed September 29, 2010.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.