")
Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 7
AMPK activation: a therapeutic target for type 2 diabetes?
Authors Coughlan K, Valentine R, Ruderman N, Saha A
Received 18 March 2014
Accepted for publication 16 April 2014
Published 24 June 2014 Volume 2014:7 Pages 241—253
DOI https://doi.org/10.2147/DMSO.S43731
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Kimberly A Coughlan, Rudy J Valentine, Neil B Ruderman, Asish K Saha
Endocrinology and Diabetes, Department of Medicine, Boston University Medical Center, Boston, MA, USA
Abstract: Type 2 diabetes (T2D) is a metabolic disease characterized by insulin resistance, β-cell dysfunction, and elevated hepatic glucose output. Over 350 million people worldwide have T2D, and the International Diabetes Federation projects that this number will increase to nearly 600 million by 2035. There is a great need for more effective treatments for maintaining glucose homeostasis and improving insulin sensitivity. AMP-activated protein kinase (AMPK) is an evolutionarily conserved serine/threonine kinase whose activation elicits insulin-sensitizing effects, making it an ideal therapeutic target for T2D. AMPK is an energy-sensing enzyme that is activated when cellular energy levels are low, and it signals to stimulate glucose uptake in skeletal muscles, fatty acid oxidation in adipose (and other) tissues, and reduces hepatic glucose production. There is substantial evidence suggesting that AMPK is dysregulated in animals and humans with metabolic syndrome or T2D, and that AMPK activation (physiological or pharmacological) can improve insulin sensitivity and metabolic health. Numerous pharmacological agents, natural compounds, and hormones are known to activate AMPK, either directly or indirectly – some of which (for example, metformin and thiazolidinediones) are currently used to treat T2D. This paper will review the regulation of the AMPK pathway and its role in T2D, some of the known AMPK activators and their mechanisms of action, and the potential for future improvements in targeting AMPK for the treatment of T2D.
Keywords: adenosine monophosphate-activated protein kinase, type 2 diabetes, insulin resistance, drug therapy
Introduction
Obesity, type 2 diabetes (T2D), and metabolic syndrome have reached epidemic proportions worldwide and the prevalence of these conditions continues to grow.1 Although there are several medications currently available to help manage T2D, there is an increasing need for more effective treatments than those currently available. T2D is associated with many comorbidities, such as cardiovascular disease and certain cancers, and it was estimated that diabetes caused 5.1 million deaths in 2013.1
The biological pathways involved in maintaining energy homeostasis have been targeted for pharmacological manipulation to combat the insulin resistance (IR) and metabolic dysfunction caused by chronic nutrient excess.2 One such pathway is that of AMP-activated protein kinase (AMPK), an enzyme that has come to be known as a master regulator of metabolism.2 This nutrient-sensing serine/threonine kinase is activated when cellular energy levels are low (ie, the intracellular AMP:adenosine triphosphate [ATP] ratio is high). Upon activation, AMPK signals through its downstream substrates to restore normal energy levels by stimulating processes that generate ATP (such as fatty acid [FA] oxidation) and inhibiting those that use ATP (such as triglyceride and protein synthesis).3 Overall, AMPK activation improves insulin sensitivity and glucose homeostasis, making it an attractive target for T2D and metabolic syndrome.
Interestingly, several drugs that have long been used for the treatment of diabetes, such as metformin and thiazolidinediones (TZDs), were later found to exert some of their beneficial effects through the indirect activation of AMPK.3 In addition to pharmaceutical agents, numerous natural compounds and hormones can also activate AMPK. Despite being a seemingly promising target for drug development, no direct AMPK activators have reached clinical use for the treatment of metabolic disease. Perhaps further research on AMPK’s regulation will lead to new activation strategies or to the development of compounds with isoform specificity or better pharmacokinetic profiles, finally unlocking the potential for a clinically efficacious AMPK activator. This paper reviews AMPK’s role and dysregulation in T2D, the currently known activators of AMPK, and the potential for a direct AMPK activator reaching the clinic.
Type 2 diabetes
T2D is a metabolic disease characterized by elevated blood glucose levels in the presence of peripheral IR.1 According to the International Diabetes Federation, more than 350 million people worldwide had diabetes in 2013.1 It is projected that this number will rise to nearly 600 million by 2035.1 T2D is associated with a number of complications and comorbidities, including cardiovascular disease, blindness, kidney failure, and lower limb amputation.1 The number one risk factor for T2D is obesity, in which the chronic overconsumption of food leads to hyperglycemia, IR, and impaired metabolic function.1
Excess exposure to glucose, free FAs (FFA), or amino acids can be toxic to cells. To protect themselves from this toxicity, cells use a mechanism of IR to avoid taking up too many nutrients in environments of over-nutrition.4 However, this protective mechanism leads to pathological changes in the setting of prolonged exposure to nutrient overload. In this state of chronic IR, reduced glucose uptake in the muscles, liver, and adipose tissue, impaired suppression of hepatic gluconeogenesis, and impaired suppression of lipolysis lead to hyperglycemia, hyperinsulinemia, and hyperlipidemia.4 At first, the pancreatic β-cells can compensate by secreting even more insulin, but eventually these cells become dysfunctional, causing the patient to become dependent on injections of exogenous insulin. In normally insulin-responsive tissues (for example, muscle, liver, and adipose tissue), the combination of impaired metabolism in the presence of excess glucose, insulin, and FFA causes pathological changes in gene and protein expression and activity.4 While there are many proteins and biological pathways involved in metabolic homeostasis that are dysregulated in IR and T2D, the remainder of this paper will focus on the AMPK pathway.
AMPK
AMPK is a phylogenetically conserved serine/threonine kinase that functions as a master metabolic regulator. It exists as a heterotrimer, consisting of a catalytic α-subunit and regulatory β- and γ-subunits. Each subunit has multiple isoforms (α1, α2, β1, β2, γ1, γ2, γ3), making a total of 12 possible heterotrimer combinations. Whether there are functional differences between the different isoforms remains unclear; however, some isoforms are tissue-specific. For example, heterotrimers containing the α1 isoform predominate in the liver and adipose tissue, whereas those containing α2 predominate in the brain, heart, and skeletal muscles.5,6
The activation of AMPK requires both an increase in the intracellular AMP:ATP ratio and phosphorylation of Thr172 on the “activation loop”7 of the α-subunit by one of its three upstream kinases: the tumor-suppressor liver kinase B1 (LKB1);8,9 the calcium-dependent calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ);10 or transforming growth factor-β activated protein kinase-1 (TAK1).11 An inhibitory site at Ser485 of the α1 subunit also exists and has been shown to be phosphorylated by Akt,12,13 protein kinase A (PKA),14,15 or autophosphorylation14 in various cell types and tissues, such as the heart,12 adipocytes,15,16 and vascular smooth muscle cells.13 Similarly, Ser491 of the α2 subunit can be phosphorylated by PKA,14,15 p70S6K,17 or autophosphorylation18 in tissues such as the adipocytes,15 hypothalamus,17 heart,12 and HEK293 cells,18 resulting in reduced AMPK activity. Although previous studies suggested that Ser491 is also an Akt phosphorylation site,12,13 a recent study by Hawley et al18 showed that Akt does not phosphorylate Ser491 in a cell-free assay. The role and regulation of this site in insulin-responsive tissues and in T2D is not yet understood. Several other phosphorylation sites on the α subunit also exist, though their functional importance is not yet known.
The γ subunit contains four cystathionine-beta-synthase (CBS) domains (each pair is referred to as a Bateman domain) to which adenine nucleotides bind.19 Three of the four CBS domains bind adenine nucleotides; site three primarily has AMP bound, however this can be replaced by ATP under specific conditions.20 The other two binding domains can bind AMP, adenosine diphosphate (ADP), or ATP, depending on their relative concentrations.19 Under normal conditions, ATP is bound to these domains; however, when the AMP:ATP ratio is increased, AMP replaces ATP at the Bateman domains, causing an allosteric change that contributes to AMPK activation.19 This allosteric change makes AMPK a better substrate for its upstream kinases to phosphorylate it at αThr172 and inhibits dephosphorylation of this site by the protein phosphatases, PP2A and PP2C.21,22 The combination of allosteric activation and phosphorylation at αThr172 leads to a greater than 1,000-fold increase in kinase activity in cell-free assays,23 although the changes under physiological conditions are likely much smaller.24 Recently, it has been proposed that ADP, as well as AMP, may be able to activate AMPK by binding to the Bateman domains,25,26 although whether this occurs under normal physiological conditions remains under debate, as AMP is a much more potent allosteric activator.24
Upon activation, AMPK phosphorylates its downstream targets, a main one being acetyl-CoA carboxylase (ACC).27 AMPK phosphorylates ACC at Ser79 (an inhibitory site), preventing the conversion of acetyl-CoA to malonyl CoA, which allows long-chain FAs to enter the mitochondria for oxidation. Other downstream targets of AMPK include TSC2, which inhibits mammalian target of rapamycin complex 1 (mTORC1) and protein synthesis;28 HMG-CoA reductase, which leads to the inhibition of cholesterol synthesis;29 peroxisome proliferator-activated receptor-gamma coactivator (PPARα) 1α, which stimulates mitochondrial biogenesis,30,31 and many others. A more comprehensive list of AMPK’s actions can be found in a recent review by Ruderman et al.3
AMPK activation has effects on a multitude of tissues (Figure 1). In skeletal muscles, its activation stimulates glucose uptake, FA oxidation, glucose transporter type (GLUT)4 translocation, and mitochondrial biogenesis, while inhibiting protein and glycogen synthesis.6 Similarly, in cardiac muscle, AMPK activation stimulates glucose uptake, FA oxidation, and glycolysis.32 AMPK stimulates glucose uptake and FA oxidation in liver, while inhibiting gluconeogenesis, as well as cholesterol, FA, and protein synthesis.3 In adipose tissue, it stimulates FA oxidation and reduces FA synthesis and lypolysis.6 AMPK inhibits insulin secretion from pancreatic β-cells,6 and it signals to increase food intake in the hypothalamus.33 Nearly all of the physiological effects of peripheral AMPK activation would be beneficial for a patient with T2D. For this reason, the pharmacological activation of AMPK has been a seemingly promising target for drug discovery and development during the past 2 decades.
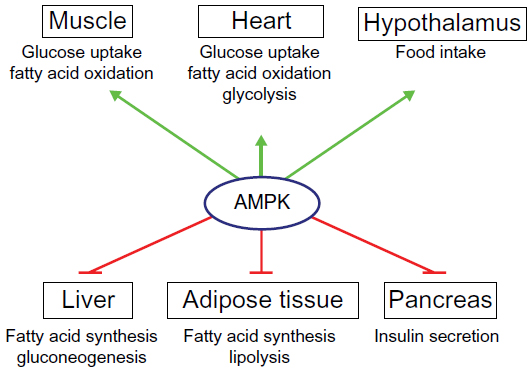 | Figure 1 Roles of AMPK in the control of whole-body energy metabolism. |
AMPK and T2D
The regulation of AMPK is of great interest in the study of T2D and metabolic syndrome due to accumulating evidence suggesting that the dysregulation of AMPK plays an important role in the development of IR and T2D, and that AMPK activation (either physiological or pharmacological) can prevent and/or ameliorate some of the pathologies of IR and T2D.3 Multiple animal models with a metabolic syndrome phenotype have exhibited decreased AMPK activity in muscle,2 and evidence exists that AMPK activity is diminished in the skeletal muscle34 or adipose35 of humans with T2D or obesity.
AMPK activators
Numerous physiological, pharmacological, natural, and hormone activators of AMPK are known. Some of these are currently used clinically for the treatment of T2D. The following is a noncomprehensive list of some of the most established and newly identified AMPK activators (and their mechanisms of action, if known) that may have positive effects in patients with T2D (Figure 2). A more thorough list of AMPK activators can be found in other recent reviews by Steinberg and Kemp6 and Fogarty and Hardie.36
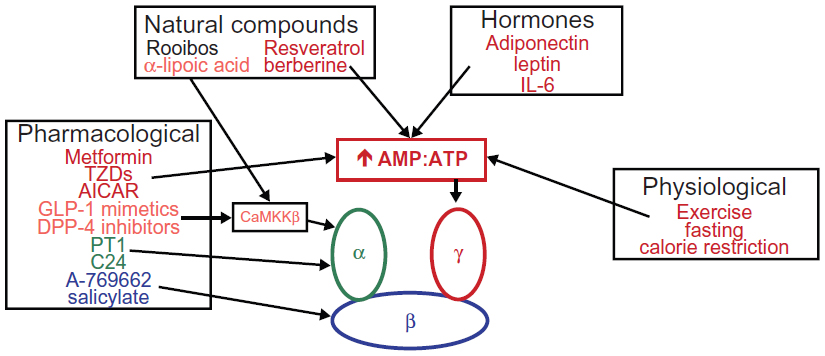 | Figure 2 Physiological, pharmacological, natural, and hormonal activators of AMPK. |
Physiological
Exercise and calorie restriction
Exercise and calorie restriction exert beneficial effects on metabolic health and decrease one’s risk for a variety of diseases, including T2D and cardiovascular disease.37 Both exercise and caloric restrictions are metabolic stresses that increase the AMP:ATP ratio in an organism’s cells, and thus can activate AMPK.37 Studies conducted in the past 2 decades have revealed that AMPK is stimulated by muscle contractions in both rodents38–42 and humans,43–45 and that it is a crucial enzyme through which exercise imparts many of its positive effects. ATP turnover in skeletal muscle is elevated over 100-fold during exercise, causing a rapid rise in AMP and ADP levels in an intensity-dependent manner.46 High-intensity muscle contractions preferentially activate α1-containing heterotrimers; while α2 activity is stimulated by low-intensity exercise and increases progressively with intensity.5
Although AMPK activation has not been proven to be the mechanism by which exercise exerts its positive metabolic effects, several studies have shown that pharmacological AMPK activation mimics the effects of endurance training (for example, through increased FA oxidation or mitochondrial biogenesis) in rodents,37 suggesting that AMPK may mediate the effects of exercise. A recent study showed that AMPK β1/β2 skeletal muscle knockout mice not only have a reduced exercise capacity, but they also have reduced contraction-stimulated glucose uptake and skeletal muscle mitochondrial content.47 Similarly, AMPK α1/α2 skeletal muscle knockout mice have reduced exercise tolerance, maximal force production, and fatigue resistance. However, in contrast to the β1/β2 knockout mice, these mice have reduced their oxidative capacity, but not their mitochondrial number.48 These findings suggest that AMPK is, at least in part, responsible for the exercise-induced stimulation of glucose uptake and mitochondrial biogenesis.
Pharmacological
5-Aminoimidazole-4-carboxamide riboside
5-Aminoimidazole-4-carboxamide riboside (AICAR) was the first compound identified to activate AMPK.49,50 It is structurally similar to adenosine and, upon entering the cells, is phosphorylated by adenosine kinase to become ZMP.49 ZMP is an AMP analog that can bind to the CBS domains on AMPK’s γ-subunit to cause allosteric activation and allow for increased phosphorylation of Thr172.49 AICAR treatment has been shown to prevent and/or reverse some aspects of metabolic syndrome in animal models, such as ob/ob mice,51 fa/fa rats,52,53 and rats fed a high-fat diet.54 For example, AICAR treatment improves glucose tolerance and whole-body glucose disposal, and reduces hepatic glucose output, as well as plasma triglyceride and FFA levels.51–54 AICAR also induces the expression of genes involved in oxidative metabolism and improves running endurance.55 For these reasons, the World Anti-Doping Code banned the use of AICAR by athletes in 2011.56
Despite these promising effects in animal models, AICAR is unlikely to be used in the treatment of human T2D or metabolic syndrome due to poor bioavailability and a short half-life.36 Additionally, AICAR can mimic other actions of AMP to exhibit AMPK-independent effects, such as the inhibition of the enzyme fructose-1,6-bisphosphatase,57 and the stimulation of muscle glycogen phosphorylase.58 However, AICAR may be useful in treating humans with acute lymphoblastic leukemia59,60 and cardiac ischemic injury.61,62 Interestingly, another recently described AMPK activator, compound 13, is taken up into the cells and converted to the AMP analog, compound 2, which is a much more potent and specific activator of AMPK when compared to ZMP.63 Compounds of this series are being optimized for oral bioavailability and pharmacokinetics; however, their clinical utility remains to be seen.63
Biguanides
Metformin, which belongs to the biguanide family of insulin-sensitizing drugs, is currently the first-line oral therapy for T2D according to national and international guidelines.64–67 The biguanides also include phenformin, buformin, and the antimalarial agent, proguanil. This class of drugs originates from the French lilac plant, which has been used in folk medicine to treat diabetes for centuries due to its glucose-lowering properties.68,69 Although phenformin and buformin are more potent insulin sensitizers than metformin, they also have a higher risk for unwanted side effects – namely, lactic acidosis.64 For this reason, they were withdrawn from the market, leaving metformin as the only biguanide available for the treatment of T2D.64 Within 12 weeks of receiving United States Food and Drug Administration approval in 1994, metformin became the most frequently prescribed oral antidiabetic drug in the US,70 and it is currently prescribed to over 100 million patients worldwide.64 Metformin reduces hemoglobin HbA1c by 1%–2% in patients with T2D, and it reduces mortality when compared to diet modifications alone.71 In addition to its hypoglycemic effects, metformin has very few side effects, is weight neutral, and recent epidemiological studies suggest that patients taking metformin may have lower risks of cardiovascular disease72 and certain types of cancer.73,74
Since metformin was discovered before the use of targeted drug discovery techniques, its mechanism of action was not known for a long time, and it is still not fully understood.64 Zhou et al reported that metformin activates AMPK, and its insulin-sensitizing actions have been attributed to AMPK.75 Metformin does not activate AMPK directly; instead, it has been shown to inhibit complex 1 of the mitochondrial respiratory chain,77 which promotes a switch from aerobic to anaerobic glycolysis, thus increasing the AMP:ATP ratio and promoting AMPK activation. This indirect mechanism is supported by the fact that metformin fails to activate AMPK in cell-free assays78 and in cells expressing AMP-insensitive (R531G) gamma 2 variants.79 Inhibition of hepatic gluconeogenesis is thought to be the primary means by which metformin exerts its effects on glucose homeostasis.77 It can also stimulate glucose uptake in adipose and skeletal muscle, although the times and concentrations needed to stimulate AMPK and glucose uptake were much larger than would be found in vivo.76,80 A recent study using liver-specific AMPK α1/α2 or liver kinase B1 knockout mice brought into question the dependence of metformin’s effects on AMPK, since metformin treatment lowered blood glucose levels in both of these mouse models.81 An even more recent study reported that metformin exerts its effects on the liver by antagonizing glucagon signaling through cyclic AMP and PKA, independent of AMPK.82 In contrast, mice with mutations in ACC1/2 that prevent phosphorylation and inactivation by AMPK are refractory to the lipid-lowering and insulin-sensitizing effects of metformin when made obese by high-fat feeding, suggesting that the inhibitory phosphorylation of ACC by AMPK is essential for metformin-induced improvements in insulin sensitivity.83 Despite these conflicting results, AMPK is likely an effecter of some of metformin’s insulin-sensitizing effects, though further studies are needed to distinguish the AMPK-dependent from AMPK-independent effects.
Thiazolidinediones
TZDs are a class of insulin-sensitizing drugs and include rosiglitazone, pioglitazone, and troglitazone. Although their primary target is the nuclear hormone receptor peroxisome proliferator-activated receptor-γ, (PPARγ) they are thought to exert some of their antidiabetic effects through AMPK activation.84 TZDs have been shown to rapidly stimulate AMPK and ACC phosphorylation in a variety of tissues, including the skeletal muscle84,85 and liver.86 Like metformin, they do so indirectly by inhibiting complex 1 of the mitochondrial respiratory chain to increase the cellular AMP:ATP ratio.79,84,87 Additionally, TZDs may indirectly activate AMPK through the effects of peroxisome proliferator-activated receptor-γ to stimulate adiponectin secretion (which will be described in more detail).88,89
In patients with T2D, TZDs improve insulin sensitivity in the muscles, liver, and adipose tissues, improve glycemic control (reduce HbA1c), enhance endothelial function, and reduce inflammation.90 However, the main drawbacks of TZDs are that they cause weight gain (particularly subcutaneous adiposity), they may increase one’s risk of bladder cancer,91 and they may worsen congestive heart failure, though they are not associated with increased mortality.90,92
Glucagon-like peptide-1 receptor agonists
Glucagon-like peptide-1 (GLP-1) is an incretin that is secreted from intestinal L-cells following the ingestion of food.93 GLP-1 stimulates insulin secretion in a glucose-dependent manner, decreases pancreatic glucagon secretion, increases β-cell mass and insulin gene expression, stimulates satiety in the brain, and increases peripheral insulin sensitivity.93 Based on these antidiabetic actions, GLP-1 mimetics, such as exenatide and liraglutide, have been developed for the treatment of T2D.94 An alternative strategy that has been undertaken to increase GLP-1 levels is the development of dipeptidyl peptidase-4 inhibitors, which prevent the inactivation of GLP-1.95 Recent studies have shown that these compounds, as well as endogenous GLP-1, can activate the AMPK pathway.96,97 For example, exenatide treatment was shown to increase AMPK phosphorylation at Thr172 in hepatocytes,96 decrease body weight, serum FFA, and triglyceride levels, and reverse the hepatic accumulation of lipids and inflammation in high-fat-fed mice, while increasing AMPK messenger ribonucleic acid (mRNA) and protein expression.97
A-769662
The first compound to be identified as a direct activator of AMPK was A-769662.6 This thienopyridone, identified by Abbott Laboratories (Abbott Park, IL, USA), activates AMPK in a similar manner to AMP; it causes allosteric activation and prevents dephosphorylation of Thr172.98–100 Unlike AMP, however, A-769662 binds in a cleft between the kinase domain of the α-subunit and the carbohydrate-binding domain of the β-subunit.101 It is specific for the β1-isoform and requires βSer108 phosphorylation.100,102 Treatment of ob/ob mice with this compound caused improvements in glucose homeostasis and lipid levels.98 Despite these benefits on the metabolic parameters, A-769662 is unlikely to be used to treat human metabolic syndrome due to its poor oral absorption and its reported AMPK-independent effects, whereby it can inhibit 26S proteasome activity and arrest cell cycle progression.103 However, A-769662 has utility as a research tool to further study the effects of AMPK activation. Recently, another compound referred to as 991, which is a cyclic benzimidazole derivative that binds to the same site as A-769662, was shown to be a much more potent AMPK activator.101 Studies regarding the efficacy of this compound are in their infancy.
Salicylate
Salicylates are natural substances produced by many plants to defend themselves against infections.104 The medicinal use of salicylate was first described thousands of years ago, when it was extracted from willow bark,105 making it one of the oldest medicines used by humans. It is often taken in the form of acetyl salicylate (trade name, aspirin; Bayer AG, Leverkusen, Germany) or the diester salsalate, both of which are rapidly converted to salicylate in vivo.106,107 It was recently reported that salicylate, but not aspirin, activates AMPK in HEK-293 cells at concentrations found in the plasma of patients treated with high doses of aspirin.108 Salicylate was determined to bind to the same site as A-769662 on the β1-subunit based on findings that the ability of both compounds to activate AMPK is greatly diminished in complexes where the β2 subunit, rather than β1 subunit, is expressed, and that the effects of both compounds are nearly abolished by an S108A mutation in β1.108 Further confirmation of a role for salicylate-induced AMPK activation in vivo was found when mice treated with salicylate had lower respiratory exchange ratios following food withdrawal, indicating a switch to fat oxidation.108 However, these effects were not seen in β1 knockout mice.108
These findings suggest that although salicylate is a less potent activator than A-769662, it may have some utility in improving metabolic parameters in patients with T2D. Indeed, two randomized controlled trials showed that oral salsalate treatment decreased plasma glucose levels and insulin C-peptide, and increased plasma adiponectin levels in obese young adults109 and in patients with impaired fasting glucose and/or impaired glucose tolerance.110 Although these findings seem promising for the use of salicylate as an AMPK-mediated antidiabetic treatment, further research is needed to fully define the role of AMPK in these outcomes. High-fat-fed wild type and AMPK β1-knockout mice treated with salicylate for 2 weeks both showed improved glucose tolerance, as well as reduced fasting glucose and insulin levels, suggesting that some of salicylate’s insulin-sensitizing effects are AMPK-independent.108
PT1 and C24
PT1 is another small molecule compound that has recently been identified as a direct activator of AMPK.111 Its mechanism of action is thought to be antagonism of the autoinhibitory (residues 313–335) domain of the α-subunit.111 Treatment with PT1 dose-dependently increased AMPK activity and ACC phosphorylation in L6 myotubes and HepG2 cells, with no significant changes in the AMP:ATP ratios.111 PT1 is not effective in vivo due to a poor pharmacokinetic profile, but its structural optimization led to the discovery of the similar, but orally bioavailable compound, C24.112,113 C24 was shown to reduce glucose production and decrease triglyceride and cholesterol contents in hepatocytes.112 Chronic oral treatment with C24 lowered blood glucose and lipid levels and improved glucose tolerance in db/db mice.112 Whether C24 or a similar compound will make it to the clinic remains to be seen.
Natural compounds
Numerous naturally occurring compounds and phytochemicals have been shown to activate AMPK in vitro and in vivo,36 and they elicit metabolic benefits that are dependent on AMPK activation.
Resveratrol
Resveratrol is a polyphenol found in red wine that has been suggested to mimic some of the effects of calorie restriction to increase one’s lifespan.114 Treatment of high-fat-fed animals with resveratrol causes improvements in insulin sensitivity and decreases markers associated with aging.115 Resveratrol has been shown to stimulate AMPK activity in multiple cell types, including hepatocytes,116–118 muscle cells,119,120 and neurons.121 The mechanism by which resveratrol activates AMPK is thought to be an increase in AMP levels due to inhibition of the mitochondrial F1 ATPase.79,122 Resveratrol treatment stimulates glucose uptake120 and mitochondrial biogenesis119 in muscle cells, and it stimulates mitochondrial biogenesis116 and reduces lipid accumulation in the liver.118 The latter effect is blocked by a dominant negative AMPK, suggesting that it is AMPK-mediated.118 Further studies are required to determine how much of resveratrol’s effects are due to AMPK activation as opposed to the activation of sirtuin 1 (SIRT1), a redox-sensitive deacetylase whose activation has been shown to increase longevity.123 Of note, however, Ruderman et al124 have shown that AMPK and sirtuin 1 can both regulate each other and share many common target molecules.
Rooibos
Rooibos (Aspalathus linearis) is a plant grown in South Africa that is popularly used in tea and has been shown to activate AMPK.125 Treatment of C2C12 myotubes with rooibos extract increases glucose uptake, mitochondrial activity, GLUT4 expression, and ATP production, and it reverses palmitate-induced IR.125 In vivo, the rooibos extract was reported to reduce serum cholesterol, triglyceride, and FFA concentrations in mice fed a high-fat diet.126 Adipocyte size and triglyceride content were also reduced and hepatic steatosis was prevented. These metabolic improvements were attributed to AMPK activation in the liver and adipose.126 Similarly, ob/ob mice fed a diet containing 0.1% rooibos extract had improved fasting blood glucose levels and improved glucose tolerance compared to mice fed a control diet.127 Furthermore, rooibos treatment decreased the expression of gluconeogenic and lipogenic hepatic genes in these animals.127
Berberine
Berberine is an isoquinoline alkaloid found in certain plants, and it has traditionally been used in Chinese and Korean cultures to treat fungal and bacterial infections, as well as T2D. Berberine has been shown to improve glucose tolerance,128 reduce body weight,128 increase the expression of the insulin receptor (IR) and low-density lipoprotein (LDL) receptor,129 lower total and LDL cholesterol levels,129 and reduce triglyceride levels128,129 in several rodent models. Berberine has also been shown to lower blood glucose, triglyceride, and cholesterol levels to nearly the same degree as metformin.130 It has been shown to potently activate AMPK in skeletal muscle,131 hepatocytes,132 and adipose tissue,133 although some of its antidiabetic effects are likely mediated through AMPK-independent mechanisms, such as dipeptidyl peptidase-4 inhibition134 and the enhancement of superoxide dismutase activity.135 Like metformin and TZDs, berberine is thought to activate AMPK by inhibiting complex 1 of the mitochondrial respiratory chain, thus increasing the AMP:ATP ratio.79,136,137 It has also been shown to increase adiponectin expression, which may contribute to both AMPK-dependent and independent effects.138
α-lipoic acid
The short-chain FA α-lipoic acid is an essential cofactor for mitochondrial respiration and a powerful antioxidant and has been shown to activate AMPK in the skeletal muscle,139,140 heart,141 and endothelium.142 It also inhibits AMPK signaling in the hypothalamus,143 thus reducing food intake. It has been shown to improve insulin sensitivity in obese rodents144 and to reduce insulin secretion and β-cell growth.145 Furthermore, ex vivo incubation of rat skeletal muscle with α-lipoic acid prevents high glucose- or leucine-induced impairments in insulin signaling,140 skeletal muscle lipid accumulation, and hepatic steatosis in obesity.144 Shen et al146 showed that the mechanism by which α-lipoic acid activates AMPK is through the CaMKKβ-mediated phosphorylation of Thr172. The authors reported that the selective inhibitor of CaMKKβ STO-609 prevented α-lipoic acid-stimulated AMPK activation and subsequent ACC phosphorylation. In addition, α-lipoic acid has also been reported to have beneficial effects on diabetic neuropathy, although whether AMPK is also involved in mediating these effects is unknown.147,148
Hormones
In addition to the exogenous pharmacological and natural compounds that can activate AMPK, endogenous hormones also exist that can activate AMPK and elicit many of the same antidiabetic effects.
Leptin
Leptin is a hormone made and secreted by adipocytes that acts on the brain to regulate food intake and body weight. It can also act directly and indirectly on peripheral tissues, as almost all tissues express the leptin receptor.149 Leptin increases the AMP:ATP ratio in skeletal muscle, thus activating AMPK and stimulating FA oxidation.150 This activation occurs only in α2-containing heterotrimers,151 although the reason for this isoform specificity is not known. In addition to directly acting on skeletal muscle to activate AMPK, leptin can also indirectly stimulate AMPK in muscles via α-adrenergic signaling from the central nervous system.152 This activation is more delayed and requires the melanocortin 4 receptor, since the intracerebroventricular administration of a melanocortin 4 receptor antagonist prevents central nervous system-mediated activation of skeletal muscle AMPK by leptin.152 In contrast to its effects in skeletal muscle, leptin inhibits AMPK in the hypothalamus to inhibit food intake.33,153 Yang et al154 reported that it does so indirectly via a mechanism involving release of an opioid from a cell that is different from that in which AMPK is located.154 However, it was recently reported that AMPK inhibition via phosphorylation of S491 on its α2-subunit by p70S6 kinase is required to mediate leptin’s anorectic effects.17
Adiponectin
Adiponectin is a protein secreted from adipose tissue, which circulates at high concentrations in the plasma in the form of low and high molecular weight multimers.155 This circulating hormone acts through its two receptors (adipoR1 and adipoR2), which are expressed in tissues such as adipose and skeletal muscle to regulate glucose levels and stimulate FA oxidation.156 Adiponectin levels are reduced in obese humans and animals.157 AMPK activation by adiponectin is dependent on signaling through adipoR1158 and requires the adaptor protein, phosphotyrosine interaction, PH domain and leucine zipper containing 1.159 Purified adiponectin from human plasma potently activates AMPK activity in C2C12 myotubes,160 and adiponectin’s ability to suppress hepatic glucose output has been shown to be AMPK-dependent.161 Adiponectin overexpression has been shown to reduce body weight, improve insulin sensitivity, and increase FA oxidation in various rodent models of genetic and diet-induced obesity.162–165 Interestingly, adiponectin trimers and hexamers, but not high molecular weight forms, stimulate food intake through AMPK activation in the hypothalamus.166
Interleukin-6
Interleukin-6 (IL-6) is a proinflammatory cytokine that is elevated in obesity.167 It is also produced and released from muscle during exercise to increase circulating levels almost 100-fold.168 Interestingly, in obesity, IL-6 is associated with IR, whereas during exercise, it may enhance glucose uptake via AMPK activation. IL-6 increases muscle glucose uptake through an AMPK-dependent mechanism in cultured cells,169 rodents,169,170 and humans.171 These effects of IL-6 are additive to those of insulin in stimulating glucose uptake.172 However, some studies have shown that these effects are only seen at super-physiological concentrations.172 AMPK activity is diminished in the muscle and adipose tissue of IL-6 knockout mice, and exercise-stimulated AMPK activity is diminished in these mice.167,170 Kelly et al169 showed that IL-6 activates AMPK by increasing the concentration of cyclic AMP and, secondarily, the AMP:ATP ratio.
Conclusion
As the T2D epidemic continues to grow, the need for safe and efficacious antidiabetic medications also increases. Current therapies leave much room for improvement, as most patients require multiple medications to get their blood glucose levels under control, and T2D is the seventh leading cause of death in the US.173 AMPK is an attractive target for T2D therapies because of its role as a master metabolic regulator. Since it is activated during calorie restriction and exercise (both of which promote insulin sensitivity), it is thought that pharmacological activation of this target could elicit many of their positive benefits.2 Although several currently used T2D medications, such as metformin and TZDs, indirectly activate AMPK, no direct AMPK activators have made it to the clinic due to poor pharmacokinetic profiles or off-target effects. Furthermore, AMPK is an attractive target because its activity is decreased in tissues such as in the muscle and adipose tissue of obese or insulin-resistant animals and humans. For a more comprehensive review of AMPK inhibition in response to over-nutrition, refer to the recent review by Coughlan et al.4
Despite accumulating evidence, both in vitro and in vivo, that AMPK activation positively affects numerous physiological processes that are dysregulated in metabolic diseases, whether the continued pursuit of direct AMPK activators is a worthwhile or promising strategy for drug development remains to be seen. An important consideration in pursuing AMPK activation as a treatment for metabolic disease is that although most of its effects are beneficial (for example, via the stimulation of glucose uptake and FA oxidation), excess activation can have unwanted consequences. For example, AMPK inhibits protein synthesis, which could be harmful, particularly to elderly patients in whom muscle wasting may be a concern.174 However, calorie restriction has been shown to delay age-related loss of muscle mass and function, and to induce a younger muscle transcription profile.175,176 Perhaps isoform-specific activators will not have the same weaknesses as currently available nonspecific activators, as they may preferentially target particular cell types or tissues. Further insights into the regulation of AMPK’s less-studied post-translational modifications may bring to light new strategies for more controlled pharmacological modulation. In summary, as a cellular energy sensor and master regulator of metabolism whose activity is diminished in states of IR, AMPK seems to be an attractive and promising target for the pharmacological treatment of T2D.
Acknowledgments
The work in the author’s laboratory was supported by US Public Health Service grants RO1DK19514, RO1DK67509, 5T32HL007224 (RJV), and 5T32GM008541 (KAC).
Disclosure
The authors report no conflicts of interest in this work.
References
International Diabetes Federation. Diabetes Atlas. 6th ed. Brussels, Belgium: International Diabetes Federation; 2013. | |
Ruderman N, Prentki M. AMP kinase and malonyl-CoA: targets for therapy of the metabolic syndrome. Nat Rev Drug Discov. 2004;3(4):340–351. | |
Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764–2772. | |
Coughlan KA, Valentine RJ, Ruderman NB, Saha AK. Nutrient excess in AMPK downregulation and insulin resistance. Journal of Endocrinology, Diabetes and Obesity. 2013;1(1):1008. | |
O’Neill HM. AMPK and exercise: glucose uptake and insulin sensitivity. Diabetes Metab J. 2013;37(1):1–21. | |
Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev. 2009;89(3):1025–1078. | |
Birnbaum MJ. Activating AMP-activated protein kinase without AMP. Mol Cell. 2005;19(3):289–290. | |
Lizcano JM, Göransson O, Toth R, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23(4):833–843. | |
Woods A, Johnstone SR, Dickerson K, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13(22):2004–2008. | |
Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19. | |
Momcilovic M, Hong SP, Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281(35):25336–25343. | |
Horman S, Vertommen D, Heath R, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281(9):5335–5340. | |
Ning J, Xi G, Clemmons DR. Suppression of AMPK activation via S485 phosphorylation by IGF-I during hyperglycemia is mediated by AKT activation in vascular smooth muscle cells. Endocrinology. 2011; 152(8):3143–3154. | |
Hurley RL, Barré LK, Wood SD, et al. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem. 2006;281(48):36662–36672. | |
Pulinilkunnil T, He H, Kong D, et al. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J Biol Chem. 2011;286(11):8798–8809. | |
Berggreen C, Gormand A, Omar B, Degerman E, Göransson O. Protein kinase B activity is required for the effects of insulin on lipid metabolism in adipocytes. Am J Physiol Endocrinol Metab. 2009;296(4):E635–E646. | |
Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab. 2012;16(1):104–112. | |
Hawley SA, Ross FA, Gowans GJ, Tibarewal P, Leslie NR, Hardie DG. Phosphorylation by Akt within the ST loop of AMPK-α1 down- regulates its activation in tumour cells. Biochem J. 2014;459(2):275–287. | |
Xiao B, Heath R, Saiu P, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449(7161):496–500. | |
Chen L, Wang J, Zhang YY, et al. AMP-activated protein kinase undergoes nucleotide-dependent conformational changes. Nat Struct Mol Biol. 2012;19(7):716–718. | |
Hawley SA, Selbert MA, Goldstein EG, Edelman AM, Carling D, Hardie DG. 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem. 1995;270(45):27186–27191. | |
Davies SP, Helps NR, Cohen PT, Hardie DG. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377(3):421–425. | |
Suter M, Riek U, Tuerk R, Schlattner U, Wallimann T, Neumann D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J Biol Chem. 2006;281(43):32207–32216. | |
Gowans GJ, Hawley SA, Ross FA, Hardie DG. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18(4):556–566. | |
Xiao B, Sanders MJ, Underwood E, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472(7342):230–233. | |
Oakhill JS, Steel R, Chen ZP, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332(6036):1433–1435. | |
Munday MR, Campbell DG, Carling D, Hardie DG. Identification by amino acid sequencing of three major regulatory phosphorylation sites on rat acetyl-CoA carboxylase. Eur J Biochem. 1988;175(2):331–338. | |
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. | |
Clarke PR, Hardie DG. Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J. 1990;9(8):2439–2446. | |
Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(29):12017–12022. | |
Zong H, Ren JM, Young LH, et al. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99(25):15983–15987. | |
Srivastava RA, Pinkosky SL, Filippov S, Hanselman JC, Cramer CT, Newton RS. AMP-activated protein kinase: an emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases. J Lipid Res. 2012;53(12):2490–2514. | |
Minokoshi Y, Alquier T, Furukawa N, et al. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428(6982):569–574. | |
Bandyopadhyay GK, Yu JG, Ofrecio J, Olefsky JM. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55(8):2277–2285. | |
Xu XJ, Gauthier MS, Hess DT, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res. 2012;53(4):792–801. | |
Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804(3):581–591. | |
Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418(2):261–275. | |
Winder WW, Hardie DG. Inactivation of acetyl-CoA carboxylase and activation of AMP-activated protein kinase in muscle during exercise. Am J Physiol. 1996;270(2 Pt 1):E299–E304. | |
Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47(8):1369–1373. | |
Hutber CA, Hardie DG, Winder WW. Electrical stimulation inactivates muscle acetyl-CoA carboxylase and increases AMP-activated protein kinase. Am J Physiol. 1997;272(2 Pt 1):E262–E266. | |
Vavvas D, Apazidis A, Saha AK, et al. Contraction-induced changes in acetyl-CoA carboxylase and 5’-AMP-activated kinase in skeletal muscle. J Biol Chem. 1997;272(20):13255–13261. | |
Park H, Kaushik VK, Constant S, et al. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002;277(36):32571–32577. | |
Wojtaszewski JF, Nielsen P, Hansen BF, Richter EA, Kiens B. Isoform-specific and exercise intensity-dependent activation of 5′-AMP-activated protein kinase in human skeletal muscle. J Physiol. 2000;528 Pt 1:221–226. | |
Chen ZP, Stephens TJ, Murthy S, et al. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. 2003;52(9):2205–2212. | |
Fujii N, Hayashi T, Hirshman MF, et al. Exercise induces isoform- specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochem Biophys Res Commun. 2000;273(3):1150–1155. | |
Sahlin K, Tonkonogi M, Söderlund K. Energy supply and muscle fatigue in humans. Acta Physiol Scand. 1998;162(3):261–266. | |
O’Neill HM, Maarbjerg SJ, Crane JD, et al. AMP-activated protein kinase (AMPK) beta1beta2 muscle null mice reveal an essential role for AMPK in maintaining mitochondrial content and glucose uptake during exercise. Proc Natl Acad Sci U S A. 2011;108(38):16092–16097. | |
Lantier L, Fentz J, Mounier R, et al. AMPK controls exercise endurance, mitochondrial oxidative capacity, and skeletal muscle integrity. FASEB J. Epub March 20, 2014. | |
Corton JM, Gillespie JG, Hawley SA, Hardie DG. 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem. 1995;229(2):558–565. | |
Sullivan JE, Brocklehurst KJ, Marley AE, Carey F, Carling D, Beri RK. Inhibition of lipolysis and lipogenesis in isolated rat adipocytes with AICAR, a cell-permeable activator of AMP-activated protein kinase. FEBS Lett. 1994;353(1):33–36. | |
Song XM, Fiedler M, Galuska D, et al. 5-Aminoimidazole-4- carboxamide ribonucleoside treatment improves glucose homeostasis in insulin-resistant diabetic (ob/ob) mice. Diabetologia. 2002;45(1):56–65. | |
Bergeron R, Previs SF, Cline GW, et al. Effect of 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside infusion on in vivo glucose and lipid metabolism in lean and obese Zucker rats. Diabetes. 2001;50(5):1076–1082. | |
Buhl ES, Jessen N, Schmitz O, et al. Chronic treatment with 5-aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside increases insulin-stimulated glucose uptake and GLUT4 translocation in rat skeletal muscles in a fiber type-specific manner. Diabetes. 2001;50(1):12–17. | |
Iglesias MA, Ye JM, Frangioudakis G, et al. AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes. 2002;51(10):2886–2894. | |
Narkar VA, Downes M, Yu RT, et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134(3):405–415. | |
Thomas A, Beuck S, Eickhoff JC, et al. Quantification of urinary AICAR concentrations as a matter of doping controls. Anal Bioanal Chem. 2010;396(8):2899–2908. | |
Vincent MF, Marangos PJ, Gruber HE, Van den Berghe G. Inhibition by AICA riboside of gluconeogenesis in isolated rat hepatocytes. Diabetes. 1991;40(10):1259–1266. | |
Longnus SL, Wambolt RB, Parsons HL, Brownsey RW, Allard MF. 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am J Physiol Regul Integr Comp Physiol. 2003;284(4):R936–R944. | |
Sengupta TK, Leclerc GM, Hsieh-Kinser TT, Leclerc GJ, Singh I, Barredo JC. Cytotoxic effect of 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on childhood acute lymphoblastic leukemia (ALL) cells: implication for targeted therapy. Mol Cancer. 2007;6:46. | |
Leclerc GM, Leclerc GJ, Fu G, Barredo JC. AMPK-induced activation of Akt by AICAR is mediated by IGF-1R dependent and independent mechanisms in acute lymphoblastic leukemia. J Mol Signal. 2010;5:15. | |
Mullane K. Acadesine: the prototype adenosine regulating agent for reducing myocardial ischaemic injury. Cardiovasc Res. 1993;27(1):43–47. | |
Mangano DT. Effects of acadesine on myocardial infarction, stroke, and death following surgery. A meta-analysis of the 5 international randomized trials. The Multicenter Study of Perioperative Ischemia (McSPI) Research Group. JAMA. 1997;277(4):325–332. | |
Gomez-Galeno JE, Dang Q, Nguyen TH, et al. A potent and selective AMPK activator that inhibits de novo lipogenesis. ACS Med Chem Lett. 2010;1:478–482. | |
Rena G, Pearson ER, Sakamoto K. Molecular mechanism of action of metformin: old or new insights? Diabetologia. 2013;56(9):1898–1906. | |
Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for the Study of Diabetes. Medical management of hyperglycaemia in type 2 diabetes mellitus: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2009;52(1):17–30. | |
Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycaemia in type 2 diabetes: a patient-centered approach. Position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2012;55(6):1577–1596. | |
Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract. 2009;15(6):540–559. | |
Witters LA. The blooming of the French lilac. J Clin Invest. 2001;108(8):1105–1107. | |
Bailey CJ, Day C. Metformin: its botanical background. Practical Diabetes International. 2004;21(3):115–117. | |
Mehnert H. Metformin, the rebirth of a biguanide: mechanism of action and place in the prevention and treatment of insulin resistance. Exp Clin Endocrinol Diabetes. 2001;109 Suppl 2:S259–S264. | |
Srinivasan B, Taub N, Khunti K, Davies M. Diabetes: glycaemic control in type 2. Clin Evid (Online). 2008;2008. pii:0609. | |
Haffner S, Temprosa M, Crandall J, et al; Diabetes Prevention Program Research Group. Intensive lifestyle intervention or metformin on inflammation and coagulation in participants with impaired glucose tolerance. Diabetes. 2005;54(5):1566–1572. | |
Buzzai M, Jones RG, Amaravadi RK, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67(14):6745–6752. | |
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330(7503):1304–1305. | |
Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. | |
Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310(5754):1642–1646. | |
Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348 Pt 3:607–614. | |
Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51(8):2420–2425. | |
Hawley SA, Ross FA, Chevtzoff C, et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010;11(6):554–565. | |
Turban S, Stretton C, Drouin O, et al. Defining the contribution of AMP-activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J Biol Chem. 2012;287(24):20088–20099. | |
Foretz M, Hébrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–2369. | |
Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494(7436):256–260. | |
Fullerton MD, Galic S, Marcinko K, et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat Med. 2013;19(12):1649–1654. | |
Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277(28):25226–25232. | |
LeBrasseur NK, Kelly M, Tsao TS, et al. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am J Physiol Endocrinol Metab. 2006;291(1):E175–E181. | |
Saha AK, Avilucea PR, Ye JM, Assifi MM, Kraegen EW, Ruderman NB. Pioglitazone treatment activates AMP-activated protein kinase in rat liver and adipose tissue in vivo. Biochem Biophys Res Commun. 2004;314(2):580–585. | |
Brunmair B, Staniek K, Gras F, et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: a common mechanism contributing to their antidiabetic actions? Diabetes. 2004;53(4):1052–1059. | |
Suzuki H, Eguchi S. Adiponectin versus angiotensin II: Key pathological role of their misbalance. Kidney Int. 2006;70(10):1678–1679. | |
Kubota N, Terauchi Y, Kubota T, et al. Pioglitazone ameliorates insulin resistance and diabetes by both adiponectin-dependent and -independent pathways. J Biol Chem. 2006;281(13):8748–8755. | |
Yau H, Rivera K, Lomonaco R, Cusi K. The future of thiazolidinedione therapy in the management of type 2 diabetes mellitus. Curr Diab Rep. 2013;13(3):329–341. | |
He S, Tang YH, Zhao G, Yang X, Wang D, Zhang Y. Pioglitazone prescription increases risk of bladder cancer in patients with type 2 diabetes: an updated meta-analysis. Tumour Biol. 2014;35(3):2095–2102. | |
Home PD, Pocock SJ, Beck-Nielsen H, et al; RECORD Study Group. Rosiglitazone evaluated for cardiovascular outcomes – an interim analysis. N Engl J Med. 2007;357(1):28–38. | |
Hwang JI, Yun S, Moon MJ, Park CR, Seong JY. Evolution of GLP1 and GLP1 receptor. J Mol Endocrinol. Epub March 5, 2014. | |
Baggio LL, Drucker DJ. Harnessing the therapeutic potential of glucagon-like peptide-1: a critical review. Treat Endocrinol. 2002;1:117–125. | |
Deacon CF, Holst JJ. Dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes: comparison, efficacy and safety. Expert Opin Pharmacother. 2013;14(15):2047–2058. | |
Svegliati-Baroni G, Saccomanno S, Rychlicki C, et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011;31(9):1285–1297. | |
Lee J, Hong SW, Chae SW, et al. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PLoS One. 2012;7(2):e31394. | |
Cool B, Zinker B, Chiou W, et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3(6):403–416. | |
Göransson O, McBride A, Hawley SA, et al. Mechanism of action of A-769662, a valuable tool for activation of AMP-activated protein kinase. J Biol Chem. 2007;282(45):32549–32560. | |
Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D. Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem. 2007;282(45):32539–32548. | |
Xiao B, Sanders MJ, Carmena D, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun. 2013;4:3017. | |
Scott JW, van Denderen BJ, Jorgensen SB, et al. Thienopyridone drugs are selective activators of AMP-activated protein kinase beta1-containing complexes. Chem Biol. 2008;15(11):1220–1230. | |
Moreno D, Knecht E, Viollet B, Sanz P. A769662, a novel activator of AMP-activated protein kinase, inhibits non-proteolytic components of the 26S proteasome by an AMPK-independent mechanism. FEBS Lett. 2008;582(17):2650–2654. | |
Reymond P, Farmer EE. Jasmonate and salicylate as global signals for defense gene expression. Curr Opin Plant Biol. 1998;1(5):404–411. | |
Jeffreys D. Aspirin: The Remarkable Story of a Wonder Drug. London, UK: Bloomsbury Publishing; 2004. | |
Miners JO, Grgurinovich N, Whitehead AG, Robson RA, Birkett DJ. Influence of gender and oral contraceptive steroids on the metabolism of salicylic acid and acetylsalicylic acid. Br J Clin Pharmacol. 1986;22(2):135–142. | |
Higgs GA, Salmon JA, Henderson B, Vane JR. Pharmacokinetics of aspirin and salicylate in relation to inhibition of arachidonate cyclooxygenase and antiinflammatory activity. Proc Natl Acad Sci U S A. 1987;84(5):1417–1420. | |
Hawley SA, Fullerton MD, Ross FA, et al. The ancient drug salicylate directly activates AMP-activated protein kinase. Science. 2012;336(6083):918–922. | |
Fleischman A, Shoelson SE, Bernier R, Goldfine AB. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care. 2008;31(2):289–294. | |
Goldfine AB, Conlin PR, Halperin F, et al. A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia. 2013;56(4):714–723. | |
Pang T, Zhang ZS, Gu M, et al. Small molecule antagonizes autoinhibition and activates AMP-activated protein kinase in cells. J Biol Chem. 2008;283(23):16051–16060. | |
Li YY, Yu LF, Zhang LN, et al. Novel small-molecule AMPK activator orally exerts beneficial effects on diabetic db/db mice. Toxicol Appl Pharmacol. 2013;273(2):325–334. | |
Yu LF, Li YY, Su MB, et al. Development of novel alkene oxindole derivatives as orally efficacious AMP-activated protein kinase activators. ACS Med Chem Lett. 2013;4(5):475–480. | |
Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5(6):493–506. | |
Howitz KT, Bitterman KJ, Cohen HY, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425(6954):191–196. | |
Um JH, Park SJ, Kang H, et al. AMP-activated protein kinase-deficient mice are resistant to the metabolic effects of resveratrol. Diabetes. 2010;59(3):554–563. | |
Hou X, Xu S, Maitland-Toolan KA, et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J Biol Chem. 2008;283(29):20015–20026. | |
Zang M, Xu S, Maitland-Toolan KA, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55(8):2180–2191. | |
Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444(7117):337–342. | |
Park CE, Kim MJ, Lee JH, et al. Resveratrol stimulates glucose transport in C2C12 myotubes by activating AMP-activated protein kinase. Exp Mol Med. 2007;39(2):222–229. | |
Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104(17):7217–7222. | |
Gledhill JR, Montgomery MG, Leslie AG, Walker JE. Mechanism of inhibition of bovine F1-ATPase by resveratrol and related polyphenols. Proc Natl Acad Sci U S A. 2007;104(34):13632–13637. | |
Cohen HY, Miller C, Bitterman KJ, et al. Calorie Restriction Promotes Mammalian Cell Survival by Inducing the SIRT1 Deacetylase. Science. 2004;305(5682):390–392. | |
Ruderman NB, Xu XJ, Nelson L, et al. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298(4):E751–E760. | |
Mazibuko SE, Muller CJ, Joubert E, et al. Amelioration of palmitate-induced insulin resistance in C2C12 muscle cells by rooibos (Aspalathus linearis). Phytomedicine. 2013;20(10):813–819. | |
Beltrán-Debón R, Rull A, Rodríguez-Sanabria F, et al. Continuous administration of polyphenols from aqueous rooibos (Aspalathus linearis) extract ameliorates dietary-induced metabolic disturbances in hyperlipidemic mice. Phytomedicine. 2011;18(5):414–424. | |
Son MJ, Minakawa M, Miura Y, Yagasaki K. Aspalathin improves hyperglycemia and glucose intolerance in obese diabetic ob/ob mice. Eur J Nutr. 2013;52(6):1607–1619. | |
Lee YS, Kim WS, Kim KH, et al. Berberine, a natural plant product, activates AMP-activated protein kinase with beneficial metabolic effects in diabetic and insulin-resistant states. Diabetes. 2006;55(8):2256–2264. | |
Kong W, Wei J, Abidi P, et al. Berberine is a novel cholesterol-lowering drug working through a unique mechanism distinct from statins. Nat Med. 2004;10(12):1344–1351. | |
Yin J, Xing H, Ye J. Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism. 2008;57(5):712–717. | |
Cheng Z, Pang T, Gu M, et al. Berberine-stimulated glucose uptake in L6 myotubes involves both AMPK and p38 MAPK. Biochim Biophys Acta. 2006;1760(11):1682–1689. | |
Brusq JM, Ancellin N, Grondin P, et al. Inhibition of lipid synthesis through activation of AMP kinase: an additional mechanism for the hypolipidemic effects of berberine. J Lipid Res. 2006;47(6):1281–1288. | |
Kim SH, Shin EJ, Kim ED, Bayaraa T, Frost SC, Hyun CK. Berberine activates GLUT1-mediated glucose uptake in 3T3-L1 adipocytes. Biol Pharm Bull. 2007;30(11):2120–2125. | |
Al-masri IM, Mohammad MK, Tahaa MO. Inhibition of dipeptidyl peptidase IV (DPP IV) is one of the mechanisms explaining the hypoglycemic effect of berberine. J Enzyme Inhib Med Chem. 2009;24(5):1061–1066. | |
Sarna LK, Wu N, Hwang SY, Siow YL, Karmin O Berberine inhibits NADPH oxidase mediated superoxide anion production in macrophages. Can J Physiol Pharmacol. 2010;88(3):369–378. | |
Turner N, Li JY, Gosby A, et al. Berberine and its more biologically available derivative, dihydroberberine, inhibit mitochondrial respiratory complex I: a mechanism for the action of berberine to activate AMP-activated protein kinase and improve insulin action. Diabetes. 2008;57(5):1414–1418. | |
Yin J, Gao Z, Liu D, Liu Z, Ye J. Berberine improves glucose metabolism through induction of glycolysis. Am J Physiol Endocrinol Metab. 2008;294(1):E148–E156. | |
Choi BH, Kim YH, Ahn IS, Ha JH, Byun JM, Do MS. The inhibition of inflammatory molecule expression on 3T3-L1 adipocytes by berberine is not mediated by leptin signaling. Nutr Res Pract. 2009;3(2):84–88. | |
Lee WJ, Song KH, Koh EH, et al. Alpha-lipoic acid increases insulin sensitivity by activating AMPK in skeletal muscle. Biochem Biophys Res Commun. 2005;332(3):885–891. | |
Saha AK, Xu XJ, Lawson E, et al. Downregulation of AMPK accompanies leucine- and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes. 2010;59(10):2426–2434. | |
Lee Y, Naseem RH, Park BH, et al. Alpha-lipoic acid prevents lipotoxic cardiomyopathy in acyl CoA-synthase transgenic mice. Biochem Biophys Res Commun. 2006;344(1):446–452. | |
Lee WJ, Lee IK, Kim HS, et al. Alpha-lipoic acid prevents endothelial dysfunction in obese rats via activation of AMP-activated protein kinase. Arterioscler Thromb Vasc Biol. 2005;25(12):2488–2494. | |
Kim MS, Park JY, Namkoong C, et al. Anti-obesity effects of alpha-lipoic acid mediated by suppression of hypothalamic AMP-activated protein kinase. Nat Med. 2004;10(7):727–733. | |
Park KG, Min AK, Koh EH, et al. Alpha-lipoic acid decreases hepatic lipogenesis through adenosine monophosphate-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. Hepatology. 2008;48(5):1477–1486. | |
Targonsky ED, Dai F, Koshkin V, et al. alpha-lipoic acid regulates AMP-activated protein kinase and inhibits insulin secretion from beta cells. Diabetologia. 2006;49(7):1587–1598. | |
Shen QW, Zhu MJ, Tong J, Ren J, Du M. Ca2+/calmodulin-dependent protein kinase kinase is involved in AMP-activated protein kinase activation by alpha-lipoic acid in C2C12 myotubes. Am J Physiol Cell Physiol. 2007;293(4):C1395–C1403. | |
Ziegler D, Gries FA. Alpha-lipoic acid in the treatment of diabetic peripheral and cardiac autonomic neuropathy. Diabetes. 1997; 46 Suppl 2:S62–S66. | |
van Dam PS. Oxidative stress and diabetic neuropathy: pathophysiological mechanisms and treatment perspectives. Diabetes Metab Res Rev. 2002;18(3):176–184. | |
Tartaglia LA, Dembski M, Weng X, et al. Identification and expression cloning of a leptin receptor, OB-R. Cell. 1995;83(7):1263–1271. | |
Minokoshi Y, Kim YB, Peroni OD, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415(6869):339–343. | |
Suzuki A, Okamoto S, Lee S, Saito K, Shiuchi T, Minokoshi Y. Leptin stimulates fatty acid oxidation and peroxisome proliferator-activated receptor alpha gene expression in mouse C2C12 myoblasts by changing the subcellular localization of the alpha2 form of AMP-activated protein kinase. Mol Cell Biol. 2007;27(12):4317–4327. | |
Tanaka T, Masuzaki H, Yasue S, et al. Central melanocortin signaling restores skeletal muscle AMP-activated protein kinase phosphorylation in mice fed a high-fat diet. Cell Metab. 2007;5(5):395–402. | |
Andersson U, Filipsson K, Abbott CR, et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004;279(13):12005–12008. | |
Yang Y, Atasoy D, Su HH, Sternson SM. Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell. 2011;146(6):992–1003. | |
Richards AA, Stephens T, Charlton HK, et al. Adiponectin multimerization is dependent on conserved lysines in the collagenous domain: evidence for regulation of multimerization by alterations in posttranslational modifications. Mol Endocrinol. 2006;20(7):1673–1687. | |
Lim S, Quon MJ, Koh KK. Modulation of adiponectin as a potential therapeutic strategy. Atherosclerosis. 2014;233(2):721–728. | |
Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J Biol Chem. 1996;271(18):10697–10703. | |
Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13(3):332–339. | |
Mao X, Kikani CK, Riojas RA, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol. 2006;8(5):516–523. | |
Hada Y, Yamauchi T, Waki H, et al. Selective purification and characterization of adiponectin multimer species from human plasma. Biochem Biophys Res Commun. 2007;356(2):487–493. | |
Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest. 2001;108(12):1875–1881. | |
Fruebis J, Tsao TS, Javorschi S, et al. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A. 2001;98(4):2005–2010. | |
Masaki T, Chiba S, Yasuda T, et al. Peripheral, but not central, administration of adiponectin reduces visceral adiposity and upregulates the expression of uncoupling protein in agouti yellow (Ay/a) obese mice. Diabetes. 2003;52(9):2266–2273. | |
Ouchi N, Kihara S, Arita Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation. 2001;103(8):1057–1063. | |
Yamauchi T, Kamon J, Waki H, et al. Globular adiponectin protected ob/ob mice from diabetes and ApoE-deficient mice from atherosclerosis. J Biol Chem. 2003;278(4):2461–2468. | |
Kubota N, Yano W, Kubota T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007;6(1):55–68. | |
Ruderman NB, Keller C, Richard AM, et al. Interleukin-6 regulation of AMP-activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes. 2006;55 Suppl 2:S48–S54. | |
Pedersen BK, Febbraio MA. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev. 2008;88(4):1379–1406. | |
Kelly M, Gauthier MS, Saha AK, Ruderman NB. Activation of AMP-activated protein kinase by interleukin-6 in rat skeletal muscle: association with changes in cAMP, energy state, and endogenous fuel mobilization. Diabetes. 2009;58(9):1953–1960. | |
Kelly M, Keller C, Avilucea PR, et al. AMPK activity is diminished in tissues of IL-6 knockout mice: the effect of exercise. Biochem Biophys Res Commun. 2004;320(2):449–454. | |
Carey AL, Steinberg GR, Macaulay SL, et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes. 2006;55(10):2688–2697. | |
Geiger PC, Hancock C, Wright DC, Han DH, Holloszy JO. IL-6 increases muscle insulin sensitivity only at superphysiological levels. Am J Physiol Endocrinol Metab. 2007;292(6):E1842–E1846. | |
Centers for Disease Control and Prevention. National Diabetes Fact Sheet: National Estimates and General Information on Diabetes and Prediabetes in the United States, 2011. Atlanta, GA: US Department of Health and Human Services, Centers for Disease Control and Prevention; 2011. | |
Drummond MJ, Dreyer HC, Fry CS, Glynn EL, Rasmussen BB. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J Appl Physiol. 2009;106(4):1374–1384. | |
McKiernan SH, Colman RJ, Lopez M, et al. Caloric restriction delays aging-induced cellular phenotypes in rhesus monkey skeletal muscle. Exp Gerontol. 2011;46(1):23–29. | |
Mercken EM, Crosby SD, Lamming DW, et al. Calorie restriction in humans inhibits the PI3K/AKT pathway and induces a younger transcription profile. Aging Cell. 2013;12(4):645–651. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.