")
Back to Journals » International Journal of Nanomedicine » Volume 11
Aminoclay–lipid hybrid composite as a novel drug carrier of fenofibrate for the enhancement of drug release and oral absorption
Received 23 November 2015
Accepted for publication 29 December 2015
Published 15 March 2016 Volume 2016:11 Pages 1067—1076
DOI https://doi.org/10.2147/IJN.S101116
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Liang Yang, Yating Shao, Hyo-Kyung Han
BK Plus Project Team, College of Pharmacy, Dongguk University, Goyang, South Korea
Abstract: This study aimed to prepare the aminoclay–lipid hybrid composite to enhance the drug release and improve the oral bioavailability of poorly water-soluble fenofibrate. Antisolvent precipitation coupled with an immediate freeze-drying method was adopted to incorporate fenofibrate into aminoclay–lipid hybrid composite (ALC). The optimal composition of the ALC formulation was determined as the ratios of aminoclay to krill oil of 3:1 (w/w), krill oil to fenofibrate of 2:1 (w/w), and antisolvent to solvent of 6:4 (v/v). The morphological characteristics of ALC formulation were determined using scanning electron microscopy, differential scanning calorimetry, and X-ray powder diffraction, which indicated microcrystalline state of fenofibrate in ALC formulation. The ALC formulation achieved almost complete dissolution within 30 minutes, whereas the untreated powder and physical mixture exhibited less than 15% drug release. Furthermore, ALC formulation effectively increased the peak plasma concentration (Cmax) and area under the curve (AUC) of fenofibric acid (an active metabolite) in rats by approximately 13- and seven-fold, respectively. Furthermore, ALC formulation exhibited much lower moisture sorption behavior than the lyophilized formulation using sucrose as a cryoprotectant. Taken together, the present findings suggest that ALC formulation is promising for improving the oral absorption of poorly soluble fenofibrate.
Keywords: aminoclay, omega-3 phospholipids, fenofibrate, drug release, oral absorption
Introduction
Pharmaceutical research and formulation development often struggle with issues related to the low aqueous solubility limiting the dissolution rate, resulting in poor oral bioavailability of new drug candidates.1 Therefore, there is need for continuous development of more efficient delivery systems to improve the dissolution and bioavailability of poorly soluble drugs. Among them, micro- or nanodrug crystals,2–6 lipid-based formulations,7–11 and aminoclay12,13 have demonstrated their potential for the effective formulation of poorly water-soluble drugs. Particularly, size reduction to the submicron range is often adopted to improve the dissolution rate by either a “top–down” approach from the large coarse drug particles (milling and high-pressure homogenization) or a “bottom–up” method from drug solution (liquid antisolvent precipitation, supercritical fluid technology, etc).14,15 For example, the dissolution of fenofibrate, a poorly water-soluble drug has been significantly enhanced by particle size reduction. Although an early formulation, Lipanthyl® (Fournier, Dijon, France) capsules, exhibited high variability in the plasma profiles and a pronounced food effect, Lipidil–Ter® (Abbott Arzneimittel, Hannover, Germany) tablets having micronized fenofibrate in a particle size of 5–15 μm leads to a higher dissolution rate.16 Consequently, Lipidil–Ter® tablets containing 160 mg of fenofibrate was bioequivalent to the 200 mg coarse powder capsule, exhibiting less pharmacokinetic variability as well as reduced food effect.16 In other formulations (Tricor® [Abbott, Lake Bluff, IL, USA], Lipidil 145 ONE® [Abbott Arzneimittel]), the particle size of fenofibrate was further reduced to less than 500 nm using a novel wet-milling technique.17 These formulations exhibited better bioavailability, and thus the formulation containing 145 mg of fenofibrate was bioequivalent to the 200 mg conventional capsule. Therefore, micro- or nanosized formulation can be an effective approach to enhance the dissolution of poorly soluble drugs. However, micro- or nanoparticles in the suspension face issues of crystal growth and agglomeration.18,19 Thus, they are often processed into solid state by spray drying, freeze-drying, palletization, or granulation.20–22
Meanwhile, several previous studies have indicated that 3-aminopropyl functionalized magnesium phyllosilicate (aminoclay) was very useful at improving the bioavailability of poorly water-soluble drugs in rats.12,13,23 When dispersed in water, aminoclay is delaminated to cationic and water-soluble nanosheets (about 30–150 nm in size and about 2–4 nm in thickness). Therefore, in water, it can interact with negatively charged drug molecules to produce the drug–clay complex and modulate in vitro drug release characteristics. To expand their application to drug molecules other than negatively charged drugs, this study developed the micro- or nanocrystals stabilized by a novel aminoclay–lipid hybrid composite (ALC) system. In this formulation, krill oil was used as the lipid matrix, which is rich in phospholipids decorated with omega-3 fatty acids (docosahexaenoic acid [DHA] and eicosapentanoic acid [EPA]). In krill oil, phospholipid has a role as a biocompatible and water dispersible surfactant, while EPA and DHA may have potential cardiovascular health benefits.24–26 More importantly, as krill oil has a negative charge, the oil-coated drug crystals may be further stabilized by cationic aminoclay nanosheets. Furthermore, micro- or nanocrystals stabilized by a novel ALC system may offer the advantages of improved physicochemical stability, such as lower moisture sorption compared to conventional sucrose-based lyophilized formulation. Therefore, ALC loaded with fenofibrate was prepared and evaluated for its physicochemical properties and in vitro drug release profiles under sink conditions as well as supersaturating conditions. In vivo performance of the formulation was also evaluated in rats.
Materials and methods
Materials
Fenofibrate, aminopropyltriethoxysilane (APTES, 99%), sucrose, polyoxyethylene (20)-sorbitan monooleate (Tween® 80), pancreatin (8× US Pharmacopeia [USP] standard), and sodium taurodeoxycholate (NaTDC) were purchased from Sigma-Aldrich (St Louis, MO, USA). Lecithin (~99.2% soybean-phosphatidylcholine) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Superba™ Krill Oil was kindly supplied by Aker BioMarine AS (Oslo, Norway). Tertiary butyl alcohol (TBA), magnesium chloride hexahydrate, sodium chloride, hydrochloric acid, potassium dihydrogen phosphate, sodium hydrogen phosphate, and phosphoric acid were obtained from Junsei Chemical Co. Ltd. (Tokyo, Japan). Acetonitrile was high-performance liquid chromatography (HPLC) grade (Merck, Darmstadt, Germany).
Preparation of aminoclay
3-Aminopropyl functionalized magnesium phyllosilicate (aminoclay) was prepared as described in a previous report.12 In brief, 3-aminopropyltriethoxysilane (2.6 mL, 11.7 mmol) was added dropwise with rapid stirring to magnesium chloride (1.68 g, 7.24 mmol) dissolved in ethanol (40 g). A white precipitate formed almost immediately and was stirred overnight. The resulting product was separated by centrifugation, washed with ethanol (3×50 mL), and dried in air at 40°C. For the exfoliation of obtained aminoclay, the bulk powder was dispersed in water and subject to ultrasonication for 10 minutes.
Preparation of ALC formulations
Drug crystals stabilized by ALC formulations were prepared by antisolvent precipitation coupled with immediate freeze-drying process.27 In brief, two separate solutions were prepared: one was aminoclay aqueous suspension, and the other was krill oil and fenofibrate dissolved in TBA. The ratios of aminoclay–krill oil, krill oil–fenofibrate, and water–TBA were varied, while the drug amount and the total volume of the solution were fixed as 40 mg and 10 mL, respectively (Table 1). The exfoliated aminoclay aqueous suspension was mixed with the TBA solution in glass vials under vigorous stirring. Immediately after mixing, it was rapidly frozen at −80°C and subsequently lyophilized. Lyophilization was performed using a lyophilizer (LYOPH-PRIDE 10R; Ilshin BioBase Co. Ltd, Dongducheon, South Korea). The frozen mixture was lyophilized at a shelf temperature of −39°C and a condenser temperature of −65°C for 24 hours. Thereafter, the shelf temperature was gradually increased to 20°C over 24 hours.
 | Table 1 Process and formulation parameters for single-factor analysis |
Scanning electron microscopy
The morphological characteristics of ALC formulation were determined using scanning electron microscopy (SEM). ALC formulation was spread on specimen stub using double-sided sticky tape, coated with platinum, and analyzed by a field-emission scanning electron microscope (S-4700; Hitachi, Tokyo, Japan).
X-ray powder diffraction
X-ray powder diffraction (XRPD) patterns were obtained on an X-ray diffractometer (X’Pert APD; PHILIPS, Amsterdam, the Netherlands) with CuKα radiation at 20 mA and 40 kV. Each sample (fenofibrate, aminoclay, and ALC) was mounted on a glass X-ray sample holder. Scans were recorded between 3° and 70° (2θ) with a step size of 0.05° and scanning speed of 2°/min. XRPD experiments were conducted at Korea Basic Science Institute (Daegu Center, Daegu, South Korea).
Differential scanning calorimetry
The differential scanning calorimetry (DSC) measurements were performed using a DSC Q2000 (TA Instruments, Ghent, Belgium). The samples were accurately weighed (5–15 mg) and placed into a hermetically sealed aluminum pan. The thermograms were obtained at the scanning rate of 2°C/min over a temperature range of −40°C to 200°C under an inert atmosphere flushed with nitrogen at a rate of 30 mL/min. The extent of relative drug crystallinity in each formulation was calculated based on the following equation:28
|
|
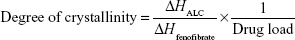
where ΔHALC and ΔHfenofibrate are the measured heat of fusion from ALC formulation and from untreated fenofibrate (100% crystalline), respectively. ΔHALC was calculated from the peak area of the melting endotherm in ALC formulation. ΔHfenofibrate was measured in a separate experiment of untreated fenofibrate.
Supersaturated dissolution study under pH-shift conditions
To select the optimal ALC formulations, drug release studies were conducted using the USP paddle method with 50 rpm at 37°C±0.5°C in the dissolution tester DT 1420 (ERWEKA, Heusenstamm, Germany). To mimic the pH transition from stomach to intestine, drug release studies were performed under varying pH conditions. Samples equivalent to 10 mg of fenofibrate were filled into hard gelatin capsules and exposed to 750 mL of 0.1 N HCl solution. After 2 hours, 250 mL of 0.2 M Na3PO4 solution preheated to 37°C±0.5°C was added to the vessel, adjusting the medium pH to approximately 6.8. One milliliter of each sample was collected at the predetermined time points (30, 60, 120, 130, 140, 150, 180, 240, 360, and 480 minutes) and filtered through syringe filters (0.2 μm). After sample collection, 1 mL of fresh medium was added to the vessel to maintain the constant volume of release media. Each filtrate was appropriately diluted with mobile phase, and the released drug amount was analyzed by an ultra-performance liquid chromatography (UPLC) assay.
Drug release characteristics of the optimal ALC formulation
After finalizing the optimal composition of ALC formulation, the dissolution characteristics of the optimal ALC formulation were examined by comparison with those of untreated drug and physical mixture (PM). Each formulation was filled into hard gelatin capsules, and drug amount in each formulation was equivalent to 40 mg of fenofibrate to reflect the commercially available dose strength. Dissolution studies were conducted as described earlier, except that the dissolution medium included 2% (w/v) Tween-80 to maintain the sink condition. Samples were collected at the predetermined time points (30, 60, 120, 130, 140, 150, 180, 240, 360, and 480 minutes), filtered, diluted, and analyzed by UPLC assay.
Dissolution studies in simulated intestinal fluid
Drug release profiles of ALC formulation were also examined in fasted state simulated intestinal fluid (FaSSIF) with/without pancreatin. Drug release studies were conducted using the USP paddle method with 50 rpm at 37°C±0.5°C in the dissolution tester DT 1420. FaSSIF with/without pancreatin was prepared as described by a previous report.29 Prewarmed media (100 mL, 37°C) was added to a 250 mL glass beaker containing ALC formulation (equivalent to 10 mg fenofibrate) and constantly stirred (50 rpm) at 37°C. The theoretical concentration of fenofibrate in the release medium was 100 μg/mL, representing a supersaturated state, as fenofibrate solubility in FaSSIF was 13.7 μg/mL.30 One milliliter of each sample was withdrawn at each selected time point (5, 15, 30, 60, 120, 180, 240, and 360 minutes), filtered through PTFE syringe filters (0.2 μm), diluted appropriately with mobile phase, and analyzed by UPLC assay.
Stability study
Storage stability of ALC formulation was evaluated at room temperature. After a 3-month storage, samples were collected and subjected to the drug release study under supersaturated condition to examine whether the drug release behavior of the ALC formulation was changed during storage.
Moisture sorption analysis
Moisture sorption analysis was performed on ALC formulation for comparison with the corresponding sucrose-based formulation (formulation composition was the same as with ALC, except replacing aminoclay by sucrose as a stabilizer). Freeze-dried sucrose-based formulation was prepared as reported previously.27 To set the 100% weight value, 100 mg of each sample was weighed in dried state. Then, each sample was exposed to 25°C and 53% relative humidity (RH), and its weight change was monitored for 10 hours.
Animal studies
The animal studies were carried out in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (USA) and the study protocol was approved by the review committee of Dongguk University. Male Sprague–Dawley rats (220–250 g) were supplied by Samtako Bio Co., Ltd (Osan, South Korea). All rats were given free access to tap water and a normal standard chow diet (Superfeed Company, Wonju, South Korea). Prior to the experiments, all rats were fasted for 18 hours and given free access to tap water. On the day of the experiment, rats were divided into two groups (six per group). Each formulation (untreated fenofibrate and ALC) was administered to rats via oral gavage at the dose equivalent to 20 mg/kg of fenofibrate. Untreated fenofibrate and ALC were dispersed in 0.5% methylcellulose solution and distilled water, respectively. Blood samples were collected from the femoral artery at 0.5, 1, 2, 3, 4, 8, 12, and 24 hours postdose and centrifuged at 9,900× g for 10 minutes. The obtained plasma samples were stored at −80°C until analyzed.
Analytical assay
In vitro samples
Drug concentration was measured by an established UPLC assay. The UPLC assay was conducted by using a Waters® ACQUITY UPLC® System (Hertfordshire, UK) and a reverse-phase C18 column (Kinetex 1.7 μ XB-C18 100A, 50×2.10 mm; Phenomenex, Torrance, CA, USA) with an injection volume of 5 μL. The mobile phase consisted of acetonitrile:water (80:20, v/v, pH adjusted to 3.5 with phosphoric acid). The flow rate was 0.2 mL/min, and the ultraviolet (UV) wavelength was set at 287 nm. Calibration curves were linear over the concentration range of 0.1–10 μg/mL (R2=0.999).
In vivo samples
The plasma concentration of fenofibric acid (an active metabolite of fenofibrate) was measured using a previously reported HPLC method.27 The HPLC system (Flexar; Perkin Elmer, Waltham, MA, USA) consisted of a UV detector, an automatic injector, two solvent delivery pumps, and an octadecylsilane column (Gemini C18, 4.6×150 mm, 5 μm; Phenomenex, Torrance, CA, USA). The mobile phase was composed of acetonitrile:0.2% phosphoric acid solution (50/50, v/v). The detection wavelength was 286 nm and flow rate was 1.0 mL/min at 40°C.
The plasma sample (150 μL) was mixed with 15 μL of ketoprofen (100 μg/mL) as an internal standard and vortexed for 5 minutes. Then, 415 μL of acetonitrile was added and mixed vigorously, followed by centrifugation at 10,000× g for 10 minutes. The supernatant was evaporated under vacuum, and the residue was reconstituted with 150 μL of the mobile phase. After vigorous mixing for 10 minutes, 20 μL of the supernatant was injected into the HPLC system. Calibration curves were linear over the concentration range of 0.05–100 μg/mL (R2=0.998). At three different concentration levels (0.1, 5.0, and 80 μg/mL), the intraday and interday variabilities were less than 9% and 12%, respectively.
Pharmacokinetic analysis and statistical analysis
Noncompartmental analysis was performed using WinNonlin® (Pharsight Co., Sunnyvale, CA, USA). All the data are expressed as mean ± standard deviation (SD). Statistical analysis was performed using Student’s t-test or one-way analysis of variance followed by Dunnett’s correction. Statistically significant difference was ascribed at P-values <0.05.
Results and discussion
Optimization of formulation variables
Fenofibrate is a highly lipophilic (log P=5.24) and neutral drug without ionizable groups,31 and its solubility is not affected by pH change.32 Fenofibrate belongs to a Biopharmaceutics Classification System (BCS) class II drug (low solubility and high permeability), and its poor aqueous solubility limits the oral absorption of fenofibrate.33,34 Therefore, in this study, ALC formulation was developed to improve the drug release profiles of fenofibrate. Single-factor analysis was used to optimize the formulation variables for the preparation of ALC formulation based on drug release profiles. The formulation variables and their compositions are summarized in Table 1.
Under the sink condition, all three test formulations released fenofibrate in a similar pattern – that is, an initial gradual release of 65%–85% in the first 60 minutes and complete drug release within 2 hours (data not shown). As the ability of drug release study under sink condition to discriminate the performance of different formulations was limited, drug release characteristics were evaluated under supersaturated condition.
The degree of supersaturation is defined as the concentration (C) of drug dissolved in the release media divided by the equilibrium solubility (Ceq) of crystalline drug in the release media (C/Ceq).35 While (C/Ceq)max indicates the maximum in vitro supersaturation levels, the area under the concentration time profile of the supersaturation drug release curve represents an overall estimate of the extent of supersaturation that takes into account both the degree and the duration of supersaturation.35 As shown in Figure 1 and Table 1, ALC formulations appeared to be useful to obtain and maintain supersaturated solution of fenofibrate. As seen in Figure 1, all the formulations had similar supersaturation profiles, reaching their maximum supersaturation at pH 1.2 and beginning to precipitate out of solution slowly after the pH shift to 6.8. Although the maximum supersaturation was not maintained after the pH shift to 6.8, supersaturation was maintained throughout the incubation period. ALC formulations could achieve supersaturation probably because of particle size reduction as well as improved wettability. As a surfactant and stabilizer during antisolvent crystallization, omega-3 phospholipids in krill oil could effectively coat the surface of drug particles and inhibit the growth of particles, leading to the smaller particles of fenofibrate.36,37 In addition, drug particles coated by the phospholipids were interdispersed and further stabilized in the aminoclay matrix to prevent aggregation during lyophilization.38,39 Aminoclay could also enhance the wettability.12,13,23 Considering the continuous drug movement across the intestinal membrane during the absorption process in vivo, the supersaturation achieved by ALC formulations could be maintained for longer time in vivo.
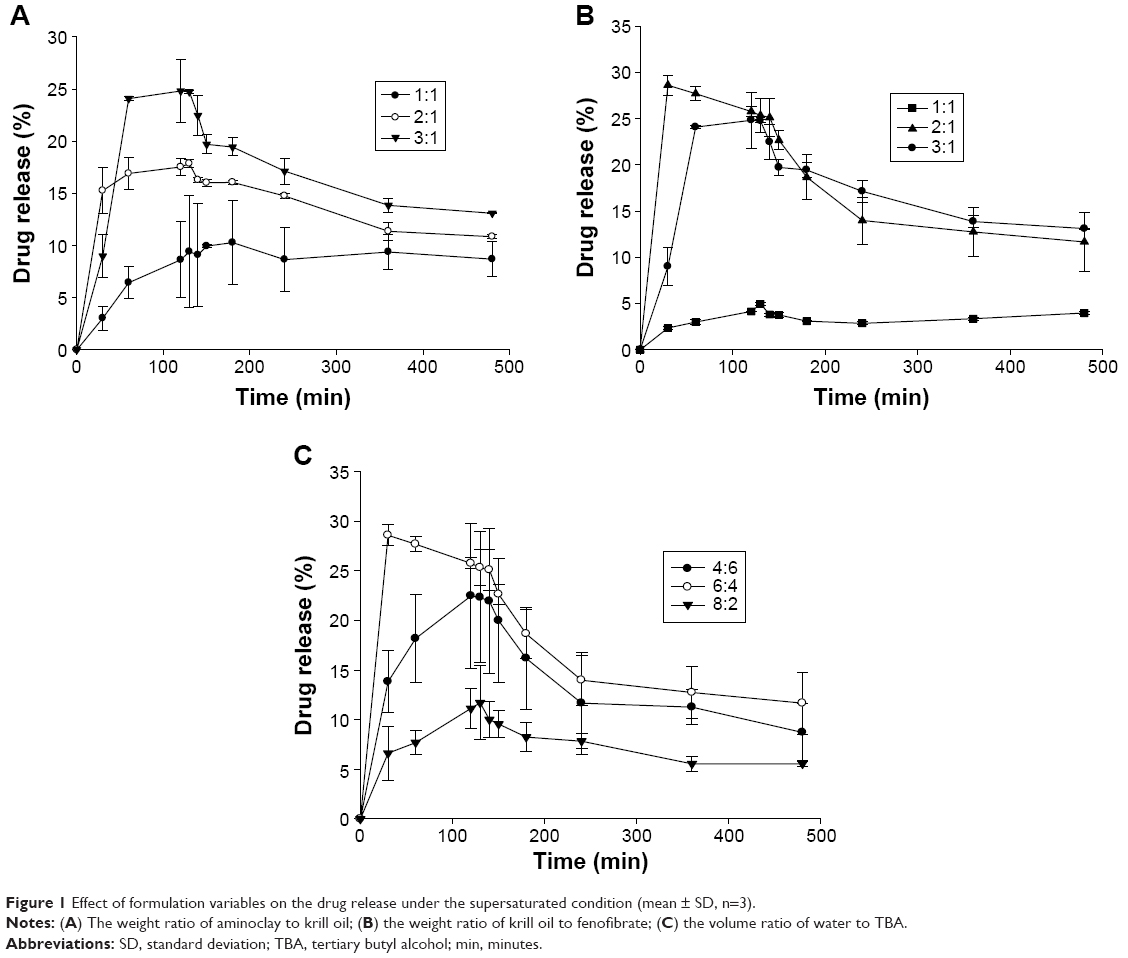 | Figure 1 Effect of formulation variables on the drug release under the supersaturated condition (mean ± SD, n=3). |
Taking together all the results of the single-factor experiments, the optimal composition of the ALC formulation was determined to be a 3:1 (w/w) ratio of aminoclay to krill oil, a 2:1 (w/w) ratio of krill oil to fenofibrate, and a 6:4 (w/w) ratio of antisolvent to solvent to maximize the drug dissolution.
Structural and morphological characteristics
The morphological characteristics of the ALC formulation were investigated by SEM. As shown in Figure 2, the ALC formulation exhibited a homogeneous blend of all ternary components in microparticles, which was about 1–2 μm, indicating that fine drug particles were formed by an antisolvent precipitation coupled with immediate freeze-drying.
 | Figure 2 SEM image of ALC formulation. |
XRPD diffractograms in Figure 3 indicated the characteristic crystalline peaks of untreated fenofibrate at 2θ of 12, 14.5, 16.2, 16.8, and 22.4. For the ALC formulation, the crystalline peaks of fenofibrate were still maintained with decreased intensity, demonstrating the presence of fenofibrate microparticles in the crystalline state. In addition, the characteristic peak of aminoclay was found at the same 2θ position, revealing that the structural framework of aminoclay was retained in the antisolvent precipitation with freeze-drying process.
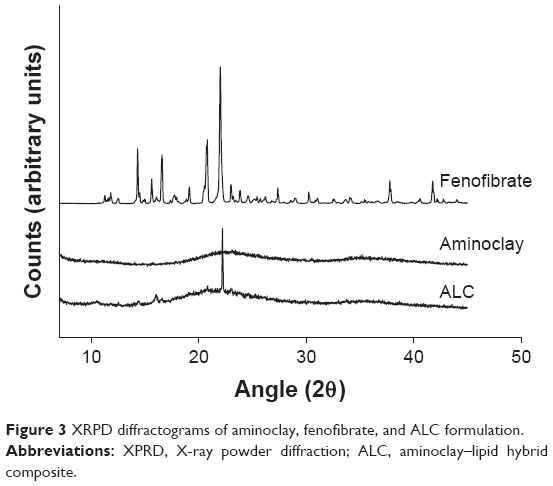 | Figure 3 XRPD diffractograms of aminoclay, fenofibrate, and ALC formulation. |
Thermograms of untreated fenofibrate, aminoclay, krill oil, and ALC formulation between −40°C and 200°C are presented in Figure 4. As can be seen, both aminoclay and ALC formulation showed an endothermic peak of about 150°C. Untreated fenofibrate exhibited a melting peak at 81°C, while the melting peak was shifted to 77°C in the ALC formulation. The degree of crystallinity of fenofibrate in the ALC formulation was calculated to be approximately 70%, indicating that fenofibrate microparticles are, to a certain extent, in amorphous state.
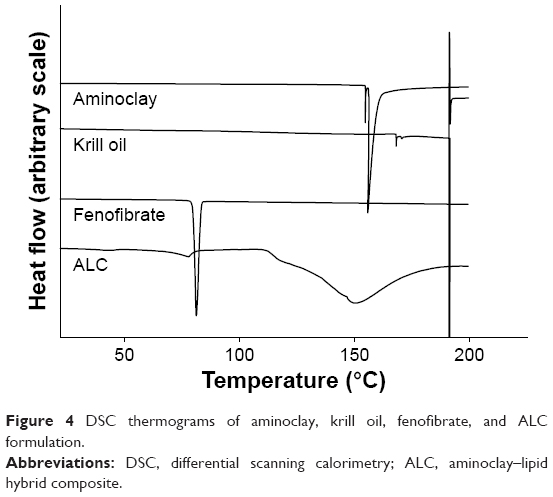 | Figure 4 DSC thermograms of aminoclay, krill oil, fenofibrate, and ALC formulation. |
Dissolution of the optimized ALC formulation
Dissolution profiles of fenofibrate from the ALC formulation were evaluated in water-under-the-sink condition and compared to those from the untreated powder formulation and the PM. As shown in Figure 5, the ALC formulation significantly improved drug dissolution rates as well as the extent of drug release. The ALC formulation achieved almost complete dissolution within 30 minutes, while the untreated drug and the PM exhibited drug release less than 15%.
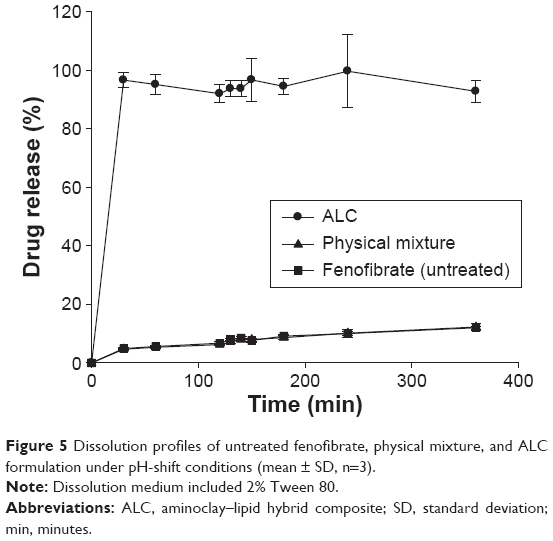 | Figure 5 Dissolution profiles of untreated fenofibrate, physical mixture, and ALC formulation under pH-shift conditions (mean ± SD, n=3). |
In the present formulation, phospholipids in krill oil could improve the solvation of dissolved drug molecules, thereby increasing the activation energy required for desolvation during nucleation and crystal growth.35 In addition, adsorption of phospholipid molecules to the surface of embryonic crystals of fenofibrate (with hydrophobic tails and anionic sulfate groups oriented toward the crystal and the surrounding water, respectively) may provide electrostatic repulsion to prevent agglomeration. Furthermore, drug particles coated by the phospholipids were dispersed in the aminoclay matrix, leading to the prevention of particle aggregation during lyophilization.38,39 Therefore, the enhanced drug dissolution from ALC formulation may be explained by the reduced particle size and increased surface area. The improved surface-wetting by the phospholipids and aminoclay in the formulation may also contribute to further enhancement in drug dissolution.12,13,23
To evaluate the impact of lipolysis, drug release profiles of ALC formulation were also examined in FaSSIF with/without pancreatin. As shown in Figure 6, the release profile of ALC formulation in FaSSIF with pancreatin was significantly lower than the corresponding release profile in FaSSIF without pancreatin. The presence of pancreatin greatly slowed down drug release. Given that phospholipids in ALC formulation would help solubilization of fenofibrate, the presence of pancreatin would facilitate the digestion of phospholipids, which affects the solubilization capacity and decreases the extent of drug release. However, as lipase activity in vivo may be lower, particularly in the fasted state, compared to that in vitro, the impact of lipolysis on the drug release can be overestimated in vitro.40 It has been reported that the specific activities of human pancreatic lipase on a test meal of triglyceride are up to 800-fold lower in vivo compared to in vitro assay conditions.41 In FaSSIF without pancreatin, supersaturation was achieved with maximum drug release at 60 minutes, followed by a gradual decline, and maintained supersaturation for 6 hours. In contrast to in vitro experiments, drug absorption process in vivo may reduce drug precipitation from supersaturated solutions via the continuous drug movement across intestinal membrane. Particularly, if absorption is fast enough to precede drug precipitation, the high degree of supersaturation may be beneficial to improve the bioavailability of highly permeable drugs such as fenofibrate.42
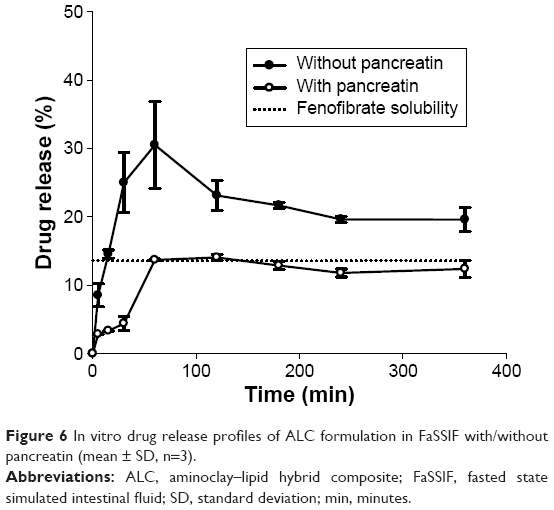 | Figure 6 In vitro drug release profiles of ALC formulation in FaSSIF with/without pancreatin (mean ± SD, n=3). |
Stability test and moisture sorption study
As shown in Figure 7, the drug release behavior of the ALC formulation was unchanged after a 3-month storage at room temperature. Although amorphous solid dispersion often suffers from recrystallization during storage and subsequent changes in drug release behavior, the drug in ALC formulation is mainly in stable crystalline state.
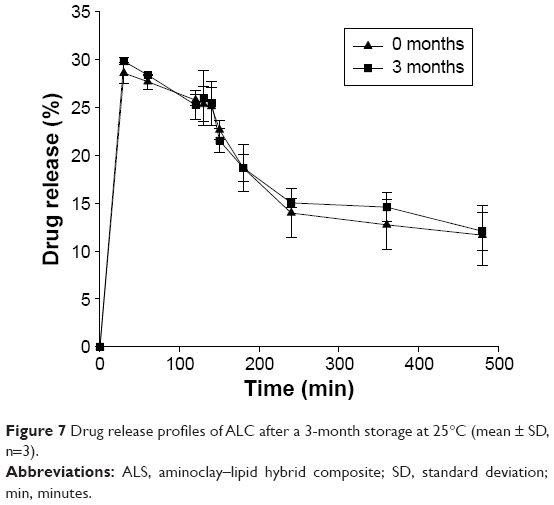 | Figure 7 Drug release profiles of ALC after a 3-month storage at 25°C (mean ± SD, n=3). |
For the freeze-drying process, sugar is commonly used as a cryoprotectant agent. However, as sugar is amorphous, hygroscopic, and tends to absorb moisture, it may negatively affect the flowability and drug release characteristics of the formulation.43 Therefore, in this study, stability of ALC formulation was compared to a conventional sucrose-based formulation, the composition of which was the same as ALC except that it had sucrose instead of aminoclay. As shown in Figure 8, ALC formulation exhibited a significantly lower moisture sorption compared to the sucrose-based formulation. Gas barrier property of aminoclay layers may also provide additional advantage.44
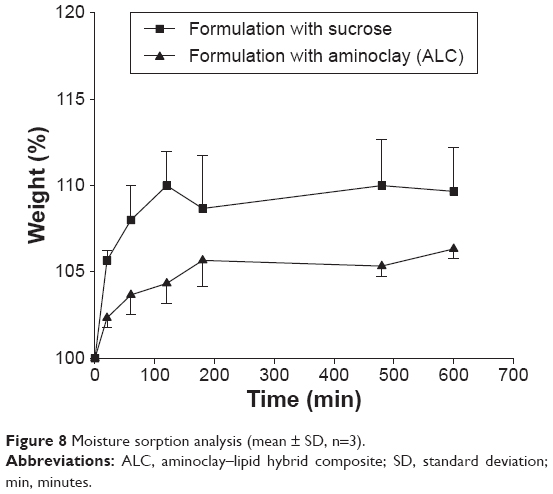 | Figure 8 Moisture sorption analysis (mean ± SD, n=3). |
In vivo pharmacokinetics
Oral pharmacokinetics of fenofibrate was evaluated based on the plasma concentration of fenofibric acid, an active metabolite, since oral fenofibrate is completely metabolized to fenofibric acid.45 The plasma concentration–time profiles of fenofibric acid after the oral administration of untreated fenofibrate or ALC formulation are illustrated in Figure 9. The pharmacokinetic parameters are summarized in Table 2.
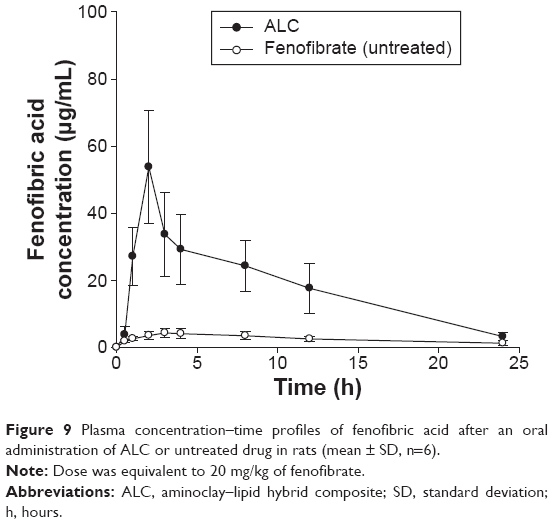 | Figure 9 Plasma concentration–time profiles of fenofibric acid after an oral administration of ALC or untreated drug in rats (mean ± SD, n=6). |
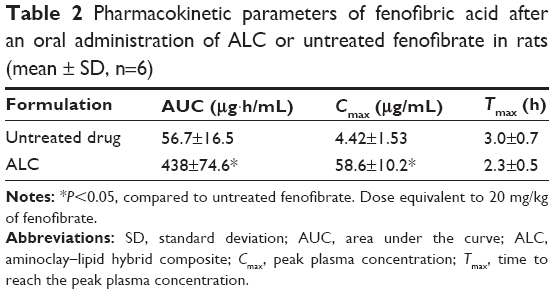 | Table 2 Pharmacokinetic parameters of fenofibric acid after an oral administration of ALC or untreated fenofibrate in rats (mean ± SD, n=6) |
Although untreated fenofibrate achieved low systemic exposure (peak plasma concentration [Cmax] and area under the curve [AUC] of 4.42±1.53 μg/mL and 56.7±16.5 μg·h/mL), ALC formulation significantly (P<0.05) increased Cmax and AUC (58.6±10.2 μg/mL and 438±74.6 μg·h/mL, respectively). Consequently, the ALC formulation achieved an approximately seven- to eight-fold higher oral exposure of fenofibric acid compared to the untreated fenofibrate. In addition, drug absorption was more rapid with the ALC formulation. In conjunction with in vitro dissolution study, rapid and enhanced drug dissolution via the ALC formulation led to faster and greater drug absorption in vivo.
Conclusion
ALC formulation of fenofibrate was prepared by the antisolvent precipitation method coupled with immediate freeze-drying. The optimal composition of the formulation was determined as the ratios of aminoclay to krill oil of 3:1 (w/w), krill oil to fenofibrate of 2:1 (w/w), and antisolvent to solvent of 6:4 (v/v). Drug in ALC formulation was mainly present in crystalline form, with the particle size of 1–2 μm. The ALC formulation of fenofibrate was effective at increasing the drug dissolution rate as well as the extent of drug release, enhancing the oral bioavailability of fenofibrate in rats.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2061289) and by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (No. 2012M3A9C1053532).
Disclosure
The authors report no conflicts of interest in this work.
References
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420:1–10. | ||
Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16:36. | ||
Mosharraf M, Nyström C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int J Pharm. 1995;122:35–47. | ||
Sarnes A, Kovalainen M, Häkkinen MR, et al. Nanocrystal-based per-oral itraconazole delivery: superior in vitro dissolution enhancement versus Sporanox® is not realized in in vivo drug absorption. J Control Release. 2014;180:109–116. | ||
Jinno J, Kamada N, Miyake M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111:56–64. | ||
De Waard H, Hinrichs W, Frijlink H. A novel bottom–up process to produce drug nanocrystals: controlled crystallization during freeze-drying. J Control Release. 2008;128:179–183. | ||
Rege BD, Kao JP, Polli JE. Effects of nonionic surfactants on membrane transporters in Caco-2 cell monolayers. Eur J Pharm Sci. 2002;16:237–246. | ||
Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev. 2008;60:625–637. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287. | ||
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11:S93–S98. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–248. | ||
Yang L, Choi S-K, Shin H-J, Han HK. 3-aminopropyl functionalized magnesium phyllosilicate as an organoclay based drug carrier for improving the bioavailability of flurbiprofen. Int J Nanomedicine. 2013;8:4147–4155. | ||
Yang L, Shao Y, Han HK. Improved pH-dependent drug release and oral exposure of telmisartan, a poorly soluble drug through the formation of drug-aminoclay complex. Int J Pharm. 2014;471:258–263. | ||
Kesisoglou F, Panmai S, Wu Y. Nanosizing – oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59:631–644. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3:785–796. | ||
Guichard JP, Blouquin P, Qing Y. A new formulation of fenofibrate: suprabioavailable tablets. Curr Med Res Opin. 2000;16:134–138. | ||
Sauron R, Wilkins M, Jessent V, Dubois A, Maillot C, Weil A. Absence of a food effect with a 145 mg nanoparticle fenofibrate tablet formulation. Int J Clin Pharmacol Ther. 2006;44:64–70. | ||
Liu Y, Kathan K, Saad W, Prud’homme RK. Ostwald ripening of β-carotene nanoparticles. Phys Rev Lett. 2007;98:036102. | ||
Verma S, Kumar S, Gokhale R, Burgess DJ. Physical stability of nanosuspensions: investigation of the role of stabilizers on Ostwald ripening. Int J Pharm. 2011;406:145–152. | ||
Chaubal MV, Popescu C. Conversion of nanosuspensions into dry powders by spray drying: a case study. Pharm Res. 2008;25:2302–2308. | ||
Van Eerdenbrugh B, Froyen L, Van Humbeeck J, Martens JA, Augustijns P, Van den Mooter G. Drying of crystalline drug nanosuspensions – the importance of surface hydrophobicity on dissolution behavior upon redispersion. Eur J Pharm Sci. 2008;35:127–135. | ||
Cerdeira AM, Mazzotti M, Gander B. Formulation and drying of miconazole and itraconazole nanosuspensions. Int J Pharm. 2013;443:209–220. | ||
Yang L, Shao Y, Han HK. Preparation and in vitro/in vivo characterization of tranilast-AMP clay complex for improving drug dissolution and bioavailability. Arch Pharm Res. 2014;37:1554–1559. | ||
Harper CR, Jacobson TA. Usefulness of omega-3 fatty acids and the prevention of coronary heart disease. Am J Cardiol. 2005;96:1521–1529. | ||
Daviglus ML, Stamler J, Orencia AJ, et al. Fish consumption and the 30-year risk of fatal myocardial infarction. N Engl J Med. 1997;336:1046–1053. | ||
Ascherio A, Rimm EB, Stampfer MJ, Giovannucci EL, Willett WC. Dietary intake of marine n-3 fatty acids, fish intake, and the risk of coronary disease among men. N Engl J Med. 1995;332:977–983. | ||
Yang L, Shao Y, Han HK. Development of omega-3 phospholipid-based solid dispersion of fenofibrate for the enhancement of oral bioavailability. Eur J Pharm Sci. 2015;78:103–110. | ||
Chen HL, Hwang JC. Some comments on the degree of crystallinity defined by the enthalpy of melting. Polymer. 1995;36:4355–4357. | ||
Jantratid E, Janssen N, Chokshi H, Tang K, Dressman JB. Designing biorelevant dissolution tests for lipid formulations: case example–lipid suspension of RZ-50. Eur J Pharm Biopharm. 2008;69:776–785. | ||
Vogt M, Kunath K, Dressman JB. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: comparison with commercial preparations. Eur J Pharm Biopharm. 2008;68:283–288. | ||
Munoz A, Guichard JP, Reginault P. Micronised fenofibrate. Atherosclerosis. 1994;110:S45–S48. | ||
Jamzad S, Fassihi R. Role of surfactant and pH on dissolution properties of fenofibrate and glipizide – a technical note. AAPS PharmSciTech. 2006;7:E17–E22. | ||
Guay DR. Update on fenofibrate. Cardiovasc Drug Rev. 2002;20:281–302. | ||
Guivarc’h P-H, Vachon MG, Fordyce D. A new fenofibrate formulation: results of six single-dose, clinical studies of bioavailability under fed and fasting conditions. Clin Ther. 2004;26:1456–1469. | ||
Overhoff KA, McConville JT, Yang W, Johnston KP, Peters JI, Williams RO III. Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm Res. 2008;25:167–175. | ||
Zu Y, Wu W, Zhao X, et al. Enhancement of solubility, antioxidant ability and bioavailability of taxifolin nanoparticles by liquid antisolvent precipitation technique. Int J Pharm. 2014;471:366–376. | ||
Zu Y, Li N, Zhao X, et al. In vitro dissolution enhancement of micronized L-nimodipine by antisolvent re-crystallization from its crystal form H. Int J Pharm. 2014;464:1–9. | ||
Strauss G, Schurtenberger P, Hauser H. The interaction of saccharides with lipid bilayer vesicles: stabilization during freeze-thawing and freeze-drying. Biochim Biophys Acta. 1986;858:169–180. | ||
Miyajima K. Role of saccharides for the freeze-thawing and freeze drying of liposome. Adv Drug Deliv Rev. 1997;24:151–159. | ||
Griffin BT, Kuentz M, Vertzoni M, et al. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: case studies with fenofibrate. Eur J Pharm Biopharm. 2014;86:427–437. | ||
Carrière F, Renou C, Lopez V, et al. The specific activities of human digestive lipases measured from the in vivo and in vitro lipolysis of test meals. Gastroenterology. 2000;119:949–960. | ||
Williams HD, Trevaskis N, Yeap YY, Anby MU, Pouton CW, Porter CJ. Lipid-based formulations and drug supersaturation: harnessing the unique benefits of the lipid digestion/absorption pathway. Pharm Res. 2013;30:2976–2992. | ||
Te Booy MP, de Ruiter RA, de Meere AL. Evaluation of the physical stability of freeze-dried sucrose-containing formulations by differential scanning calorimetry. Pharm Res. 1992;9:109–114. | ||
Datta K, Kulkarni C, Eswaramoorthy M. Aminoclay: a permselective matrix to stabilize copper nanoparticles. Chem Commun. 2010;46: 616–618. | ||
Hanafy A, Spahn-Langguth H, Vergnault G, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59:419–426. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.